
Abstract
An expeditious synthesis of 2,3-dihydroquinazolin-4(1H)-ones in the presence of oxalic acid as an environmentally friendly organocatalyst is reported. The salient features of the protocol are high yields, green reaction profile, shorter reaction times, and simple experimental and work-up procedure.
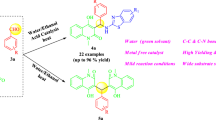
Graphical Abstract

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The recent quest towards the implementation of green methodologies in synthetic chemistry has spurred an extensive interest in the multicomponent reactions (MCRs) for the generation of highly privileged scaffolds of molecular complexity [1]. Such methodologies are able to manage dual goals such as environmental protection, as well as economic benefit. The systematic improvements in such methodologies are challenging benchmarks to organic chemist, and hence desperate attempts have been made for the development of these methodologies [2, 3]. The MCRs show remarkable significance in construction of diverse and complex organic molecules in a single step, which includes all essential parts of the starting materials. Furthermore, MCRs also have additional merits such as its exceptional synthetic efficiency, high atom economy, decrease in waste generation and formation of essential carbon–carbon and carbon-heteroatom bonds [4–8]. MCRs using environmentally benign catalyst under mild reaction conditions in eco-friendly solvent have become fundamental strategy in green synthetic methodologies [9, 10].
Recently, a great deal of attention has been focused on the use of organocatalysts in synthetic chemistry [11–13]. Organocatalyzed reaction offers handy technique for the synthesis of fundamental moieties having potential applications in the diverse fields [14]. Organocatalysts are cost-effective, stable, non-toxic, easy to handle, metal-free, and are often used in stoichiometric amounts [15]. The most exciting features of organocatalysts include minimization of waste and environmentally safe disposal [16, 17]. Oxalic acid is a strong acid since it has two carboxylic groups joined directly to each other and thus is widely used as an efficient organocatalyst in organic transformation [18–23]. Furthermore, oxalic acid is also used for the preparation of low transition temperature mixtures (LTTMs) liquids [24, 25]. The remarkable catalytic efficacy and hydrophilic nature of oxalic acid prompted us to explore its catalytic utility in the MCRs leading to synthesis of biologically active heterocyclic scaffolds.
Quinazolinones are prominent class of aza-heterocyclic compounds which acts as a core unit in the number of marketed drug products (Fig. 1). Quinazolinone derivatives such as 2,3-dihydroquinazolin-4(1H)-ones are privileged structural motifs having variety of applications in the biological, pharmacological and medicinal fields [26–32]. Owing to unique applicative profile, a number of protocols have been developed for the synthesis of 2,3-dihydroquinazolin-4(1H)-ones. The most common route for the synthesis of 2,3-dihydroquinazolin-4(1H)-ones includes MCR of isatoic anhydride, ammonium acetate with aryl aldehydes. Variety of catalysts have been reported for the synthesis of 2,3-dihydroquinazolin-4(1H)-ones using this route [33–50]. However, some of these methods suffer from drawbacks such as use of a metal-based catalyst, high reaction temperature, tedious catalyst preparation, high expense, and toxic nature of catalyst. Therefore, the development of a green and cost-effective method for synthesis is highly desirable.

Examples of some important marketed drugs having quinazolinones framework. (Color figure online)
As part of our research work in the development of sustainable methodologies for the preparation of bioactive heterocycles [51, 52], herein we report green protocol for synthesis of 2,3-dihydroquinazolin-4(1H)-ones by means of one-pot, three-component reaction of isatoic anhydride, NH4OAc with various aromatic aldehydes in the presence of oxalic acid as a catalyst.
Experimental
All chemicals were purchase from local supplier and used as received. Melting points were determined by the open capillary method. The IR spectra were measured on Bruker ALPHA FT-IR spectrometer in between the frequency range 500–4000 cm−1. The NMR spectra were recorded on Bruker AC (300 MHz for 1H NMR and 75 MHz for 13C NMR) spectrometer using TMS as an internal standard in DMSO-d 6 . Chemical shifts (δ) are expressed in ppm.
Representative procedure for synthesis of 2,3-dihydroquinazolin-4(1H)-ones
Synthesis of 2,3-dihydroquinazolin-4(1H)-ones by using three-component reaction of isatoic anhydride, NH4OAc and aryl aldehydes (method A)
In a 25-mL RB flask, a mixture of isatoic anhydride (1 mmol), aryl aldehyde (1 mmol), ammonium acetate (1.2 mmol), and oxalic acid (20 mol%) in ethanol:water (1:1, 4 mL) was stirred at 80 °C. Upon the completion of reaction as indicated by TLC, the reaction mixture was allowed to cool at room temperature, and water (5 mL) was added and stirred continuously until a solid was obtained in the reaction flask. The resultant solid was filtered, washed with water, and then dried. The solid was recrystallized by petroleum ether:ethyl acetate (70:30, v/v). All the resulting products were pure and characterized by spectroscopic techniques.
Synthesis of 2,3-dihydroquinazolin-4(1H)-ones by using anthranilamide and aryl aldehydes (method B)
In a 25-mL RB flask, a mixture of anthranilamide (1 mmol), aryl aldehyde (1 mmol), and oxalic acid (20 mol%) in ethanol:water (1:1, 4 mL) was stirred at 80 °C. Upon the completion of reaction as indicated by TLC, the reaction mixture was allowed to cool at room temperature, and water (5 mL) was added and stirred continuously until a solid was obtained in the reaction flask. The resultant solid was filtered, washed with water, and then dried. The solid was recrystallized by petroleum ether:ethyl acetate (70:30, v/v). All the resulting products were pure and characterized by spectroscopic techniques.
Selected spectral data of representative compounds
2-Phenyl-2,3-dihydroquinazolin-4(1H)-one (Table 3 , entry 1)
Mp: 225–227 °C, 1H NMR (300 MHz, DMSO d 6 ): δ (ppm) δ 8.19 (bs, 1H, NH), 7.61–7.64 (d, 1H, J = 6.9 Hz, Ar–H), 7.34–7.49 (m, 5H, Ar–H), 7.20 (s, 1H, Ar–H), 7.01 (bs, 1H, NH), 6.65–6.75 (t, 2H, J = 6.9 Hz, Ar–H), 5.75 (s, 1H, CH); 13C NMR (75 MHz, DMSO d 6 ): δ 67.1, 114.8, 115.3, 117.5, 127.2, 127.7, 128.6, 128.8, 133.6, 141.9, 148.2, 164.1; IR (KBr) (cm−1): 3305, 3180, 3059, 3031, 1652, 1609 cm−1.
2-(p-Tolyl)-2,3-dihydroquinazolin-4(1H)-one (Table 3 , entry 9)
Mp: 220–222 °C, 1H NMR (300 MHz, DMSO d 6 ): δ (ppm) δ 8.00 (s, 1H, NH), 7.61–7.64 (d, 1H, J = 7.8 Hz, Ar–H), 7.36–7.38 (d, 2H, J = 7.5 Hz, Ar–H), 7.13–7.20 (m, 3H, Ar–H), 6.61–6.83 (m, 3H, NH, Ar–H), 5.70 (s, 1H, CH), 2.29 (s, 3H, CH3); 13C NMR (75 MHz, DMSO d 6 ): δ 21.2, 67.2, 114.7, 115.2, 117.5, 127.2, 127.7, 133.5, 138.2, 138.6, 148.3, 164.4; IR (KBr) (cm−1): 3308, 3180, 3058, 3030, 2943, 1651, 1606 cm−1.
2-(1H-Indol-3-yl)-2,3-dihydroquinazolin-4(1H)-one (Table 3 , entry 11)
Mp: 218–220 °C, 1H NMR (300 MHz, DMSO d 6 ): δ (ppm) 10.87 (s, 1H, Indole-H1), 10.37 (s, 1H, NH), 7.36–7.52 (m, 1H, Ar–H), 7.03–7.22 (m, 4H, Indole-H4, H5, H6, H7), 6.88–6.96 (m, 1H, Ar–H), 6.85 (s, 1H, Indole-H2), 6.63–6.69 (m, 2H, Ar–H), 6.56 (s, 1H, NH), 5.81 (s, 1H, CH); 13C NMR (75 MHz, DMSO d 6 ): δ 55.5, 110.8, 112.1, 114.1, 121.6, 122.7, 124.9, 125.4, 126.0, 130.4, 132.3, 134.9, 141.6, 152.9, 163.9; IR (KBr) (cm−1): 3283, 3172, 3055, 2923, 1657, 1615 cm−1.
2-(Pyridine-2-yl)-2,3-dihydroquinazolin-4(1H)-one (Table 3 , entry 12)
Mp: 186–188 °C, 1H NMR (300 MHz, DMSO d 6 :CDCl3): δ (ppm) 8.51–8.53 (d, 1H, J = 3.9 Hz, Pyridine-H3), 8.27 (s, 1H, NH), 7.50–7.77 (m, 3H, Pyridine-H4, H5, H6), 7.15–7.29 (m, 3H, NH, Ar–H), 6.63–6.76 (m, 2H, Ar–H), 5.73 (s, 1H, CH); 13C NMR (75 MHz, DMSO d 6 :CDCl3): δ 67.6, 114.9, 115.2, 117.6, 120.8, 123.6, 127.7, 133.6, 137.2, 147.7, 149.2, 160.3, 164.1; IR (KBr) (cm−1): 3328, 3182, 3051, 2928, 1665, 1650 cm−1.
2-(Ferrocenyl)-2,3-dihydroquinazolin-4(1H)-one (Table 3 , entry 13)
Mp: 241–242 °C, 1H NMR (300 MHz, DMSO d 6 ): δ (ppm) 11.9 (s, 1H, NH), 8.07–8.09 (d, 1H, J = 7.5 Hz, Ar–H), 7.92 (s, 1H, NH), 7.55–7.70 (m, 1H, Ar–H), 7.15–7.41 (doublet of triplet, 1H, Ar–H), 6.62–6.74 (m, 1H, Ar–H), 5.54 (s, 1H, CH), 5.24 (s, 1H, Cp), 4.48 (s, 1H, Cp), 4.15–4.18 (m, 5H, Cp), 4.07 (s, 2H, Cp); 13C NMR (75 MHz, DMSO d 6 ): δ 63.5, 66.4, 67.9, 68.6, 70.0, 76.5, 90.5, 114.8, 117.3, 121.0, 125.7, 126.2, 127.6, 133.5, 134.6, 149.6, 155.9, 162.5; IR (KBr) (cm−1): 3428, 3158, 3075, 3023, 1641, 1006 cm−1.
Results and discussion
To establish the most appropriate reaction conditions, initially the template reaction between isatoic anhydride (1 mmol), benzaldehyde (1 mmol), and NH4OAc (1.2 mmol) in the presence of oxalic acid (20 mol%) was carried out in various solvents at 80 °C. The reaction proceeded scarcely in toluene, DMF, THF, EDC, water, and ethanol providing the desired 2-phenyl-2,3-dihydroquinazolin-4(1H)-one in lower yields (Table 1, entries 1–6). Surprisingly, it was observed that reaction marches efficiently in mixed solvent system of ethanol:water (1:1, v:v) furnishing the anticipated product in excellent yield (Table 1, entry 7).

Our next task was to optimize the catalyst loading. For this, we have carried out the template reaction using different amounts of catalyst and the results are summarized in Table 2. It was found that the quantity of catalyst had a significant impact on the yield of reaction. When the quantity of oxalic acid was increased from 5 to 15 mol%, the yield of anticipated product was elevated significantly from 45 to 80% (Table 2, entries 1–3). To our delight, the excellent yield was achieved for 20 mol% of oxalic acid (Table 2, entry 4). Further increase in catalyst quantity did not have a deep influence on yield of the desired product (Table 2, entries 5).

After the optimization of reaction conditions, we evaluated the scope and generality of protocol by reacting isatoic anhydride, NH4OAc with various aromatic aldehydes. The results are shown in Table 3. The reaction proceeded smoothly in all the cases forming the corresponding 2,3-dihydroquinazolin-4(1H)-ones in good to excellent yields (72–92%). It is worthy to note that aryl aldehyde with electron donating, as well as electron withdrawing substituents reacted efficiently with equal chemical reactivity (Table 3, entries 2–10). Interestingly, hetero-aromatic aldehydes such as indole-3-carboxaldehyde (Table 3, entry 11) and sterically congested pyridine-2-carboxaldehyde (Table 3, entry 12) were also well-tolerated providing the desired products in moderate yields (81–72%). To our delight, ferrocenecarboxaldehyde (Table 3, entry 13) could also serve as a substrate providing moderate yield (75%) of the expected product.

To broaden further the scope of the protocol, oxalic acid was scrutinized for analogous synthesis of 2,3-dihydroquinazolin-4(1H)-ones using reaction of anthranilamide and aryl aldehydes under the optimized reaction conditions (Scheme 1). It was observed that oxalic acid efficiently catalyzes the reaction between anthranilamide and aryl aldehydes providing the corresponding products in good to excellent yield (Table 4).

Oxalic acid catalyzed reaction between anthranilamide and aromatic aldehydes
The plausible mechanism for the synthesis of 2,3-dihydroquinazolin-4(1H)-ones using both methods (method A & B) is shown in Scheme 2. In method A, initial protonation of the carbonyl group of isatoic anhydride (I) by oxalic acid, followed by the attack of NH4OAc results in the subsequent ring opening, and decarboxylation provides anthranilamide intermediate (II). Further, activated aldehyde reacts with (II) followed by removal of water to give imine intermediate (III). The intramolecular nucleophilic attack of carboximidate nitrogen on imine carbon followed by cyclization furnishes the anticipated 2,3-dihydroquinazolin-4(1H)-ones (IV). Thus, we conclude that reaction proceeds through the formation of anthranilamide intermediate [45]. In method B, oxalic acid activates the carbonyl group of aromatic aldehyde, which then reacts with anthranilamide following the similar pathway as depicted for method A.

Mechanistic pathway for the synthesis of 2,3-dihydroquinazolin-4(1H)-ones
The reusability of oxalic acid was tested for the template reaction. After the separation of product, the resulting filtrate was extracted with chloroform, the aqueous layer containing oxalic acid was separated, and water was evaporated in vacuo to get crude oxalic acid. The recovered oxalic acid was reused three times without significant decrease in the yield of desired product. The slight decrease in the observed yield (Table 5) is attributed to inadequate recovery of the catalyst due to the attrition during filtration.
In order to show the advantages of oxalic acid in comparison with other catalysts, we have summarized several results for the preparation of 2,3-dihydroquinazolin-4(1H)-ones (Table 6). The comparison of results reveals that oxalic acid is a highly effective catalyst in terms of reaction time and yield than other reported catalysts.
Conclusion
We have described a one-pot, simple and efficient method for the multi-component synthesis of 2,3-dihydroquinazolin-4(1H)-ones using isatoic anhydride, NH4OAc with diverse aryl aldehydes using a catalytic amount of oxalic acid at 80 °C. The present environmentally benign methodology follows the tenets of green chemistry for synthesis of important scaffolds by using an easily available, inexpensive, water soluble, and non-toxic organocatalyst. The method offers several advantages such as high yields, cleaner reaction profiles, minimum waste generation, less environmental pollution, operational simplicity, shorter reaction times, and simple experimental and work-up procedure.
References
H. Koga, T. Kitaoka, A. Isogai, Molecules 20, 1495 (2015)
R.A. Sheldon, I. Arends, U. Hanefeld, Green Chemistry and Catalysis (Wiely-VCH, Weinheim, 2007)
P. Anastas, N. Eghbali, Chem. Soc. Rev. 39, 301 (2010)
M.S. Singh, S. Chowdhury, RSC Adv. 2, 4547 (2012)
D.J. Ramon, M. Yus, Angew. Chem. Int. Ed. 44, 1602 (2005)
A. Domling, Chem. Rev. 106, 17 (2006)
R.K. Singh, R. Bala, R. Duvedi, S. Kumar, Iran. J. Catal. 5, 187 (2015)
A. Domling, I. Ugi, Angew. Chem. Int. Ed. 39, 3168 (2000)
A.J. Wangelin, H. Neumann, D. Gordes, S. Klaus, D. Strubing, M. Beller, Chem. Eur. J. 9, 4286 (2003)
M.B. Gawande, V.D.B. Bonifacio, R. Luque, P.S. Branco, R.S. Varma, Chem. Soc. Rev. 42, 5522 (2013)
K. Aghapoor, F. Mohsenzadeh, H.R. Darabi, H. Sayahi, Y. Balavar, Res. Chem. Intermed. 42, 407 (2016)
X. Liu, Y. Wang, D. Yang, J. Zhang, D. Liu, W. Su, Angew. Chem. Int. Ed. 55, 8100 (2016)
J. Zhang, X. Liu, X. Maa, R. Wang, Chem. Commun. 49, 9329 (2013)
S. Nazari, M. Keshavarz, B. Karami, N. Iravani, M. Vafaee-Nezhad, Chin. Chem. Lett. 25, 317 (2014)
S. Khaksar, S.M. Vahdat, R.N. Moghaddamnejad, Monatsh. Chem. 143, 1671 (2012)
A.R. Khorrami, P. Kiani, A. Bazgir, Monatsh. Chem. 142, 287 (2011)
X.Z. Lian, Y. Huang, Y.Q. Li, W.J. Zheng, Monatsh. Chem. 139, 129 (2008)
S. Chandrasekhar, K. Gopalaiah, Tetrahedron Lett. 44, 7437 (2003)
N.D. Kokare, J.N. Sangshetti, D.B. Shinde, Chin. Chem. Lett. 19, 1186 (2008)
J.N. Sangshetti, N.D. Kokare, D.B. Shinde, J. Heterocycl. Chem. 45, 1191 (2008)
N.D. Kokare, J.N. Sangshetti, D.B. Shinde, Chin. Chem. Lett. 18, 1309 (2007)
N.D. Kokare, J.N. Sangshetti, D.B. Shinde, Synthesis 18, 2829 (2007)
J.N. Sangshetti, N.D. Kokare, D.B. Shinde, Chin. J. Chem. 26, 1506 (2008)
D.R. Chandam, A.G. Mulik, D.R. Patil, A.P. Patravale, D.R. Kumbhar, M.B. Deshmukh, J. Mol. Liq. 219, 573 (2016)
D.R. Chandam, A.G. Mulik, D.R. Patil, M.B. Deshmukh, Res. Chem. Intermed. 42, 1411 (2016)
V.K. Pandey, S. Tusi, Z. Tusi, R. Raghubir, M. Dixit, M.N. Joshi, Indian J. Chem. B. 43, 180 (2004)
M.J. Hour, L.J. Huang, S.C. Kuo, Y. Xia, K. Bastow, Y. Nakanishi, E. Hamel, K.H. Lee, J. Med. Chem. 43, 4479 (2000)
R.J. Alaimo, H.E. Russel, J. Med. Chem. 15, 335 (1972)
D.A. Erlanson, R.S. McDowell, T. O’Brien, J. Med. Chem. 47, 3463 (2004)
E. Hamel, C.M. Lin, J. Plowman, H.K. Wang, K.H. Lee, K.D. Paull, Biochem. Pharmacol. 51, 53 (1996)
K. Ozaki, Y. Yamada, T. Oine, T. Ishizuka, Y. Iwasawa, J. Med. Chem. 28, 568 (1985)
E. Jafari, M.R. Khajouei, F. Hassanzadeh, G.H. Hakimelahi, G.A. Khodarahmi, Res. Pharm. Sci. 11, 1 (2016)
A. Magyar, Z. Hell, Catal. Lett. 146, 1153 (2016)
B.H. Chen, J.T. Li, G.F. Chen, Ultrason. Sonochem. 23, 59 (2015)
J. Safari, S. Gandomi-Ravandi, J. Mol. Struct. 1072, 173 (2014)
A. Davoodnia, M. Khashi, N. Tavakoli-Hoseini, Chinese J. Catal. 35, 1054 (2014)
H.R. Shaterian, F. Rigi, Res. Chem. Intermed. 40, 2983 (2014)
J. Safari, S. Gandomi-Ravandi, J. Mol. Catal. A: Chem. 371, 135 (2013)
P. Yerram, R. Chowrasia, S. Seeka, S.J. Tangenda, Eur. J. Chem. 4, 462 (2013)
Z. Song, L. Liu, Y. Wang, X. Sun, Res. Chem. Intermed. 38, 1091 (2012)
S. Zhaleh, N. Hazeri, M.T. Maghsoodlou, Res. Chem. Intermed. 42, 6381 (2016)
J. Chen, D. Wu, F. He, M. Liu, H. Wu, J. Ding, W. Su, Tetrahedron Lett. 49, 3814 (2008)
S. Rostamizadeh, A.M. Amani, G.H. Mahdavinia, H. Sepehrian, S. Ebrahimi, Synthesis 8, 1356 (2010)
S. Rostamizadeh, A.M. Amani, R. Aryan, H.R. Ghaieni, N. Shadjou, Synth. Commun. 38, 3567 (2008)
M.A.B. Fard, A. Mobinikhaledi, M. Hamidinasab, Synth. React. Inorg. Metal Org. Nano Met. Chem. 44, 567 (2014)
Z. Karimi-Jaberi, R. Arjmandi, Monatsh. Chem. 142, 631 (2011)
K. Niknam, M.R. Mohammadizadeh, S. Mirzaee, Chin. J. Chem. 29, 1417 (2011)
M. Dabiri, P. Salehi, M. Bahramnejad, M. Alizadeh, Monatsh. Chem. 141, 877 (2010)
S. Li, Q. Zhang, Y. Peng, Monatsh. Chem. 146, 1859 (2015)
M.T. Maghsoodlou, N. Khorshidi, M.R. Mousavi, N. Hazeri, S.M. Habibi-Khorassani, Res. Chem. Intermed. 41, 7497 (2015)
S. Karhale, K. Patil, C. Bhenki, G. Rashinkar, V. Helavi, Res. Chem. Intermed. 42, 7257 (2016)
D.N. Survase, H.V. Chavan, S.B. Dongare, V.B. Helavi, Synth. Commun. 46, 1665 (2016)
Acknowledgements
One of the author (SSK) thankful to UGC, New Delhi, for Teacher Fellowship [F. No. 30-35/14 (WRO) dated: 11th June 2014] under the Faculty Development Programme. Author is also thankful to Principal, Abasaheb Marathe Arts & New Commerce, Science College, Rajapur, Ratnagiri, for encouragement and Head, Department of Chemistry, Rajaram College, Kolhapur for providing necessary facilities.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Karhale, S., Survase, D., Bhat, R. et al. A practical and green protocol for the synthesis of 2,3-dihydroquinazolin-4(1H)-ones using oxalic acid as organocatalyst. Res Chem Intermed 43, 3915–3924 (2017). https://doi.org/10.1007/s11164-016-2855-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11164-016-2855-6