
Abstract.
A consortium comprised of two engineered microorganisms was assembled for biodegradation of the organophosphate insecticide parathion. Escherichia coli SD2 harbored two plasmids, one encoding a gene for parathion hydrolase and a second carrying a green fluorescent protein marker. Pseudomonas putida KT2440 pSB337 contained a p-nitrophenol-inducible plasmid-borne operon encoding the genes for p-nitrophenol mineralization. The co-culture effectively hydrolyzed 500 μM parathion (146 mg l−1) and prevented the accumulation of p-nitrophenol in suspended culture. Kinetic analyses were conducted to characterize the growth and substrate utilization of the consortium members. Parathion hydrolysis by E. coli SD2 followed Michaelis–Menten kinetics. p-Nitrophenol mineralization by P. putida KT2440 pSB337 exhibited substrate-inhibition kinetics. The growth of both strains was inhibited by increasing concentrations of p-nitrophenol, with E. coli SD2 completely inhibited by 600 μM p-nitrophenol (83 mg l−1) and P. putida KT2440 pSB337 inhibited by 1,000 μM p-nitrophenol (139 mg l−1). Cultivation of the consortium as a biofilm indicated that the two species could cohabit as a population of attached cells. Analysis by confocal microscopy showed that the biofilm was predominantly comprised of P. putida KT2440 pSB337 and that the distribution of E. coli SD2 within the biofilm was heterogeneous. The use of biofilms for the construction of degradative consortia may prove beneficial.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Successful detoxification of recalcitrant organic chemicals may require the concerted effort of multispecies bacterial consortia. There are several recent examples of multispecies consortia isolated by enrichment culture that degrade structurally diverse organic compounds, including endosulfan (Sutherland et al. 2000), chloronitrobenzene (Park et al. 1999), 1,3-dichloropropene (Katsivela et al. 1999), atrazine (Alvey and Crowley 1996), and nitrate esters (Ramos et al. 1996). However, it may not be possible to isolate a natural bacterial consortium capable of degrading some organic compounds. Some of the factors that can prevent the enrichment of a degradative consortium include natural infrequency of an essential degradative gene (Shapir et al. 1998), production of recalcitrant intermediates (Van Hylckama Vlieg and Janssen 2001), molecular structural features of the target compound that limit its degradability (e.g. polyhalogenated compounds; Wackett et al. 1994), poor bioavailability (e.g. 5-ring polycyclic aromatic hydrocarbons; Bastiaens et al. 2000), and the natural dominance of a nonproductive metabolic pathway (Oh and Bartha 1997).
An alternative strategy to isolating natural degradative consortia is to combine bacteria with complementary metabolic pathways into functional assemblages. This approach has been used for biological degradation of several recalcitrant compounds, including 4,4'-dichlorobiphenyl (Adriaens et al., 1989), chlorinated dibenzofurans (Arfmann et al. 1997; Wittich et al. 1999), 9-fluorenone (Casellas et al. 1998), and mixtures of volatile organic hydrocarbons (Komukai-Nakamura et al. 1996; Oh and Bartha 1997). The use of multiple organisms with distinct metabolic capabilities may be advantageous in some cases over introducing the requisite degradative genes into a single microorganism. This is particularly true when biochemical intermediates inhibit the activity of one or more steps in the catabolic pathway, as was reported for aerobic polychlorinated biphenyl metabolism (Adams et al. 1992).
In the present work, a dual-species consortium was developed for biological detoxification of the insecticide parathion. One member of the consortium was responsible for hydrolyzing the parent compound, yielding two metabolites: p-nitrophenol and diethylthiophosphate. The other member of the consortium was responsible for mineralizing the stable intermediate p-nitrophenol. A kinetic analysis of the processes involved in parathion biodegradation was conducted and the ability of the consortium to be cultivated as a biofilm was investigated.
Materials and methods
Strains and plasmids
Escherichia coli SD2 was constructed by transformation of E. coli DH10B (Life Technologies) with pWM513 (Mulbry and Karns 1989) and pMAG1. Plasmid pWM513 is a pUC19-based vector carrying opd, which encodes parathion hydrolase, and bla, encoding beta-lactamase (Mulbry et al. 1986). Plasmid pMAG1 was constructed by introducing gfpuv, encoding a UV-excitable green fluorescent protein (Clontech, Palo Alto, Calif.), into pUCP27 (Schweizer 1994), a tetracycline-resistant vector. Pseudomonas putida KT2440, an ampicillin-resistant strain, was transformed with the pRK415-based vector pSB337, encoding the p-nitrophenol biodegradation operon from P. fluorescens ENV2030 and tetracycline resistance (Bang and Zylstra 1996). Both strains were maintained on Luria–Bertani (LB) medium containing 100 μg ampicillin ml−1 and 20 μg tetracycline ml−1 and were transformed by electroporation (Cho et al. 1995).
Chemicals
Parathion (O,O-diethyl O-4-nitrophenyl phosphorothioate) was obtained from Chem Service (West Chester, Pa.). p-Nitrophenol was purchased from Sigma. All solvents were high pressure liquid chromatography (HPLC) grade or better.
Batch culture study
Parathion was added directly to six flasks containing 50 ml autoclaved LB medium, resulting in an initial concentration of 500 μM parathion (146 mg l−1). Three of the flasks were inoculated with E. coli SD2 only. The remaining three flasks were inoculated with both E. coli SD2 and P. putida KT2440 pSB337. Both strains were inoculated to an initial optical density (at 600 nm) of 0.04. All cultures were grown in a shaking water bath at 23 °C. At each timepoint, a 1-ml aliquot was withdrawn from each flask and the optical density was measured. Subsequently, each aliquot was centrifuged and the supernatant was stored at −20°C for p-nitrophenol analysis by HPLC.
Influence of p-nitrophenol on growth
The effect of p-nitrophenol on the growth of E. coli SD2 and P. putida KT2440 pSB337 was determined. Both strains were grown at 23 °C in LB broth containing 0–2 mM p-nitrophenol. Optical density measurements (at 600 nm) were made over a 22-h period.
Kinetic analyses
To determine the effect of substrate concentration on p-nitrophenol degradation kinetics, resting cells of P. putida KT2440 pSB337 were prepared from mid- to late-exponential phase cultures grown in LB medium containing 100 μg ampicillin ml−1, 20 μg tetracycline ml−1, and 50 μM p-nitrophenol (7 mg L−1) to induce gene expression. Cells were washed twice in 50 mM phosphate buffer (pH 7.2) and were resuspended to an optical density (at 600 nm) of 1.4–1.7. For each sample, 1 ml cell suspension was added to a 1.7-ml microfuge tube and p-nitrophenol was added from a stock solution to the requisite initial concentration. Samples were prepared in triplicate. After a 4-h incubation at 23 °C, each sample was centrifuged and the resulting supernatant was stored at −20 °C prior to analysis by HPLC. In order to account for any potential loss of p-nitrophenol by abiotic processes, cell-free samples were assayed following the same protocol.
The rate of parathion hydrolysis by E. coli SD2 over 0–2,000 μM was determined by measuring the rate of p-nitrophenol accumulation (since one mole of p-nitrophenol is produced for each mole of parathion hydrolyzed). Requisite parathion concentrations were prepared from a 10% stock solution of parathion in methanol. Each sample received 1 ml of resting cells prepared from mid- to late-exponential phase cultures distributed in 1.7-ml microfuge tubes. Samples were prepared in triplicate and p-nitrophenol was collected for analysis by HPLC, as described above for P. putida, following 4 h incubation at 23 °C. For each strain, growth data were converted from optical density measurements to milligrams protein per milliliter, using an empirically determined conversion factor. Kinetic parameters were determined by nonlinear curve-fitting, using SigmaPlot 2001 (SPSS, Chicago, Ill.).
HPLC analysis of p-nitrophenol
p-Nitrophenol determinations were made using a Hewlett-Packard HPLC 1100 (Agilent, Palo Alto, Calif.), operated at ambient temperature. The mobile phase was 50% acetonitrile, 50% 40 mM acetic acid, and was pumped at a flow rate of 1 ml min−1. An Alltech C18 column [250×4.6 mm (length × internal diameter), 5 μm particle size; Deerfield, Ill.] was used to measure p-nitrophenol, which was detected at 402 nm, with a retention time of 5.7 min. The lower detection limit for p-nitrophenol was 1 μM (0.14 mg l−1) and there was a linear response for concentrations as high as 1,000 μM (139 mg l−1).
Protein determination
Protein contents were determined by the biuret method (Munkres and Richards 1965).
Biofilm growth and confocal microscopy
Biofilms were cultured in flow cells as described by Cowan et al. (2000a). Briefly, cells were inoculated into LB medium containing ampicillin and tetracycline and were recirculated through a flow cell at a rate of 0.862 ml min−1 for 21 h. Biofilms were rinsed, stained, and imaged using a Bio-Rad MRC1024 confocal laser scanning microscope, as described by Cowan et al. (2000b).
Results
Parathion hydrolysis and p-nitrophenol accumulation
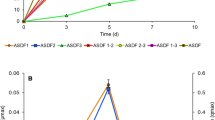
Parathion hydrolysis by E. coli SD2 led to steady p-nitrophenol accumulation during growth as a batch monoculture, reaching a concentration of 131±25 μM (18±3 mg l−1) in 52 h (Fig. 1). When E. coli SD2 grew in co-culture with P. putida KT2440 pSB337, 23±5 μM p-nitrophenol (3.2±0.7 mg l−1) accumulated during the first 23 h, which was subsequently degraded over the remainder of the experiment.

p-Nitrophenol (PNP) accumulation and metabolism during parathion biodegradation by suspended cells. Diamonds Escherichia coli SD2, triangles mixed culture of E. coli SD2 and Pseudomonas putida KT2440 pSB337
Effect of p-nitrophenol on growth
The growth of both members of the co-culture was inhibited by p-nitrophenol. The maximum specific growth rate of E. coli SD2 in LB broth at 23 °C was 0.19 h−1. The growth rate of strain SD2 decreased steadily at p-nitrophenol concentrations greater than 200 μM (28 mg l−1); and growth was completely inhibited by 600 μM p-nitrophenol (83 mg l−1). The growth of P. putida KT2440 pSB337 in LB at 23 °C (maximum specific growth rate of 0.41 hr−1) was only minimally affected by concentrations of p-nitrophenol less than 300 μM (42 mg l−1), but was more than 98% inhibited by 1 mM p-nitrophenol (139 mg l−1).
Kinetic analysis
A kinetic analysis was performed to evaluate the capacity of the co-culture to hydrolyze parathion and mineralize its hydrolysis product, p-nitrophenol. Parathion hydrolysis by E. coli SD2 was determined to be a concentration-dependent function (Fig. 2a) and could be characterized by a Michaelis–Menten expression:

Parathion biodegradation kinetics. Parameter estimates for each model are given in the Results section. a Parathion hydrolysis kinetics for E. coli SD2. Diamonds Measured rates, solid line best fit curve. b p-Nitrophenol metabolism kinetics for P. putida KT2440 pSB337. Circles Measured rates, solid line linear fit for p-nitrophenol concentrations <200 μM, dashed line first-order fit for p-nitrophenol concentrations at 200–1,000 μM
Here, q para is the parathion-utilization rate, q max is the maximum rate of parathion hydrolysis, S para is the parathion concentration, and K para is the half-saturation constant for parathion utilization. At 23 °C, the maximum parathion-utilization rate, q max, is 0.175 μmol parathion hydrolyzed h−1 mg−1 protein (6.0 mg parathion g−1 dry cells h−1) and the half-saturation constant for parathion utilization is estimated to be 163 μM parathion (47 mg l−1).
p-Nitrophenol degradation by P. putida KT2440 pSB337 was also a concentration-dependent function, with substrate inhibition evident in replicate experiments (Fig. 2b). The data could not be described by several previously reported substrate inhibition models, including the models of Andrews (1968), Edwards (1970), and Luong (1987). Consequently, two functions were used to describe the increasing and decreasing components of the data. A linear model was used to characterize the initial increase in the rate of p-nitrophenol biodegradation up to 200 μM (58 mg l−1; slope = 5.2×10−4 l h−1 mg−1 protein; intercept = 2.5×10−3; r 2 = 0.973). Rates measured at p-nitrophenol concentrations greater than 200 μM fit a first-order expression:
Here k PNP_hi is the rate of p-nitrophenol biodegradation, S PNP is the p-nitrophenol concentration, and a and b are constants. Measured values for a and b were 0.132 and 1.6×10−3, respectively; and r 2 for the best fit curve was 0.875.
Biofilm development and distribution of organisms
Cultivation of the co-culture as a biofilm indicated that the two strains were heterogeneously mixed in both the horizontal and vertical directions (Fig. 3). P. putida KT2440 pSB337 (Fig. 3, red) was present in greater proportion than E. coli SD2 (Fig. 3, yellow), which was located in small clusters distributed throughout the biofilm. Under the conditions of the experiment, the biofilm attained a thickness of approximately 50 μm over a 48-h period and substantially colonized the glass substratum.

Dual-species biofilm composed of E. coli SD2 expressing gfpuv (yellow) and P. putida KT2440 pSB337 (red). Upper panel Horizontal (x–y) cross-section 15 μm above the substratum. Lower panel Vertical (x–z) section, 80 μm thickness. 200× magnification
Discussion
Combining metabolically complementary bacteria in a consortium for xenobiotic bioremediation is an alternative to constructing a complete degradative pathway in a single microorganism. The ability to combine and organize specific microorganisms into a functional consortium was proposed through the use of surface-expressed antigens (Kjaergaard et al. 2000) or surface adhesives (Cowan and Keasling 2001). In the present work, two organisms that were genetically engineered to each carry out part of the pathway for parathion catabolism effectively worked together to break down the target compound and prevent accumulation of p-nitrophenol, a stable, toxic intermediate. The two organisms could be cultivated together in a biofilm, a feature that could be beneficial for the long-term survival of the consortium.
Kinetic analysis indicated that the concentration of the metabolic intermediate, p-nitrophenol, was central to the process of parathion biodegradation. The concentration of p-nitrophenol affected the growth rates of both member species and the rate of its own biodegradation. Additionally, p-nitrophenol biodegradation required induction, a process that depended on the accumulation of p-nitrophenol. An element that was essential to the growth of the co-culture as a biofilm was the ability of the member species to adhere to a surface. E. coli DH10B colonizes an exposed glass surface relatively slowly (Cowan and Keasling 2001); and its use necessitated coupling it with an organism that would condition the surface and facilitate its adhesion. P. putida KT2440 efficiently colonized the flow-cell surface, allowing the adhesion of E. coli, and promoting the development of a mixed-species biofilm. The difference in the abilities of the two strains to adhere to the substratum was a contributing factor to the significantly higher proportion of Pseudomonas that was observed in the biofilm.
The application of metabolically engineered microorganisms to the remediation of environmental toxicants has numerous potential benefits, including the ability to overexpress genes involved in detoxification of the target compound (Richins et al. 1997; Wang et al. 2000) and establishing catabolic pathways that have not been identified in nature (Haro and de Lorenzo 2001). However, an issue to be considered with regard to engineered microorganisms is the stable maintenance of introduced genetic elements in the absence of selective pressure. In this work, antibiotics assisted in maintaining the necessary plasmids. Under real-world conditions, it is expected that the target compounds of concern would be present only intermittently and would not provide a constant selective pressure that could contribute to sustaining engineered plasmids over the long term. Approaches for maintaining genes in the absence of selective pressure include the incorporation of "stabilizing elements" into engineered plasmids (Wang et al. 2000), inserting genes into the bacterial chromosome via transposons, and the use of high-stability, low-copy-number plasmids (Jones and Keasling 1998), all of which have been shown to significantly reduce the loss of introduced genes.
Metabolically engineered microorganisms for the treatment of specific target compounds could potentially be introduced to biofilm-based wastewater-treatment processes to augment the removal of specific compounds. In the case of engineered consortia, well mixed populations are most likely essential for efficient catabolism. Additional research will be required to evaluate the effect of spatial heterogeneity within the biofilm on biodegradation by the E. coli–Pseudomonas co-culture.
References
Adams RH, Huang CM, Higson FK, Brenner V, Focht DD (1992) Construction of a 3-chlorobiphenyl-utilizing recombinant from an intergeneric mating. Appl Environ Microbiol 58:647–654
Adriaens P, Kohler HPE, Kohler-Staub D, Focht DD (1989) Bacterial dehalogenation of chlorobenzoates and coculture biodegradation of 4,4'-dichlorobiphenyl. Appl Environ Microbiol 55:887–892
Alvey S, Crowley DE (1996) Survival and activity of an atrazine-mineralizing bacterial consortium in rhizosphere soil. Environ Sci Technol 30:1596–1603
Andrews JF (1968) A mathematical model for the continuous culture of microorganisms utilizing inhibitory substrates. Biotechnol Bioeng 10:707–723
Arfmann H-A, Timmis KN, Wittich R-M (1997) Mineralization of 4-chlorodibenzofuran by a consortium consisting of Sphingomonas sp. strain RW1 and Burkholderia sp. strain JWS. Appl Environ Microbiol 63:3458–3462
Bang S-W, Zylstra GJ (1996) Cloning and characterization of genes involved in p-nitrophenol degradation by Pseudomonas fluorescens ENV2030. Abstr Annu Meet Am Soc Microbiol 96:414
Bastiaens L, Springael D, Wattiau P, Harms H, De Wachter R, Verachtert H, Diels L (2000) Isolation of adherent polycyclic aromatic hydrocarbon (PAH)-degrading bacteria using PAH-sorbing carriers. Appl Environ Microbiol 66:1834–1843
Casellas M, Grifoll M, Sabata J, Solanas AM (1998) Isolation and characterization of a 9-fluorenone-degrading bacterial strain and its role in synergistic degradation of fluorene by a consortium. Can J Microbiol 44:734–742
Cho J-H, Kim E-K, So J-S (1995) Improved transformation of Pseudomonas putida KT2440 by electroporation. Biotechnol Tech 9:41–44
Cowan SE, Keasling JD (2001) Development of engineered biofilms on poly-l-lysine patterned surfaces. Biotechnol Lett 23:1235–1241
Cowan SE, Gilbert E, Khlebnikov A, Keasling JD (2000a) Dual labeling with green fluorescent proteins for confocal microscopy. Appl Environ Microbiol 66:413–418
Cowan SE, Gilbert E, Liepmann D, Keasling JD (2000b) Commensal interactions in a dual-species biofilm exposed to mixed organic compounds. Appl Environ Microbiol 66:4481–4485
Edwards VH (1970) The influence of high substrate concentration on microbial kinetics. Biotechnol Bioeng 12:679–712
Haro M-A, Lorenzo V de (2001) Metabolic engineering of bacteria for environmental applications: construction of Pseudomonas strains for biodegradation of 2-chlorotoluene. J Biotechnol 85:103–113
Jones KL, Keasling JD (1998) Construction and characterization of F plasmid-based expression vectors. Biotechnol Bioeng 59:659–665
Katsivela E, Bonse D, Krueger A, Stroempl C, Livingston A, Wittich RM (1999) An extractive membrane biofilm reactor for degradation of 1,3-dichloropropene in industrial waste water. Appl Environ Microbiol 52:853–862
Kjaergaard K, Schembri MA, Hasman H, Klemm P (2000) Antigen 43 from Escherichia coli induces inter- and intraspecies cell aggregation and changes in colony morphology of Pseudomonas fluorescens. J Bacteriol 182:4789–4796
Komukai-Nakamura S, Sugiura K, Yamauchi-Inomata Y, Toki H, Venkateswaran K, Yamamoto S, Tanaka H, Harayama S (1996) Construction of bacterial consortia that degrade Arabian light crude oil. J Ferment Bioeng 82:570–574
Luong JHT (1987) Generalization of Monod kinetics for analysis of growth data with substrate inhibition. Biotechnol Bioeng 29:242–248
Mulbry W, Karns JS (1989) Purification and characterization of three parathion hydrolases from gram-negative bacterial strains. Appl Environ Microbiol 55:289–293
Mulbry WW, Karns JS, Kearney PC, Nelson JO, McDaniel CS, Wild JR (1986) Identification of a plasmid-borne parathion hydrolase gene from Flavobacterium sp. by Southern hybridization with opd from Pseudomonas diminuta. Appl Environ Microbiol 51:926–930
Munkres KD, Richards FM (1965) The purification and properties of Neurospora malate dehydrogenase. Arch Biochem Biophys 109:466–479
Oh YS, Bartha R (1997) Construction of a bacterial consortium for the biofiltration of benzene, toluene and xylene emissions. World J Microbiol Biotechnol 13:627–632
Park H-S, Lim S-J, Chang YK, Livingston AG, Kim H-S (1999) Degradation of chloronitrobenzenes by a coculture of Pseudomonas putida and a Rhodococcus sp. Appl Environ Microbiol 65:1083–1091
Ramos JL, Haidour A, Duque E, Pinar G, Calvo V, Oliva J-M (1996) Metabolism of nitrate esters by a consortium of two bacteria. Nat Biotechnol 14:320–322
Richins RD, Kaneva I, Mulchandani A, Chen W (1997) Biodegradation of organophosphorus pesticides by surface-expressed organophosphorus hydrolase. Nat Biotechnol 15:984–987
Schweizer HP (1994) A method for construction of bacterial hosts for lac-based cloning and expression vectors: alpha-complementation and regulated expression. Biotechniques 17:452–4, 456
Shapir N, Mandelbaum RT, Jacobsen CS (1998) Rapid atrazine mineralization under denitrifying conditions by Pseudomonas sp. strain ADP in aquifer sediments. Environ Sci Technol 32:3789–3792
Sutherland TD, Horne I, Lacey MJ, Harcourt RL, Russell RJ, Oakeshott JG (2000) Enrichment of an endosulfan-degrading mixed bacterial culture. Appl Environ Microbiol 66:2822–2828
Van Hylckama Vlieg JET, Janssen DB (2001) Formation and detoxification of reactive intermediates in the metabolism of chlorinated ethenes. J Biotechnol 85:81–102
Wackett LP, Sadowsky MJ, Newman LM, Hur HG, Li S (1994) Metabolism of polyhalogenated compounds by a genetically engineered bacterium. Nature 368:627–629
Wang CL, Maratukulam PD, Lum AM, Clark DS, Keasling JD (2000) Metabolic engineering of an aerobic sulfate reduction pathway and its application to precipitation of cadmium on the cell surface. Appl Environ Microbiol 66:4497–4502
Wittich RM, Stroempl C, Moore ERB, Blasco R, Timmis KN (1999) Interaction of Sphingomonas and Pseudomonas strains in the degradation of chlorinated dibenzofurans. J Ind Microbiol Biotechnol 23:353–358
Acknowledgements.
This work was supported by the National Science Foundation (BES-9814088), the Office of Naval Research (N00014-99-1-0182), and the University of California Center for Water Resources (W-946). The plasmid pSB337 was a gift from S. Bang and G. Zylstra, Rutgers University. We thank Carolyn Larabell, Lawrence Berkeley National Laboratories, for assistance with confocal microscopy, and acknowledge Samir Davila and Richard Tsai for their assistance.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gilbert, E.S., Walker, A.W. & Keasling, J.D. A constructed microbial consortium for biodegradation of the organophosphorus insecticide parathion. Appl Microbiol Biotechnol 61, 77–81 (2003). https://doi.org/10.1007/s00253-002-1203-5
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00253-002-1203-5