
Abstract
A consortium comprised of an engineered Escherichia coli DH5α and a natural pentachlorophenol (PCP) degrader, Sphingobium chlorophenolicum ATCC 39723, was assembled for degradation of hexachlorobenzene (HCB), a persistent organic pollutant. The engineered E. coli strain, harbouring a gene cassette (camA + camB + camC) that encodes the F87W/Y96F/L244A/V247L mutant of cytochrome P-450cam (CYP101), oxidised HCB to PCP. The resulting PCP was then further completely degraded by ATCC 39723. The results showed that almost 40 % of 4 μM HCB was degraded by the consortium at a rate of 0.033 nmol/mg (dry weight)/h over 24 h, accompanied by transient accumulation and immediate consumption of the intermediate PCP, detected by gas chromatography. In contrast, in the consortium comprised of Pseudomonas putida PaW340 harbouring camA + camB + camC and ATCC 39723, PCP accumulated in PaW340 cells but could not be further degraded, which may be due to a permeability barrier of Pseudomonas PaW340 for PCP transportation. The strategy of bacterial co-culture may provide an alternative approach for the bioremediation of HCB contamination.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In past decades, mass production and intensive use of hexachlorobenzene (HCB) in agriculture and industrial processes has led to widespread contamination of the environment. HCB is of great concern as one of the persistent organic pollutants (POPs) named in the Stockholm Convention, due to its tendency to accumulate along the food chain and its resistance to degradation, together with its harmful effects on human beings and the environment. Although HCB production has been prohibited in most countries for many years, it is still being generated inadvertently as a by-product in several chemical processes, such as the manufacture of chlorinated solvents, chlorinated aromatics and pesticides (Jacoff et al. 1986) and is also released to the environment by incomplete combustion. HCB levels in human tissues and pine needles appear to have increased in China recently, particularly in the industrialised north-east of China (Kunisue et al. 2004; Xu et al. 2004).
Microbial biodegradation of HCB, as a potential means of transforming HCB to harmless compounds, has been investigated in the last few decades. In particular, microbial reductive dechlorination of HCB has been intensively studied. Pure culture of Dehalococcoides sp. CBDB1 (Jayachandran et al. 2003) and several mixed cultures (Chang et al. 1998; Fathepure et al. 1988; Yeh and Pavlostathis 2001) have been reported to reductively dechlorinate HCB to less chlorinated benzenes, such as 1,3,5-trichlorobenzene, and 1,2-, 1,3- and 1,4-dichlorobenzene under anaerobic conditions. However, the resulting products could not be further degraded, and moreover, they may lead to serious secondary environmental contamination. There is little information available on microbial metabolism of HCB under aerobic conditions apart from one HCB-mineralising bacterium (Nocardioides sp. PD653) (Takagi et al. 2009). Nevertheless, the F87W/Y96F/L244A/V247L mutant of CYP101 (cytochrome P-450cam, encoded by camC) has been shown to be capable of catalysing the oxidation of HCB to PCP in the presence of its physiological electron transfer co-factors putidaredoxin reductase (PdR, encoded by camA +) and putidaredoxin (Pd, encoded by camB +) which mediate the transfer of electrons from NADH co-factor to CYP101 (Chen et al. 2002). In our previous work, a gene cassette (camA + camB + camC) encoding the F87W/Y96F/L244A/V247L cytochrome P-450cam variant was introduced into the natural PCP utiliser Sphingobium chlorophenolicum ATCC 39723, to degrade HCB (Yan et al. 2006). However, the engineered S. chlorophenolicum strain was not appropriate for HCB bioremediation because of its low degradation activity. Therefore, construction of a complete HCB catabolic pathway by combining bacteria with complementary metabolic pathways into functional assemblages could be an alternative strategy to dispose of HCB contamination. In this study, a consortium comprised of engineered Escherichia coli harbouring the camA + B + C cassette and the natural PCP utiliser ATCC 39723 was assembled for biodegradation of HCB without any accumulation of toxic intermediates (Fig. 1).

A scheme for the proposed catabolic pathway for degradation of HCB by an engineered bacterial consortium
Materials and methods
Enzymes and chemicals
Pyrobest™ DNA polymerase, restriction endonucleases and T4 DNA ligase were purchased from Takara Company (Dalian, China). HCB, PCP, 1,3,5-tribromobenzene, 2,4,6-tribromophenol were obtained from Sigma-Aldrich (St. Louis, MO, USA). Ethanol and acetone were of high-performance liquid chromatography (HPLC) grade and all other chemicals were commercially available and of analytical grade.
Strains and media
The bacterial strains and plasmids used in this study are listed in Table 1. E. coli strains were grown at 37 °C in Luria–Bertani (LB) medium or mineral M9 medium (Sambrook and Russell 2001) supplemented with glucose (0.2 %, w/v) as a carbon source. Pseudomonas putida PaW340 was grown at 30 °C in LB or mineral M9 medium supplemented with glucose (0.2 %, w/v) and tryptophan (40 μg/mL). S. chlorophenolicum ATCC 39723 was grown at 30 °C in glutamate (0.4 %) mineral salts (GMS) medium (Saber and Crawford 1985). When necessary, ampicillin, kanamycin and spectinomycin were added at final concentrations of 100, 50, and 1 mg/mL, respectively.
DNA preparation and manipulation
Plasmid DNA was isolated using the alkaline lysis method, with the exception of plasmid pVLT33 and its derivatives which were isolated using the boiling lysis method (Walker and Keasling 2002). A DNA gel extraction kit (Hangzhou Vitagene Biochemical Technique Co. Ltd, Hangzhou, China) was used for recovery of DNA fragments from agarose gels. E. coli was transformed with the CaCl2 method (Sambrook and Russell 2001). Transfer of pVLT33 and its derivatives into strain PaW340 was performed with triparental matings (Williams and Murray 1974). Nucleotide sequences were determined by Sangon Biotech Co. Ltd (Shanghai, China).
Construction of recombinant plasmids
The promoterless camA + B + C catabolic cassette was PCR-amplified from the plasmid pCWSGB-camC using Pyrobest™ DNA polymerase with primers A1 (5′-TAAGAATTCATGAACGCAAACGACA-3′; EcoRI restriction site underlined) and A2 (5′-TTCAAGCTTTTATACCGCTTTGGTAGT-3′; HindIII restriction site underlined). The amplified PCR product (2,974 bp) was cut with EcoRI and HindIII and ligated into the same digested pVLT33, placing the camA + B + C cassette downstream of the tac promoter to form the plasmid pZWY010.
Resting cell preparation
Single colonies of transformants or transconjugants were used to inoculate 10 mL of LB supplemented with the appropriate antibiotics. The cultures were grown at 37 °C for E. coli or at 30 °C for P. putida PaW340 overnight and then inoculated into 1 L of LB. After recombinant strains were grown to an optical density at 600 nm (OD600) of 0.6, protein expression was induced at 30 °C by the addition of 0.4 mM isopropyl thiogalactoside (IPTG) for DH5α [pCWSGB-camC], or 1 mM IPTG for both DH5α [pZWY010] and PaW340 [pZWY010]. After incubation for 16 h, the cells were harvested by centrifugation at 5,000×g for 10 min, and then washed twice in M9 medium, before they were resuspended in 500 mL of the same medium to an OD600 of 3.0. Control experiments were done with strains transformed with corresponding plasmids without insert.
Biotransformation of HCB
The substrate HCB was added into 500 mL M9 medium as 1 mM stock in ethanol to a final concentration of 4 μM to initiate whole cell biotransformation and the culture was incubated at 30 °C with agitation (200 rpm).
After incubation for 24 h, 1/10 of the original volume of ATCC 39723 culture with OD600 of 3.0 was added to each mixture and sampled at two-hourly intervals. At each sample time, two 20-mL aliquots of the cultures were withdrawn from each flask to 50-mL centrifuge tubes (test tube with a Teflon-lined screw cap) for qualitative and quantitative analysis.
The samples were further derivatized and analysed with gas chromatography (GC) and GC-mass spectrometry (GC–MS) as previously described (Yan et al. 2006), using the scanning model (Scan) and selective ion monitoring (SIM). Internal standards—1,3,5-tribromobenzene for HCB and 2,4,6-tribromophenol for PCP—were added to the mixture to standardise both the extraction and analysis steps.
Results
E. coli DH5α and P. putida Paw340 expressing camA + B + C transform HCB to PCP
The promoterless camA + B + C cassette was PCR-amplified from the plasmid pCWSGB-camC and cloned into a broad-host range expression vector pVLT33, resulting in the plasmid pZWY010. To characterise the expression of camA + B + C in different genetic backgrounds, strains E. coli DH5α [pCWSGB-camC], E. coli DH5α [pZWY010] and P. putida Paw340 [pZWY010] were constructed and whole cell HCB biotransformation was conducted after IPTG induction. The samples were collected and analysed by GC to monitor the progress of HCB degradation. The product of HCB oxidisation was identified to be PCP by GC–MS analysis using an NIST02 MS Data Library, based on comparisons of mass spectra of the derivative of the product with those of the derivative of authentic PCP.
Molecular ion peak of the derivative of the product was observed at 306 (M+) m/z, and ions observed at 308 (M+2), 310 (M+4) and 312 (M+6) m/z were identified as isotopic ions containing 1, 2 and 3 of 37Cl(s), respectively (Fig. 2). Their relative intensities are consistent with a natural abundance of 76 % for 35Cl and 24 % for 37Cl. Major fragments were observed at 264 and 235 m/z. The ion observed at 264 m/z is ascribed to the loss of CH2=C=O from the molecular ion of 306 m/z and the ion at 235 m/z corresponds to the loss of HCO from the ion of 264 m/z. The mass fragmentation patterns of the derivative of intermediate are identical to those of the derivative of authentic PCP, pentachlorophenyl acetate in the NIST02 mass spectral database. Whereas in the control experiments with strains E. coli DH5α or PaW340 harboring pVLT33 vector, the amount of HCB has little change and the corresponding peak of the PCP derivative was not detected in either case. These data clearly indicated that all the engineered strains could convert HCB to PCP in vivo.

GC–MS analysis of acetic anhydride-derivatized supernatant of engineered E. coli DH5α [pCWSGB-camC] culture with feeding of HCB. Comparison of the mass spectra of intermediate PCP (a) and the derivative of authentic PCP (b) in the NIST02 mass spectral database. The molecular ion is indicated at 306 (M+) m/z. For MS diagrams, the ordinates show the abundance (%) of each ionic species, while the abscissas correspond to m/z values
Both HCB removal and PCP formation was quantified by GC analysis; the results showed that PCP was formed concomitantly during the oxidation of HCB. In three different cultures, the amount of substrate HCB oxidised by E. coli DH5α [pZWY010], P. putida PaW340 [pZWY010] and E. coli DH5α [pCWSGB-camC] reached concentrations of 0.74, 0.82 and 1.79 μΜ over 24 h, respectively. The oxidation rates of HCB increased in the following order: E. coli DH5α [pZWY010] (0.014 nmol/mg(dry weight)/h), PaW340 [pZWY010] (0.015 nmol/mg(dry weight)/h) and DH5α [pCWSGB-camC] (0.033 nmol/mg(dry weight)/h).
Degradation of HCB via PCP intermediate
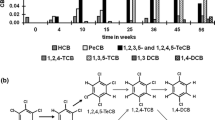
The amount of PCP produced by monoculture DH5α [pZWY010], PaW340 [pZWY010] and DH5α [pCWSGB-camC] reached concentrations of 0.41, 0.52 and 0.78 μM in 24 h, respectively. To completely degrade HCB, S. chlorophenolicum ATCC 39723 with PCP-mineralising capability was added into each mixture. As shown in Fig. 3, when DH5α [pCWSGB-camC] or DH5α [pZWY010] was co-cultured with strain ATCC 39723, the intermediate PCP accumulated during the first 24 h was completely degraded within 2–3 h after the addition of ATCC 39723. These data indicated that co-culture of engineered strains DH5α and ATCC 39723 could function cooperatively to degrade HCB (Fig. 3a, b). Almost 40 % of 4 μM HCB was degraded by the consortium of DH5α [pCWSGB-camC] and ATCC 39723 over 24 h at a rate of 0.033 nmol/mg(dry weight)/h. However, when PaW340 [pZWY010] was co-cultured with strain ATCC 39723, the intermediate PCP could not be further degraded (Fig. 3c).

Time course of HCB degradation by engineered bacterial consortia. The substrate HCB was added as 1 mM stock in ethanol to a final concentration of 4 μM to initiate whole cell biotransformation. After 24 h incubation, 1/10 of the original volume of ATCC 39723 culture with OD600 of 3.0 was added to each mixture and sampled at 2-hourly intervals. Experiments were performed in triplicate and data are presented as average values. a The consortium of E. coli DH5α [pCWSGB-camC] and ATCC 39723. b The consortium of E. coli DH5α [pZWY010] and ATCC 39723. c The consortium of PaW340 [pZWY010] and ATCC 39723
Low permeability of PCP in Pseudomonas may prevent PCP from being further degraded by ATCC 39723
A previous study showed that the PCP-degrading gene cluster in strain ATCC 39723 was inducible by PCP (Cai and Xun 2002) and our preliminary study showed that PCP could trigger degradation at a low concentration of 0.2 μM. The concentration of PCP formed from HCB oxidation in the supernatant of whole cell biotransformation was greater than the threshold for PCP induction in strain ATCC 39723 (Fig. 3). PCP subsequently disappeared after addition of the PCP degrader ATCC 39723 in the two cases where E. coli strains were involved, but it was not degraded in the third case where a Pseudomonas strain was used as an expression host. The potential reason for this may lie in limitation of the mass transport of substrates across the cell wall and membrane due to the lower permeability of PCP in Pseudomonas.
To confirm this hypothesis, the culture samples were centrifuged at 6,000×g for 10 min. The supernatants were collected and adsorbed onto an Argonaut/IST ISOLUTE C18 (EC) column (500 mg of matrix, 6 mL) to be concentrated, while the bacterial cells in the pellets were disrupted by ultrasonification and then extracted with a 1:1 (v/v) mixture of acetone and n-hexane. PCP could be detected after it was released from the lysed cells during the procedure of sonication and extraction. For samples from strain PaW340 [pZWY010], PCP was detected in the pellets but not in the supernatant by GC analysis. However, PCP was detected in both the supernatant and pellets from samples with strains DH5α [pCWSGB-camC] and DH5α [pZWY010]. Thus, it was proved that the permeability barrier of strain PaW340 prevented the PCP produced from moving out of cells, making it undegradable by the PCP utiliser.
Similar rates of HCB degradation were also observed in all cases if monoculture E. coli DH5α [pCWSGB-camC] or E. coli DH5α [pZWY010] or PaW340 [pZWY010] was mixed with ATCC 39723 when the reaction first started, and PCP was detected only when PaW340 [pZWY010] was involved.
Discussion
With the rapid development of modern industry, numerous recalcitrant chemicals with structural elements or substituents that do not (or seldom) occur in nature, have been artificially synthesised. But microorganisms have not yet evolved appropriate metabolic pathways to degrade these kinds of foreign compounds (Pieper and Reineke 2000). To date, several strategies have been developed for use of microorganisms for bioremediation. Genetically engineered microorganisms and gene bioaugmentation technologies have been applied to bioremediate xenobiotics in place of indigenous degrading microorganisms. These approaches involve rational design and genetic manipulation of metabolic routes, including the regulation of gene expression circuits to improve catalytic activities, the combination of series of metabolic modules to create novel metabolic routes, and the creation of engineered microorganisms with better performance in elimination of environmental pollutants. All of these metabolic engineering strategies aim to design effective biocatalysts for the target xenobiotics to avoid the formation of dead-end products or toxic metabolites. A large number of highly toxic compounds, including parathion (Walker and Keasling 2002), 2-chlorotoluene (Haro and de Lorenzo 2001), and HCB (Yan et al. 2006) have been successfully biodegraded using these strategies.
An alternative strategy to constructing a complete degradative pathway in a single microbe is to combine bacteria with complementary metabolic pathways into functional assemblages. Several highly toxic compounds have been biodegraded using this approach, including 4-chlorodibenzofuran (Arfmann et al. 1997), parathion (Gilbert et al. 2003) and methyl parathion (Li et al. 2008).
To address the resistance to degradation of HCB, particularly in soil environments, other technologies like phytoremediation have also been adopted. The rhizosphere of certain plant species, where the physicochemical and biological processes induced by the interaction between plants, microorganisms, the soil and pollutants take place, may be important for facilitating microbial degradation of HCB in soil (Zhou et al. 2013). This approach has important implications for detoxifying or transforming the higly toxic HCB to less toxic compounds even though HCB is metabolised to several toxic dechlorination products including pentachlorobenzene (PeCB), 1,2,3,4-, 1,2,3,5- and 1,2,4,5-tetrachlorobenzene, and 1,2,3-, 1,2,4- and 1,3,5-trichlorobenzene, rather than completely metabolized (Zhou et al. 2013).
To our knowledge, the use of defined bacterial co-culture to degrade HCB has not previously been reported. Tricistronic pCWSGB-camC has been constructed previously by cloning the genes encoding the three proteins of the CYP101 system in the order camA +, camB + and camC into the pCWori +vector. It was chosen for our study because this system has two powerful tac promoters in tandem and has been successfully used to express the triple fusion CYP101 system (Bell et al. 2001). In this study, a consortium comprised of an engineered E. coli strain and a natural PCP degrader was assembled for complete degradation of HCB without the accumulation of any toxic intermediate to non-toxic final products. The HCB was first transformed into PCP by the engineered strain harbouring camA + camB + camC, and then PCP was subsequently mineralised by its natural degrader S. chlorophenolicum ATCC 39723.
Although the consortium in this study was able to degrade HCB, it could not grow with HCB as the sole carbon and energy source. The specific activity of mutant cytochrome P-450cam (CYP101) on the substrate HCB may not have been enough to couple its substrate conversion levels productively to funnel sufficient PCP into the lower pathway. This problem could be resolved by increasing the enzyme-specific activity via gene shuffling or genome shuffling (Bosma et al. 2002; Dai and Copley 2004). This, however, still may not account entirely for the lack of growth observed. Another serious potential problem is the limitation of mass transport of substrates across the cell membrane. Due to their polarity or low water solubility resulting in a permeability barrier, substrates may be inhibited from interacting with the enzymes residing within the cell. This phenomenon appeared when PaW340 [pZWY010] grew in co-culture with S. chlorophenolicum ATCC 39723, and the intermediate PCP accumulated during the first 24 h could not be degraded, as shown in Fig. 3c. The complex cell envelope structure of P. putida PaW340, consisting of a cytoplasmic membrane, cell wall and outer membrane, prevented the intermediate PCP from contact with strain ATCC 39723. To enhance solubilisation-limited bioavailability, surfactants or specific PCP transporters could be used. It is important for a consortium to effectively transport substrates of interest to their degraders and this problem should be taken into consideration in the case of biodegradation with co-culture.
Initially, we were aiming to improve the degradation rate of HCB beyond that of single-culture ATCC 39723-containing gene cassette (camA + B + C) by using a constructed microbial consortium, particularly considering the advantage of homologous expression of the gene cassette originally from Pseudomonas. Our preliminary study has demonstrated that cultures of a bacterial consortium can degrade HCB, even though the degradation activity is not as high as expected. The work was still indispensable in identifying the need for future investigations on how to improve the bioavailability and the permeability of the substrates before this technology can become an effective and competitive approach for bioremediation of HCB. The data presented here not only open up a new opportunity for bioremediation of HCB in the environment, but also provide a principle for the potential application of metabolic engineering in the biodegradation of aromatic pollutants, particularly POPs.
Abbreviations
- HCB:
-
Hexachlorobenzene
- PCP:
-
Pentachlorophenol
References
Arfmann H, Timmis KN, Wittich R (1997) Mineralization of 4-chlorodibenzofuran by a consortium consisting of Sphingomonas sp. strain RW1 and Burkholderia sp. strain JWS. Appl Environ Microbiol 63:3458–3462
Bell SG, Harford-Cross CF, Wong LL (2001) Engineering the CYP101 system for in vivo oxidation of unnatural substrates. Protein Eng 14:797–802
Bosma T, Damborsky J, Stucki G, Janssen DB (2002) Biodegradation of 1,2,3-trichloropropane through directed evolution and heterologous expression of a haloalkane dehalogenase gene. Appl Environ Microbiol 68:3582–3587
Boyer HW, Roulland-Dussoix D (1969) A complementation of analysis of the restriction and modification of DNA in Escherichia coli. J Mol Biol 41:459–472
Cai M, Xun L (2002) Organization and regulation of pentachlorophenol-degrading genes in Sphingobium chlorophenolicum ATCC 39723. J Bacteriol 184:4672–4680
Chang BV, Su CJ, Yuan SY (1998) Microbial hexachlorobenzene dechlorination under three reducing conditions. Chemosphere 36:2721–2730
Chen X, Christopher A, Jones JP, Bell SG, Guo Q, Xu F, Rao Z, Wong LL (2002) Crystal structure of the F87W/Y96F/V247L mutant of cytochrome P-450cam with 1,3,5-trichlorobenzene bound and further protein engineering for the oxidation of pentachlorobenzene and hexachlorobenzene. J Biol Chem 277:37519–37526
Dai M, Copley SD (2004) Genome shuffling improves degradation of the anthropogenic pesticide pentachlorophenol by Sphingobium chlorophenolicum ATCC 39723. Appl Environ Microbiol 70:2391–2397
de Lorenzo V, Eltis L, Kessler B, Timmis KN (1993) Analysis of Pseudomonas gene products using lacI q /Ptrp-lac plasmids and transposons that confer conditional phenotypes. Gene 123:17–24
Fathepure BZ, Tiedje JM, Boyd SA (1988) Reductive dechlorination of hexachlorobenzene to tri- and dichlorobenzenes in anaerobic sewage sludge. Appl Environ Microbiol 54:327–330
Figurski DH, Helinski DR (1979) Replication of an origin-containing derivative of plasmid RK2 dependent on a plasmid function provided in trans. Proc Natl Acad Sci USA 76:1648–1652
Gilbert ES, Walker AW, Keasling JD (2003) A constructed microbial consortium for biodegradation of the organophosphorus insecticide parathion. Appl Microbiol Biotechnol 61:77–81
Haro MA, de Lorenzo V (2001) Metabolic engineering of bacteria for environmental applications: construction of Pseudomonas strains for biodegradation of 2-chlorotoluene. J Biotechnol 85:103–113
Jacoff FS, Scarberry R, Rosa D (1986) Source assessment of hexachlorobenzene from the organic chemical manufacturing industry. IARC Sci Publ 77:31–37
Jayachandran G, Görisch H, Adrian L (2003) Dehalorespiration with hexachlorobenzene and pentachlorobenzene by Dehalococcoides sp. strain CBDB1. Arch Microbiol 180:411–416
Jeenes DJ, Williams PA (1982) Excision and integration of degradative pathway genes from TOL plasmid pWW0. J Bacteriol 150:188–194
Kunisue T, Someya M, Kayama F, Jin Y, Tanabe S (2004) Persistent organochlorines in human breast milk collected from primiparae in Dalian and Shenyang, China. Environ Pollut 131:381–392
Li L, Yang C, Lan W, Xie S, Qiao C, Liu J (2008) Removal of methyl parathion from artificial off-gas using a bioreactor containing a constructed microbial consortium. Environ Sci Technol 42:2136–2141
Pieper DH, Reineke W (2000) Engineering bacteria for bioremediation. Curr Opin Biotech 11:262–270
Saber DL, Crawford RL (1985) Isolation and characterization of Flavobacterium strains that degrade pentachlorophenol. Appl Environ Microbiol 50:1512–1518
Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual, 3rd edn. Cold Spring Harbor Laboratory Press, New York
Takagi K, Iwasaki A, Kamei I, Satsuma K, Yoshioka Y, Harada N (2009) Aerobic mineralization of hexachlorobenzene by newly isolates pentachloronitrobenzene-degrading Nocardioides sp. strain PD653. Appl Environ Microbiol 75:4452–4458
Walker AW, Keasling JD (2002) Metabolic engineering of Pseudomonas putida for the utilization of parathion as a carbon and energy source. Biotechnol Bioeng 78:715–721
Williams PA, Murray K (1974) Metabolism of benzoate and the methylbenzoates by Pseudomonas putida (arvilla) mt-2: evidence for the existence of a TOL plasmid. J Bacteriol 120:416–423
Xu D, Zhong W, Deng L, Chai Z, Mao X (2004) Regional distribution of organochlorinated pesticides in pine needles and its indication for socioeconomic development. Chemosphere 54:743–752
Yan DZ, Liu H, Zhou NY (2006) Conversion of Sphingobium chlorophenolicum ATCC 39723 to a hexachlorobenzene degrader by metabolic engineering. Appl Environ Microbiol 72:2283–2286
Yeh DH, Pavlostathis SG (2001) Development of hexachlorobenzene-dechlorinating mixed cultures using polysorbate surfactants as a carbon source. Water Sci Technol 43:43–50
Zhou Y, Tigane T, Li X, Truu M, Truu J, Mander U (2013) Hexachlorobenzene dechlorination in constructed wetland mesocosms. Water Res 47:102–110
Acknowledgments
This work was supported by the Natural Science Foundation of Hubei Province of China (2008CDB067) and the National Natural Science Foundation of China (NSFC, Project No. 31270112). We thank Luet-Lok Wong for the gift of plasmid pCWSGB-camC and Luying Xun for providing Sphingobium chlorophenolicum ATCC 39723.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Yan, DZ., Mao, LQ., Li, CZ. et al. Biodegradation of hexachlorobenzene by a constructed microbial consortium. World J Microbiol Biotechnol 31, 371–377 (2015). https://doi.org/10.1007/s11274-014-1789-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11274-014-1789-7