
Abstract
The Amentotaxus argotaenia (Taxaceae) species complex is comprised of four relict and endangered gymnosperms. Amentotaxus argotaenia and A. yunnanensis are scattered throughout South and Central China, and A. formosana, and A. poilanei are restricted to Taiwan and Vietnam. In the present study, we examined genetic divergence and biogeographical patterns in the species complex based on chloroplast DNA atpI-atpH and rpS16-trnK intergenic spacers, the mitochondrial DNA nad1 intron, and microsatellite markers. Genealogical analyses revealed systematic inconsistencies between organellar DNA markers, with paraphyletic species inferred based on cpDNA, versus a monophyletic A. formosana, and clustering of A. poilanei within A. yunnanensis at mtDNA. AMOVA revealed that most organellar DNA variants resided among species and populations, a pattern also supported by clustering with STRUCTURE analyses on microsatellites. Higher levels of genetic diversity in the Yongxin population of A. argotaenia reflected features of a refugium. Most speciation events were rather recent, with divergence no earlier than 2.50 MYA, although many ancient lineages derived 4.12–13.84 MYA were maintained within species. Significant genetic structuring within A. argotaenia was detected among three geographical regions—a phylogeographical pattern likely attributable to past fragmentations. In contrast to paraphyletic A. argotaenia based on the maintenance of ancestral polymorphisms, interspecific hybridization likely contributed to polyphyly in A. yunnanensis. Furthermore, despite the low likelihood of recurrent gene flow due to geographical isolation, IM analyses revealed continued interspecific gene flow after species divergence, reflecting a parapatric speciation model.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Amentotaxus (Taxaceae) represents an ancient lineage of six relic species (Farjon 2010). Tertiary fossil evidence indicates this genus was once widespread in the Northern Hemisphere (Harris 1976). However, currently Amentotaxus is restricted to subtropical, southeast Asia, from Taiwan west across southern China to Assam in the eastern Himalaya, and south to Vietnam (Ferguson et al. 1978; Royer et al. 2003). Four species of Amentotaxus, namely A. argotaenia Pilg., A. formosana H. L. Li, A. yunnanensis H. L. Li, and A. poilanei D. K. Ferguson are considered a species complex, which remains a taxonomic challenge due to the existence of intermediate morphotypes based on species diagnostic traits, i.e., stomatal band width and leaf shape (Cheng 1978; Zhou 2001). All four species are characteristic of subtropical forests, and exhibit an allopatric distribution with clear habitat differentiation. A. yunnanensis is confined to southwest China, growing in limestone habitats; A. poilanei is restricted to moist montane forests of Vietnam; A. formosana is an element of temperate broadleaf forests in Taiwan; and A. argotaenia is restricted to limestone mountains, forests, ravines, shady and damp stream banks across 11 provinces of South and Central China. Demographically, all populations remain in small, isolated fragments, primarily with fewer than 100 individuals; and only a single A. formosana population remains in southern Taiwan (Li and Keng 1994). According to IUCN categories (IUCN 2013), A. argotaenia and A. poilanei are under vulnerable conditions (VU), and A. formosana and A. yunnanensis are under serious threats (EN), all with small population numbers and sizes.
Geographical barriers, such as the Taiwan Strait and the Himalayan mountain ranges might have played an integral role in genetic differentiation and speciation of this complex. Nevertheless, species boundaries among gymnosperms remain poorly explored. Although allopolypoid and homoploid speciation has been found much more common than expected in conifers (Chiang et al. 2006; Wachowiak and Prus-Glowacki 2008; Wang et al. 2012; Zhou et al. 2010), gene flow levels among species remain poorly understood. Occurrence of natural hybrids does not necessarily reject allopatric speciation—a long-accepted model in which gene flow ceases immediately following speciation—as it has long been suggested hybrids can act as a reproductive barrier between progenitor species (Wang et al. 2000). In fact, the existence of natural hybrids is not equivalent to the occurrence of interspecific gene flow. In this study, we examined allopatric speciation in Amentotaxus by examining the extent of gene flow since species divergence.
Extant and historical distributions of Amentotaxus species show a demographic history attributable to fluctuating climates. Based on the palynoflora, Quaternary glaciers largely removed most temperate species, like Pinus massoniana and P. hwangshanensis, in the north regions of China (Zhou et al. 1994, 2010; Ge et al. 2012), resulting in range shifts that forced plants into southern refugia, and a rich south China flora (Hewitt 1999; Tong et al. 1992). In contrast to North Europe and North America, where many of the Tertiary plant species went extinct, refugia in central and southwestern China (below 31° N) supported many southward migrants during the last glacial maximum (LGM) (Ge et al. 2012; Harrison et al. 2001; Liu 1991; Ray and Adams 2001; Wang et al. 2011; Xu et al. 2008). Furthermore, Del Tredici et al. (1992) suggested another refugium located in Eastern China. Geographically, primarily east–west oriented mountain ranges buffered climate change effects across a latitudinal gradient. Tertiary relicts, such as Cathaya, Ginkgo, Metasequoia, and Taiwania, which were once widespread across Europe and North America, survived in this region, with a scattered distribution and small population sizes (Traula 1968). However, subsequent postglacial recolonization did not substantially recover the species ranges, due largely to dry climate in northern China, where vegetation has since been dominated by steppe (Tong et al. 1999). Accordingly, postglacial colonization resulted in a range of phylogeographical patterns in species from different latitudes (Hewitt 1996; Chiang and Schaal 2006).
The Amentotaxus argotaenia complex, which spans the subtropical mainland, and a continental island provides an ideal model with which to examine genetic divergence among sister species, and genetic structure among populations across species geographical ranges, and to further elucidate speciational processes. Species evolving through glacial cycles are of particular interest to evolutionists, as climate changes during the ice ages had a dramatic influence on species, causing isolation, migration, and extinction, as well as accelerating speciation (Taberlet et al. 1998). Under conditions of small population sizes shaped by random genetic drift, rare species possess low levels of genetic variation, and display evident geographic structuring due to the stochastic loss of genetic polymorphisms from populations. Ge et al. (2005) reported significant genetic differentiation among species of the A. argotaenia complex, where all species populations grow under forest canopies, with a scattered and patch-like distribution.
In this study, we reconstructed phylogenies and population structure of the Amentotaxus species complex by investigating the genetic variation of organellar DNAs and microsatellite fingerprinting. The following questions were addressed: (1) Are the four species genetically distinct entities? (2) Did these taxa experienced interspecific gene flow since divergence? (3) A. argotaenia exhibits a much larger geographic area than the other three species; therefore, does it possess higher levels of genetic diversity? And (4) what is the phylogeographic pattern of relict Amentotaxus species in hypothetical refugia of China?
Materials and Methods
Population, Sampling and DNA Extraction
Seventeen populations throughout the whole geographical range of distribution of the Amentotaxus argotaenia complex were surveyed. In total, 98 individuals were sampled from 11 populations of A. argotaenia, 12 individuals from the single population of A. formosana, 38 individuals from four populations of A. yunnanensis, and 11 individuals from the single population of A. poilanei. Rare distributions of this complex pose a difficulty in sampling (Tran 2011) (Table 1). Within the distributional range of A. argotaenia, three regions can be discerned based on the geographical topography. The East region was separated from West and Central regions by the Mt. Nanling and the Mt. Wuyi; West and Central regions were separated from each other by the Yangtze River (Fig. 1). Identical materials were previously used for an ISSR fingerprinting analyses (Ge et al. 2005), except for Bamianshan (HU) population of A. argotaenia in China, and Lao Va Chai Municipality (VL) of A. poilanei in Vietnam. Young, healthy leaves were collected and dried in silica gel until DNA extraction. Total genomic DNA was extracted from leaves following a CTAB methodology (Doyle and Doyle 1987).

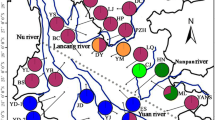
Map showing distributions and frequencies of clades of organellar DNA in the populations of Amentotauxs species. Clades for cpDNA: dark blue A, brown B, green C, purple D, blue E, orange F, light blue G, light orange H; clades for mtDNA: dark blue I, brown II, green III, purple IV, blue V, orange VI, light blue VII. For abbreviations of populations see Table 1. A. hatuyenensis was not included in this study
Organelle DNA Analyses
The cpDNA (chloroplast DNA) spacers and mtDNA (mitochondrial DNA) intron were amplified by the polymerase chain reaction (PCR) using primers atpI 5′ TAT TTA CAA GCG GTA TTC AAG CT 3′ and atpH 5′ CCA ACC CAG CAG CAA TAA C 3′, trnK 5′ TTA AAA GCC GAG TAC TCT ACC 3′ and rpS16 5′ AAA GTG GGT TTT TAT GAT CC 3′, and nad1/F 5′ GAT CGG CCA TAA ATG TAC TCC 3′ and nad1/R 5′ CCC CAT ATA TTC CCG GAG C 3′. PCR products were electrophoresed on agarose gels and the DNA fragment was excised from the gel and purified. Eluted PCR products were sequenced directly in both directions on an ABI 377XL automated sequencer (Applied Biosystem, Foster City, CA).
Microsatellite Genotyping
Fifteen variable microsatellite loci were amplified and genotyped (Ho et al. 2012). PCR amplification was carried out in a 50 uL reaction, with forward primers for PCR labelled with fluorescent dye. PCR products were separated with an ABI 3100 automated sequencer, and fragment sizes were assessed using GENEMAPPER software version 3.7 (Applied Biosystems).
Phylogenetic and Phylogeographic Analyses
Nucleotide sequences were aligned with the program BlastAlign (Belshaw and Katzourakis 2005). Indels were coded as binary characters with SeqState version 1.4.1 (Müller 2005). Maximum-likelihood (ML) analyses were performed using PHYLIP v. 3.67 (Felsenstein 2007). Confidence of the lineages reconstructed was tested by bootstrapping (Felsenstein 1985) with 1,000 replicates using unweighted characters. Genetic distances between haplotypes were calculated based on the transition/transversion ratio using MEGA, version 5.1 (Tamura et al. 2011), and were used to construct a minimum spanning network with MINSPNET software (Excoffier and Smouse 1994).
Population Genetic Aanalyses
To investigate the genetic structure of Amentotaxus populations, several genetic analyses within and between populations were conducted. Genetic analyses within and between populations were conducted for cp- and mt-DNA sequences. The two cpDNA fragments were combined, and treated as a single marker. Statistics such as the number of segregating sites (S), and genetic diversity (Hd) were determined (Nei 1987). The level of genetic diversity within populations was quantified by measures of nucleotide divergence, θ (Watterson 1975) and π (Tajima 1989), using DnaSP (Rozas et al. 2003). In order to make inferences about demographic dynamics of Amentotaxus, we employed both mismatch distributions (Slatkin and Hudson 1991) and statistical tests of neutrality. We calculated Tajima’s D (Tajima 1989), Fu and Li’s D* statistic (Fu and Li 1993), and Fu’s Fs statistics (Fu 1997). Mismatch distributions have a unimodal pattern when the population expands from a single source or recovers from a bottleneck; mismatch distributions are multimodal in populations with a stable effect size (Rogers and Harpending 1992). A hierarchical analysis of molecular variance (AMOVA, Excoffier et al. 1992) was conducted to summarize the variation among and within populations with the computer program Arlequin (Excoffier et al. 2005). The Ф statistics were computed using haplotype frequencies alone, and the significance of departures from zero of Ф statistics and genetic variance components was tested using 10,000 permutations. Patterns of geographical subdivision were estimated hierarchically using DnaSP. Statistical significance of Фst estimates was assessed using software Arlequin with permutations. The differentiation parameter of Gst was also estimated for the total data set and for each species using PerMut 1.0 (Pons and Petit 1996).
The xmfa2struct program (http://www.xavierdidelot.xtreemhost.com/clonalframe.htm) was used to convert sequence data of cpDNA and mtDNA into genotype data for HP-RARE 1.0 (Kalinowski 2005) and FSTAT V2.9.3.2 (Goudet 1995). HP-RARE 1.0 was used to calculate standardized haplotype richness (H r) and private allelic richness (H p), equalizing to a sample size using rarefaction. The permutation tests of significance for allelic richness were implemented in FSTAT V2.9.3.2.
A Bayesian estimate of group delimitation and the ages of TMRCA of the Amentotaxus clades was obtained using *BEAST (Melo-Ferreira et al. 2012) and BEAST v. 1.6 (Drummond et al. 2006). For estimating divergence between populations or species, a well-documented evolutionary rate is needed. The fossil record of the Taxads extends from the Lower Jurassic to the present (Stewart and Rothwell 1993). Even so, it is difficult to assess the divergence times using the fossil record. In this study, evolutionary rates for cp DNA and mtDNA have been shown to reach 1.10–2.90 × 10−9, and 2.00–3.00 × 10−10 substitutions per site per year (Wolfe et al. 1987; Chiang et al. 2006). Posterior estimates of age of the TMRCA were obtained by BEAST v. 1.6, with samples drawn every 500 steps over a total of 1,000,000 steps. Adequate sampling and convergence to the stationary distribution were checked using TRACER v. 1.6 (Rambaut and Drummond 2004). Posterior estimates of parameters were all found to be distinctly unimodal (although with wide 95 % highest posterior densities), and all parameters appeared to be identifiable, despite the relatively low information content in the sequences and the small age range of the sequences.
For microsatellite loci, the genetic diversity within species/populations was estimated by calculating allele number, observed, and expected heterozygosities of microsatellites with Arlequin (Excoffier et al. 2005). The program was also performed to calculate population differentiation, and molecular variance. The standardized allelic richness (A r) and private allelic richness (A p), with significance for allelic richness via permutation tests, and differentiation parameters (Gst) were assessed using programs HP-RARE 1.0, FSTAT V2.9.3.2, and PerMut 1.0. Program STRUCTURE version 2.1 (Pritchard and Wen 2003) applies a Bayesian method to infer the number of clusters (K) without using prior information of individual sampling locations. Program STRUCTURE v. 2.1 (Pritchard and Wen 2003) applies a Bayesian method to infer the number of clusters (K = 1–17) without using prior information of individual sampling locations and was estimated by the Markov chain Monte Carlo (MCMC) method with 20 separate runs for each number of clusters to evaluate the consistency of the results. Each run was pursued for 1,000,000 MCMC interactions, with an initial burn-in of 100,000 and an ancestry model that allowed for admixture. The final posterior probability of K, Pr (X|K), was computed using the runs with highest probability for each K (Pritchard et al. 2000). However, as indicated in the STRUCTURE documentation, Pr (X|K) usually plateaus or increases slightly after the ‘right’ K is reached. Thus, following Evanno et al. (2005), ΔK, where the modal value of the distribution is located at the real K, was calculated by STRUCTURE HARVESTER v. 0.6.8 (Earl and vonHoldt 2012).
IM Estimation of Demographic Dynamics
To determine the demographic history that the species experienced, we used the simulation program IMa (Hey and Nielsen 2004) to investigate the isolation with migration model in microsatellites and organelle DNAs. Using coalescent simulations and Bayesian computation procedures, IM yields six demographic parameters, including the divergence time (t), bidirectional migration rates (m 1 and m 2), and the effective population sizes of the ancestral (θA) and descendent populations (θ1 and θ2). The stepwise mutation model (SMM) and Hasegawa-Kishino-Yano (HKY) models were applied to microsatellite and organelle sequence data, respectively. The posterior probability densities of these parameters are generated by MCMC simulations, and simulations were run with individual simulations being updated 100,000 times. Within each simulation, we used the procedure to swap among ten heated chains (Metropolis coupling) and observed sufficient swapping rate while the simulation was running. These simulations were carried out using ten independent runs, with each chain started at a different starting point and initiated with a burn-in period of 100,000 updates. Convergence upon the stationary distribution was assessed by estimating the effective sample size (ESS) for the parameters, based on the autocorrelation of parameter values measured over the course of the run. The analysis was considered to have converged upon a stationary distribution if the independent runs generated similar posterior distributions with a minimum ESS of 100. Each simulation yielded a marginal density histogram for the six parameters of interest. The peaks of the resulting distributions were considered as the maximum likelihood estimates (MLE) of the parameter with credibility intervals equaling the 95 % highest posterior density (HPD) intervals. Demographic parameters were scaled using the neutral mutation rate (u). For loci of cpDNA and mtDNA, evolutionary rates have been shown to reach 1.10–2.90 × 10−9, and 2.00–3.00 × 10−10 substitutions per site per year (Wolfe et al. 1987; Chiang et al. 2006). Then we used the geometric mean (u =4.55 × 10−7 substitutions/locus/year) based on the above rate for rescaling parameters. For microsatellites, 6.3 × 10−4 mutations per locus were applied (O’Connell and Ritland 2004). The effective population size for ancestral and present times were estimated by N = (θ/2u) and N = (θ/4u) for organelle and SSR data.
Results
Sequence Variation and Genetic Diversity
We examined organellar DNA genetic diversity in the Amentotaxus species complex (Tables 2–3). In total, 38 haplotypes (C1–C38) of the cpDNA were identified (GenBank accession numbers: HG424279–HG424291 for atpI-atpH spacer; HG424292–HG424316 for rpS16-trnK spacer), only five of which (C5, 9, 10, 23, and 24) were shared among species. Twenty-six mtDNA haplotypes were identified (M1–M26, GenBank accession numbers: EF418631–EF418656), and almost all were private, i.e., none were shared among species (Fig. 2b).

Unrooted maximum likelihood tree of haplotypes in Amentotaxus. Numbers at nodes represent bootstrap values and divergence times between major lineages. Distribution of each clade is indicated in parentheses: ARG A. argotaenia, ARG (C) Central region, ARG (E) East region, ARG (W) West region), FOR A. formosana, YUN A. yunnanensis, POI A. poilanei. a cpDNA, b mtDNA
Population level genetic diversity (Hd) based on cpDNA was 0.78, and nucleotide diversity (θ) was 4.00 × 10−3. Amentotaxus yunnanensis exhibited the highest nucleotide diversity (θ = 3.21× 10−3), followed by A. formosana (θ = 2.57× 10−3), A. poilanei (θ = 1.86× 10−3), and A. argotaenia (θ = 1.75× 10−3). The A. argotaenia population sampled from Yongxin had the highest nucleotide diversity; and A. yunnanensis collected from Xichou exhibited the highest nucleotide diversity level (Table 2). The standardized haplotype richness (Hr) and private haplotype richness (Hp) of populations ranged from 1.00 to 1.38 and 0.00 to 0.21, respectively. Higher private allelic richness was found in A. formosana, and A. yunnanensis (Table 2). The mtDNA population genetic diversity (Hd) was 0.91, and nucleotide diversity (θ) was 7.86× 10−3. Amentotaxus argotaenia possessed the highest nucleotide diversity level (θ = 6.03× 10−3), followed by A. yunnanensis (θ = 2.10× 10−3), and A. formosana (θ = 1.40× 10−3), while no variation was detected in A. poilanei. Amentotaxus argotaenia exhibited 16 of 22 haplotypes private to populations with the highest private allelic richness (Hp = 0.33), while three haplotypes were restricted to a single A. formosana population (Hp = 0.17). Amentotaxus argotaenia from Yongxin had the highest nucleotide diversity and haplotype richness, whereas the Anlong population of A. argotaenia was fixed at haplotype M26 (Table 3). No significant differences in the allelic richness were detected among species (P = 0.19, cpDNA; P = 0.43, mtDNA).
Gene Genealogy of CpDNA and MtDNA Haplotypes
A maximum likelihood tree was reconstructed based on cpDNA haplotypes, and eight major lineages (A–H) were identified. Haplotype data indicated paraphyly in all species in the complex. No distinct geographical subdivisions were detected among lineages. A dominant basal haplotype C5 (46.35 %) was resolved in Cluster A, which occurred exclusively in A. argotaenia and A. yunnanensis (Fig. 2a). A minimum spanning network was also reconstructed based on mutational changes. Eight major lineages (A–H) were identified, and lineages C, D, and G were nested as the interior node, independently linked to A, B, E, F, and H (Fig. 3a).

Minimum spanning network of haplotypes in Amentotaxus. Geographical distribution of each haplotype and lineage is indicated. Distribution of each clade is indicated in parentheses: ARG A. argotaenia, ARG (C) Central region, ARG (E) East region, ARG (W) West region, FOR A. formosana, YUN A. yunnanensis, POI A. poilanei. a cpDNA, b mtDNA
Seven major mtDNA lineages (I–VII) were identified (Fig. 2b), with I–V forming a cluster, and sister to VI + VII. Lineages II and VI occurred exclusively in A. formosana, and A. yunnanensis and A. poilanei, respectively; while the remainder occurred in A. argotaenia. A. yunnanensis and A. poilanei were nested within A. argotaenia (Fig. 3b). Furthermore, A. argotaenia and A. yunnanensis were respectively paraphyletic and polyphyletic in genetic composition; monophyly was observed in A. formosana and A. poilanei. Amentotaxus argotaenia was monophyletic in populations distributed in the East region, included in lineage VII; while paraphyletic in both Central and West regions. A minimum spanning network of mtDNA, comprised of two major lineages I + II + III + IV + V and VI + VII, corresponded to clusters of the maximum likelihood tree (Fig. 3b).
The *BEAST population tree based on the combined organelle makers in the species complex identified two major groups, i.e., A. argotaenia vs. other three species (Supplementary Fig. 1). Within the A. argotaenia group, geographical subdivision nevertheless was not supported. Admixture of haplotypes across geographical populations, plus a low posterior possibility (Pp = 0.61) for the internal node of coalescence, indicated effects of ancestral lineage sorting or occurrence of gene flow.
Genetic Diversity at Microsatellite Loci
The species complex was polymorphic at all 15 microsatellite loci, and the number of alleles for each locus ranged from 1.93 to 5.47 (Table 4). Amentotaxus argotaenia exhibited the highest number of alleles per locus, while the lowest number was resolved in A. poilanei. The eastern geographic region had a higher number of alleles per locus than other regions within A. argotaenia. Observed (H O) and expected (H e) heterozygosity varied respectively from 0.05 to 0.29, and 0.24 to 0.61 (Table 4). The standardized allelic richness (Ar) and private allelic richness (Ap) of populations ranged from 1.91 to 4.90 and 0.00 to 0.65, respectively. Higher private allelic richness was found in A. argotaenia, and A. formosana. Overall, four loci deviated from Hardy-Weinberg expectations in A. formosana, A. yunnanensis, and A. argotaenia in all populations due to heterozygote deficiencies, the exception being A. poilanei. At the population level, heterozygote deficiencies were also detected in most populations. Like organellar DNA, no significant differences of allelic richness among species was detected in microsatellites (P = 0.08).
Subsequently, data were analyzed to examine genetic structure at a finer resolution using STRUCTURE (Pritchard et al. 2000). The best, second, and third fit numbers of groupings were inferred as 2, 3, and 6 based on ΔK, i.e. ΔK = 8.46 at K = 2, ΔK = 3.64 at K = 3, and ΔK = 2.84 at K = 6, based on Bayesian clustering analyses on microsatellites (Fig. 4a). At K = 2, the A. argotaenia species complex was divided into two differentiated groups; A. formosana and A. argotaenia were allied in one cluster, and A. yunnanensis and A. poilanei were grouped in the other cluster (Fig. 4b). Each individual possessed a high percentage of component 1 (green segment in Fig. 4b) or component 2 (blue segment in Fig. 4b), with the exception of 11 A. argotaenia individuals, which showed > 20 % of component 2, revealing genetic admixture composition. Similarly, at K = 3, A. formosana and A. argotaenia were separated from an A. yunnanensis and A. poilanei cluster (Fig. 4b); at K = 6, A. argotaenia was divided into four clusters, which did not correspond to geographic subdivisions (Fig. 4b).

Bayesian inference of the number of clusters (K) of Amentotaxus based on microsatellites. a The scatter plots of ΔK. The ΔK is based on the rate of change of ln P (X/K) between successive K values. b The two, three, and six clusters (K = 2, 3, and 6) detected from STRUCTURE analyses
Population Genetic Structure and Demographic Dynamics
Hierarchical and nonhierarchical AMOVAs were conducted to determine the spatial apportionment of genetic variants among Amentotaxus species and populations (Table 5). The AMOVA of cpDNA revealed only 13.48 % of the total variants within populations (ФSC = 0.82, P < 0.01) and the largest level of variation (59.87 %) was explained by differences among populations (ФST = 0.87, P < 0.01). However, 26.65 % of the total variation was due to differences among species (ФCT = 0.27, P < 0.01) (Table 5). A hierarchical AMOVA was conducted based on three geographic subdivisions of A. argotaenia, i.e., west, central, and east regions. Results showed 82.54 % of the total variation was among regions, and 17.46 % within populations. Most of genetic differentiation within A. argotaenia and A. yunnanensis was partitioned among populations, with respective values of 82.54 % and 89.92 %. The mtDNA AMOVA results yielded similar patterns. Results indicated the highest level of variation was among populations (63.49 %, ФST = 0.92, P < 0.01), with only 8.22 % within population variation (ФSC = 0.89, P < 0.01) (Table 5), and 28.30 % of the variance was explained by differences among species (ФCT = 0.32, P < 0.01). In contrast to organellar DNA loci, AMOVA of microsatellite loci revealed that the largest level of variation (51.13 %) was due to differences within populations (ФSC = 0.22, P < 0.01) (Table 5), and only 14.68 % of the total variation was among populations within species (ФST = 0.49, P < 0.01). Nonetheless, 34.19 % of the variance was explained by differences among species (ФCT = 0.34, P < 0.01). The estimates of Gst for the total data set and for each species group were presented. High levels of Gst were detected in the total data set (cpDNA: 0.45; mtDNA: 0.64; microsatellites: 0.41), and similar patterns were detected in A. argotaenia (cpDNA: 0.40; mtDNA: 0.55; microsatellites: 0.24) and A. yunnanensis (cpDNA: 0.55; mtDNA: 0.67; microsatellites: 0.19).
Pairwise comparisons of genetic differentiation between species and between regions were conducted using DnaSP. Significant genetic differentiation was detected between species. Amentotaxus yunntanensis was closely related to A. poilanei, as indicated by the lowest Fst values for all loci. Amentotaxus argotaenia populations from the Central region were related most closely to populations from the West at all loci but cpDNA (Table 6).
We used mismatch distribution analyses to infer the long-term demographic history of populations. A bimodal mismatch distribution was displayed in populations H, T, and YX for the cpDNA, and populations X, Q, T, and YX for mtDNA. Unimodal distributions were detected in F, X, YM, and VL for cpDNA, and in H, HU, J, E, and QI populations for mtDNA (Tables 2–3). All loci displayed a bimodal or unimodal mismatch in most regions at the geographical level. The data used to generate mismatch distributions are not independent (Groth and Barrowclough 1999), therefore Tajima’s D, Fu and Li’s D*, and Fu’s Fs statistics were applied to detect departures from population equilibrium. In this study, most of Tajima’s D, Fu and Li’s D* values yielded nonsignificant statistics for the three markers (Tables 2–3). The Bayesian skyline plot was applied to infer past demographic patterns; all three loci revealed a long history of constant population sizes for each species, followed by subsequent demographic expansion (Fig. 5).

Bayesian skyline plots combing organelle DNA datasets for the effective population size fluctuation through time. Y-axes Population size × generation time; x-axes time [years before present (million years ago)]. a Amentotaxus complex, b A. argotaenia, c A. formosana, d A. yunnanensis, e A. poilanei. Solid lines Median estimations; area between gray dashed lines 95 % confidence intervals
Effective Population Size and Gene Flow
The IM program was used to refine six demographic parameters in Amentotaxus based on organellar DNA and the microsatellite data matrix. All pairwise comparisons indicated that ancestral effective population sizes were larger than the present, suggesting demographic shrinking (Supplementary Table S1). SSR fingerprints estimated an ancestral effective population size hundreds of times greater than the present size. The migration parameters further revealed a peak at the lower resolution limits in one direction, i.e., from A. yunnanensis to A. poilanei, from A. argotaenia to A. poilanei, and from A. formosana to A. poilanei in organellar DNAs. The population migration rates [M12 = N1 m 12 = m12*(θ 1/4)] estimated from A. poilanei to the three other species were substantially lower than vice versa. However, approximating estimates between ancestral and recent gene flow were obtained based on SSRs. Between inland and island species, the estimates of gene flow were not consistent among markers (Supplementary Table S1), revealing extremely low, recent gene flow based on SSRs. Altogether, at the species level, nonzero and asymmetric migrations were detected ever since species divergence.
Molecular Dating
Based on the maximum likelihood reconstructed tree, the node coalescence times were estimated using the BEAST program. For cpDNA, all haplotypes coalesced to approximately 3.90–10.28 MYA, while lineages A + B + C + D + E and F coalesced to 1.97–5.20 MYA. The coalescence of two primary mtDNA lineages (I + II + III + IV + V and VI + VII) in Amentotaxus was dated to 14.93–22.40 MYA. Similarly, A. formosana lineage VI and A. argotaenia lineage VII diverged 13.93–20.09 MYA; while A. yunnanensis lineage II and A. argotaenia lineage I did not coalesce until recently, approximately 2.76–4.14 MYA (Fig. 2).
Between-species divergence times estimated by organellar DNAs were calculated based on the isolation with migration model using the T = t/u formula. Pairwise comparisons revealed divergence times between species ranging from 1.10 to 13.84 MYA, and divergence between island and inland species ranging from 2.40 to 13.84 MYA (Supplementary Table 1). Divergence times between inland species were estimated from 1.10 to 2.31 MYA, with the longest divergence between A. yunnanensis and A. poilanei.
Discussion
Species Delimitation in Amentotaxus
Amentotaxus is an ancient genus with origins that date to the early Tertiary, and this genus diverged from Torreya ca. 55 MYA (Leslie et al. 2012). Members of the genus survived Pleistocene climatic oscillations, and must have experienced range contractions, species extinctions, and subsequent diversification. Molecular dating revealed a long period of isolation among Amentotaxus species. Monophyletic species are therefore expected. However, cp- and mtDNAs revealed para- and polyphyletic Amentotaxus species (Fig. 2). The absence of reciprocal monophyly might result from lineage sorting (Chiang and Schaal 2006; Chiang et al. 2009), or hybridization/introgression (Chiang et al. 2001). Stochastic effects of genetic drift might have also resulted in the systematic inconsistencies between gene trees at cp- and mtDNA markers (Chiang et al. 2001, 2009).
Hennigian cladistists would recognize the four taxa as a single species, as reciprocal monophyly is the only criterion for grouping taxa (Dias et al. 2005; Potter and Freudenstein 2005). Nevertheless, Chiang and Schaal (2006) indicated that paraphyly/polyphyly is a rule rather than an exception for the plant species complexes of East Asia, and is not even rare between/among well-defined species. In addition, gymnosperm species with East Asian geographic divisions revealed shared haplotypes due to ancient ancestral polymorphisms (Chiang et al. 2006, 2009). Various approaches to species delimitations have been developed. Wiens and Penkrot’s (2002) methodology serves to identify species exclusivity instead of reciprocal monophyly, where the term “exclusive” was used to describe haplotype phylogenies. If the haplotypes were not exclusive within species, and instead paraphyletic or polyphyletic with respect to other species, the focal species would likely represent multiple species. In the present study, mtDNA genetic analyses, which showed stronger geographic structuring and deeper divergence in phylogenetic gene tree clustering than other loci, was adequate to assess species delimitations and phylogeographical patterns (Fig. 2b). Case study of the Pinaceae species in central and southern China also suggested that maternally inherited mtDNA markers are more prone to introgression than paternally inherited cpDNA markers, and the latter therefore provide better resolution when delimitating species (Zhou et al. 2010; Wang et al. 2011; Peng et al. 2012). The populations of A. formosana were relatively unique, with no genetic compositions shared with other species (Figs. 2b and 4b). Lineage VI was detected in A. yunnanensis and A. poilanei; however the H13 haplotype nested in A. argotaenia (lineage III) indicated polyphyletic origins of A. yunnanensis. The assignment test of microsatellite fingerprints also indicated that genetic compositions of A. yunnanensis tended to be similar to A. poilanei (Fig. 4b). Following the Wiens-Penkrot methodology, A. formosana represents a well-defined species. In contrast, A. argotaenia, A. yunnanensis, and A. poilanei might possibly represent identical species. Significant genetic differentiation between species revealed by STRUCTURE analyses of microsatellite fingerprints (Fig. 4), and *BEAST population tree of organelle DNA (Supplementary Fig. 1), consistent with ISSR fingerprinting (Ge et al. 2005), further supported A. argotaenia and A. yunnanensis represent independent species; however, data support the recognition of A. yunnanensis and A. poilanei as one species.
Divergence times from 1.10 to 13.84 MYA among the four allopatric species (Supplementary Table S1) coincided with late Tertiary and Quaternary glaciations (Wang et al. 2013). The complex landscapes and fragmented habitats had significant effects on spatial and temporal patterns of genetic polymorphisms (Hewitt 1996), and speciation in Amentotaxus. In addition, divergence between A. formosana in Taiwan, and A. argotaenia can be traced back 2.40 MYA, a time period that approximates the coalescence of haplotypes resolved in A. formosana, suggesting that genetic divergence following colonization in the island may have occurred. The progenitor–derivative speciation scenario also indicated that island species Picea morrisonicola derived from continental species P. wilsonii within the more recent past (Zou et al. 2013). In this study, significant genetic differentiation was detected between A. argotaenia and A. formosana at all markers (Table 6), including ISSR fingerprints (Fst = 0.761 – 0.850, Ge et al. 2005). The facts no mtDNA haplotypes were shared among species, and private cpDNA haplotypes were detected within species decreased the possibility of recurrent interspecific gene flow.
Apparently, cpDNA provided evidence for species paraphyly, and the maintenance of long-term ancestral polymorphisms within species. Such a phenomenon, regulated by random genetic drift across population fragments, often occurred in once widespread relict species in Pliocene forests, which subsequently became fragmented during the Quaternary. Vendramin et al. (2000) reported population-specific haplotypes as a general attribute in long diverged conifer species. Furthermore, recent population fragmentation accelerated genetic differentiation among populations, and fixation of alleles within populations/species.
In contrast to the inferred long divergence time between A. argotaenia and A. formosana, divergence between A. argotaenia, A. yunnanensis, and A. poilanei was more recent, between 1.10 to 2.31 MYA in the inland species. The three mainland species exhibit high morphological similarities; however different evolutionary processes can serve to explain the shared character traits. Only one haplotype (M9) was shared by A. poilanei and A. yunnanensis, indicating a descent-descendent relationship. A polyphyletic origin of A. yunnanensis suggests the divergence time was overestimated, with three haplotypes (M9, 10 and 17) representing lineage II and one haplotype (M26) in lineage V (Fig. 2b). Lineage II, including A. yunnanensis was sister to lineage I, which included A. argotaenia populations. Accordingly, the coalescence of Yunnan’s haplotypes was even more recent. In contrast, the phylogenetically distant haplotype M26 likely represents immigrants from population X (M19 and 23) of A. argotaenia (Fig. 3b) (cf. Matos and Schaal 2000). All A. yunnanensis populations (including S) showed evidence of clustering based on ISSR (Ge et al. 2005) and microsatellite fingerprinting, plus mismatch analyses displaying a bimodal distribution, which supports a scenario of two lineages merging within A. yunnanensis due to obtaining the foreign M26 haplotype. This hybrid origin hypothesis, nevertheless, remains to be tested.
Recurrent gene flow via seeds and pollen, between A. yunnanensis and A. argotaenia is constrained by geographical barriers and ecological preferences, as indicated by significant genetic differentiation based on all markers in this study, and ISSR fingerprinting at the species level (Ge et al. 2005). The strong genetic structure at the species level (Fig. 4), and extreme low levels of recent migration (Supplementary Table S1) inferred from microsatellites suggest again that historical gene flow was likely between the species. Providing a low possibility of recurrent genetic exchanges, coancestry instead accounts for the lower level of genetic differentiation. Accordingly, morphological similarities in Amentotaxus are likely attributable to high developmental constraints. Nevertheless, diversification and allopatric distribution in Amentotaxus do not guarantee a model of allopatric speciation. Nonzero and asymmetric gene flow among species inferred from the three organellar markers revealed that gene flow lasted for a certain period following species divergence, particularly given historical connectivity among species (Wang et al. 2000). This finding further confirms the recognition of paraphyletic species in a recently evolving species complex.
Low Genetic Diversity within Species versus High Genetic Differentiation between Species/Populations
Organellar DNA genetic variation in the Amentotaxus complex was low compared with many other species (Chiang et al. 2004, 2009; Ge et al. 2012). Similarly, for microsatellite loci, all populations/species in this complex exhibited lower observed heterozygosity than other conifers (Moriguchi et al. 2009; Parchman et al. 2011). Rare species tend to possess low levels of genetic diversity due to stochastic losses of genetic polymorphisms resulting from genetic drift. Low levels of genetic variability at all loci examined suggested demographic fragmentation caused the notable loss of genetic variation. Nevertheless, the loci did not show congruent variability. For example, A. argotaenia possessed the highest levels of mtDNA variation and microsatellite allele number, while having the lowest level of cpDNA diversity (Tables 2–4). In contrast, there were no significant differences among species for the haplotype richness for all loci (P = 0.19, cpDNA; P = 0.43, mtDNA; P = 0.08, microsatellites). The larger numbers of private alleles led A. argotaenia to have higher levels of mtDNA and microsatellites variations than cpDNA (Tables 2–4). These results indicated stochastic effects of genetic drift.
Hierarchical AMOVA revealed significant genetic differentiation between species and among geographical regions/populations in this complex (Tables 5–6), a pattern congruent with the population structuring based on ISSR fingerprinting (Ge et al. 2005). The reconstructed phylogeographical patterns suggested colonization likely occurred at the time of Taiwan’s formation via long distance dispersal in A. formosana (Lin 1966) from F or LQ populations of the East region that are geographically near the Taiwan population, and characterized by fixation of the M1 haplotype, closely related to M14 (Figs. 2–3). Geological evidence shows Taiwan was connected to the Asian landmass by a land bridge during the LGM as sea levels dropped to 120 m lower than present (Liu 1988). The land bridge enhanced gene flow in many plants (Chiang and Schaal 2006). Nevertheless, in this study, results suggested gene flow between populations of the mainland and Taiwan during the Holocene were very restricted or unlikely to have occurred. The vicariant event was the formation of the Taiwan Strait, which we can conclude prevented gene flow between mainland and island populations. Subsequently, as no foreign alleles were introgressed into Taiwan species, many ancestral polymorphisms were stochastically lost and replaced by newly mutated haplotypes, resulting in monophyly within A. formosana and its divergence from A. argotaenia in mtDNA.
Spanning a large distributional range, A. argotaenia exhibited the highest genetic diversity levels in the species complex, evidenced by two major mtDNA lineages I + II + III + IV + V and VI + VII, which diverged about 22.4 MYA (Fig. 2b). Furthermore, geographical subdivision, i.e., mtDNA clades I + II + III + IV + V of West and Central regions, and VI + VII of the East region and population X (Central region), also contributed to high diversity levels. The divergence times between A. argotaenia and other three species ranging from 1.10 to 2.39 MYA indicated a large effective population size of widespread species based on IMa analysis (Supplementary Table 1). Nevertheless, most populations today remain small in the wild, and possess low levels of nucleotide diversity (Table 3). Data suggested habitat fragmentation triggered genetic differentiation among populations. Even so, only three populations of A. argotaenia were fixed at a single allele, while most populations were polymorphic, a pattern also occurring in A. formosana and A. yunnanensis. In contrast, A. poilanei was fixed at a single allele, likely resulting from stochastic losses. Furthermore, limited geographic distribution of low-frequency alleles represents footprints of recent demographic expansion (Chiang et al. 2006).
A general assumption inherent to most phylogeographic studies of postglacial colonization is that glacial refugia are expected to harbor more genetic diversity than recolonized areas (Comes and Kaderaeit 1998; Hewitt 1996, 1999, 2000; Petit et al. 2003). Rapid range expansions out of refugia involving serial population bottlenecks in founder events are expected to exhibit decreasing genetic diversity, particularly in allele or haplotype number, as sampling extends away from source populations (Hewitt 1996). Increased genetic diversity in refugia often reflects stable population dynamics and large population sizes (Hewitt 1996). Therefore, populations with high levels of genetic variation are generally recognized as refugia (DeChaine and Martin 2005). Here, population X with high ISSR fingerprinting diversity was also among the highest genetic levels based on all molecular markers (Tables 2–4) in Amentotaxus populations (Ge et al. 2005), reflecting features of a refugium.
Nevertheless, a large population size alone would not explain sustained maintenance of long diverged lineages. Bimodal mismatch distributions for mtDNA (Table 3) suggested that population X was likely a melting pot that maintained genetic diversity. In addition, populations H and E might represent other Quaternary refugia based on mismatch distributions, and higher genetic diversity levels in most genetic markers (Table 2). Phylogeographical analyses of mtDNA variant spatial distributions in Amentotaxus identified refugia in southwest (Sichuan Province) and central (Hunan Province) China (Fig. 1), congruent with fossil evidence (Axelrod et al. 1996). However, we did not detect refugia in eastern China (Del Tredici et al. 1992).
Amentotaxus yunnanensis exhibited very low mtDNA diversity levels, consistent with small populations (Table 4). However, all individuals from population S possessed the M26 haplotype—the haplotype delimiting lineage V—and clustered within A. argotaenia lineages III and IV (Figs. 2–3). If population S was composed of foreign migrants, the level of genetic diversity across the remaining populations would be reduced dramatically in mtDNA (π = 1.12× 10−3 and θ = 0. 72× 10−3)—a level even lower than in A. formosana (Table 2). Consequently, colonization via small founders must have occurred; leading not only to low genetic variation, but also to a unique genetic composition in each population. Interestingly, although population S was fixed in organellar DNA, microsatellites and ISSR fingerprinting displayed levels of variation that exceeded other populations (Ge et al. 2005). A demographic bottleneck might have occurred in this population, driving the cpDNA and mtDNA loci towards fixation, followed by subsequent expansion that recovered genetic polymorphisms at nuclear loci. Lower haplotype numbers and levels of genetic diversity were detected in A. poilanei compared with other species, indicating effects of recent demographic bottlenecks. Dominant haplotype fixation within species for most loci contributed to the depletion of genetic diversity in A. poilanei, and the absence of genetic differentiation between A. yunnanensis and A. poilanei. However, increased levels of genetic diversity were detected in microsatellites, showing a similar demographic history in A. yunnanensis (Table 5). Sample size is one of the critical factors affecting the accuracy of the estimation of population genetic diversity. Here all markers suggested similar patterns. Nucleotide diversity of A. poilanei (θ =1.86 × 10−3) in cpDNA was close to that of A. argotaenia (θ = 1.75 × 10−3), both with larger sample size, whereas lower than that of A. yunnanensis (θ = 3.21 × 10−3), altogether indicating that the lower genetic diversity of A. poilanei likely resulted from genetic drift rather than from small sample size. In summary, long diverged Amentotaxus species exhibited significant genetic differentiation across the species’ complex geographic range, but low genetic diversity within species/populations, likely due to past fragmentation and recent habitat degradation. In contrast to ecologically pioneering gymnosperms, e.g., Pinus, Amentotaxus species are subordinate in well-developed forests, where forest canopies prevent seed and pollen exchange among populations. Most gene flow therefore is restricted within populations. In addition, human overexploitation has accelerated isolation among population fragments, leading to threats of extinction for these relic species.
References
Axelrod I, Ai-Shehba I, Raven PH (1996) History of the modern flora of China. In: Zhang A, Wu A (eds) Floristic characteristics and diversity of East Asian plants. Springer, New York, pp 43–55
Belshaw R, Katzourakis A (2005) BlastAlign: a program that uses blast to align problematic nucleotide sequences. Bioinformatics 21:122–123
Cheng WC (1978) Flora Republicae Popularis Sinicae. vol. 7, The genus Amentotaxus, vol 7. Science, Beijing
Chiang TY, Schaal BA (2006) Phylogeography of plants in Taiwan and the Ryukyu Archipelago. Taxon 55:3–41
Chiang TY, Chiang YC, Chou CH, Havanond S, Hong TN, Huang S (2001) Phylogeography of Kandelia candel in East Asiatic mangroves based on nucleotide variation of chloroplast and mitochondria DNAs. Mol Ecol 10:2697–2710
Chiang TY, Hung KH, Hsu TW, Wu WL (2004) Lineage sorting and phylogeography in Lithocarpus formosanus and L. dodonaeifolius (Fagaceae) from Taiwan. Ann Mo Bot Gard 91:207–222
Chiang YC, Hung KH, Schaal BA, Ge XJ, Hsu TW, Chiang TY (2006) Contrasting phylogeographical patterns between mainland and island taxa of the Pinus luchensis complex. Mol Ecol 15:765–779
Chiang YC, Hung KH, Moore SJ, Ge XJ, Huang S, Hsu TW, Schaal BA, Chiang TY (2009) Paraphyly of organelle DNAs in Cycas sect. Asiorientales due to ancient ancestral polymorphisms. BMC Evol Biol 9:161
Comes HP, Kaderaeit JW (1998) The effect of Quaternary climatic changes on plant distribution and evolution. Trends Plant Sci 3:432–438
DeChaine EG, Martin AP (2005) Marked genetic divergence among sky island populations of Sedum lanceolatum (Crassulaceae) in the Rocky Mountains. Am J Bot 92:477–486
Del Tredici P, Lin G, Yang G (1992) The Ginkgos of Tian Mu Shan. Conserv Biol 4:202–209
Dias P, Assis LCS, Udulutsch RG (2005) Monophyly vs paraphyly in plant systematics. Taxon 54:1039–1040
Doyle JJ, Doyle JL (1987) A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull 19:11–15
Drummond AJ, Ho SY, Phillips MJ, Rambaut A (2006) Relaxed phylogenetics and dating with confidence. PLoS Biol 4:e88
Earl DA, vonHoldt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Smouse PE (1994) Using allele frequencies and geographic subdivision to reconstruct gene trees within a species: molecular variance parsimony. Genetics 136:343–359
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA data. Genetics 131:479–491
Excoffier L, Laval G, Schneider S (2005) Arlequin (version 3.0): an integrated software package for population genetics data analysis. Evol Bioinform Online 1:47–50
Farjon A (2010) A handbook of the world’s conifers. Brill, Leiden
Felsenstein J (1985) Confidence limits on phylogenies: an approach using the bootstrap. Evolution 39:783–791
Felsenstein J (2007) PHYLIP (Phylogeny Inference Package), version 3.67, Department of Genetics, University of Washington, Seattle.
Ferguson DK, Jahnichen H, Alvin KL (1978) Amentotaxus Pilger from the European Tertiary. Feddes Repert 89:379–410
Fu YX (1997) Statistical tests of neutrality of mutations against population growth, hitchhiking, and background selection. Genetics 147:915–925
Fu YX, Li WH (1993) Statistical tests of neutrality of mutations. Genetics 133:693–709
Ge XJ, Zhou XL, Li ZC, Hsu TW, Schaal BA, Chiang TY (2005) Genetic structure of the relict Amentotaxus argotaenia complex (Taxaceae) based on ISSR fingerprinting. J Plant Res 118:415–422
Ge XJ, Hsu TW, Hung KH, Lin CJ, Huang CC, Huang CC, Chiang YC, Chiang TY (2012) Inferring multiple refugia and phylogeographical patterns in Pinus massoniana based on nucleotide sequence variation and fingerprinting. PLoS One 7:e43717
Goudet J (1995) FSTAT Version 2.9.3.2: a computer program to calculate F-statistics. J Hered 86:485–486
Groth JG, Barrowclough GF (1999) Basal divergences in birds and the phylogenetic utility of the nuclear RAG-1 gene. Mol Phylogenetd Evol 12:115–123
Harris TM (1976) Two neglected aspects of fossil conifers. Am J Bot 63:902–910
Harrison SP, Yu G, Takahar H, Prentice IC (2001) Diversity of temperate plants in east Asia. Nature 413:129–130
Hewitt GM (1996) Some genetic consequences of ice ages, and their role in divergence and speciation. Biol J Linn Soc 58:247–276
Hewitt GM (1999) Post-glacial re-colonization of European biota. Biol J Linn Soc 68:87–112
Hewitt GM (2000) The genetic legacy of the Quaternary ice ages. Nature 405:907–913
Hey J, Nielsen R (2004) Multilocus methods for estimating population sizes, migration rates, and divergence time, with applications to the divergence of Drosophila pseudoobscura and D. persimilis. Genetics 167:747–760
Ho CS, Chen CT, Ko YZ, Ge XJ, Hung KH, Hsu TW, Chiang YC (2012) Isolation and characterization of 15 microsatellite loci in four endangered Amentotaxus species (Taxaceae). Am J Bot 99:e157–159
IUCN (2013) The IUCN Red List of Threatened Species. Version 2013.2 http://www.iucnredlist.org
Kalinowski ST (2005) HP-RARE 1.0: a computer program for performing rarefaction on measures of allelic richness. Mol Ecol Notes 5:187–189
Leslie AB, Beaulieu JM, Rai HS, Crane PR, Donoghue MJ, Mathews S (2012) Hemisphere-scale differences in conifer evolutionary dynamics. Proc Natl Acad Sci USA 109:16217–16221
Li HL, Keng H (1994) Amentotaxaceae. In: Editorial Committee of the Flora of Taiwan (ed) Flora of Taiwan, 2nd edn. Vol. 1. Department of Botany, National Taiwan University, Taipei, pp 553–554
Lin CC (1966) An outline of Taiwan’s Quaternary geohistory with a special discussion of the relation between natural history and cultural history in Taiwan. Bull Dept Archaeol Anthropol 23:7–44
Liu KB (1988) Quaternary history of the temperate forests of China. Q Sci Rev 7:1–20
Liu HP (1991) The spore-pollen communities and paleoenvironment since Quaternary in the reach of Chang Jiang River. J Huazhong Normal Univ Nat Sci 2:236–241
Matos JA, Schaal BA (2000) Chloroplast evolution in the Pinus montezumae complex: a coalescent approach to hybridization. Evolution 54:1218–1233
Melo-Ferreira J, Boursot P, Carneiro M, Esteves PJ, Farelo L, Alves PC (2012) Recurrent introgression of mitochondrial DNA among hares (Lepus spp.) revealed by species-tree inference and coalescent simulations. Syst Biol 61:367–381
Moriguchi Y, Kang KS, Lee KY, Lee SW, Kim YY (2009) Genetic variation of Picea jezoensis populations in South Korea revealed by chloroplast, mitochondrial and nuclear DNA markers. J Plant Res 122:153–160
Müller K (2005) SeqState - primer design and sequence statistics for phylogenetic DNA data sets. Appl Bioinform 4:65–69
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York
O’Connell LM, Ritland K (2004) Somatic mutations at microsatellite loci in western red cedar (Thuja plicata: Cupressaceae). J Hered 95:172–176
Parchman TL, Benkman CW, Jenkins B, Buerkle CA (2011) Low levels of population genetic structure in Pinus contorta (Pinaceae) across a geographic mosaic of co-evolution. Am J Bot 98:669–679
Peng Y, Yin S, Wang J, Tian B, Ren G, Guo Q, Liu J (2012) Phylogeographic analysis of the fir species in southern China suggests complex origin and genetic admixture. Ann Forest Sci 69:409–416
Petit RJ, Aguinagalde I, de Beaulieu JL, Bittkau C, Brewer S, Cheddadi R, Ennos R, Fineschi S, Grivet D, Lascoux M, Mohanty A, Müller-Starck G, Demesure-Musch B, Palmé A, Martín JP, Rendell S, Vendramin GG (2003) Glacial refugia: hotspots but not melting pots of genetic diversity. Science 300:1563–1565
Pons O, Petit RJ (1996) Measuring and testing genetic differentiation with ordered versus unordered alleles. Genetics 144:1237–1245
Potter D, Freudenstein JV (2005) Character-based phylogenetic Linnaean classification: taxa should be both ranked and monophyletic. Taxon 54:1033–1035
Pritchard J, Wen W (2003) Documentation for STRUCTURE Software, version 3.2. http://pritch.bsd.uchicago.edu/structure.html
Pritchard J, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Rambaut A, Drummond A (2004) Tracer–MCMC trace analysis tool. University of Oxford, Oxford
Ray N, Adams JM (2001) A GIS-based vegetation map of the world at the last glacial maximum (25,000-15,000 BP). Internet Archaeology 11. Available from: http://www.ncdc.noaa.gov/paleo/pubs/ray2001/ray_adam_2001.pdf
Rogers AR, Harpending H (1992) Population growth makes waves in the distribution of pairwise genetic differences. Mol Biol Evol 9:552–569
Royer DL, Hickey LJ, Wing SL (2003) Ecological conservatism in the “living fossil” Ginkgo. Paleobiology 29:84–104
Rozas J, Sánchez-DelBarrio JC, Messeguer X, Rozas R (2003) DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19:2496–2497
Slatkin M, Hudson RR (1991) Pairwise comparisons of mitochondrial DNA sequences in stable and exponentially growing populations. Genetics 129:555–562
Stewart WN, Rothwell GW (1993) Paleobotany and the evolution of plants, 2nd edn. Cambridge University Press, Cambridge
Taberlet P, Fumagalli L, Wust-Saucy AG (1998) Comparative phylogeography and postglacial colonization routes in Europe. Mol Ecol 7:453–464
Tajima F (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123:585–595
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739
Tong G, Zhang J, Fan S (1992) Distribution of Quaternary palynoflora in China. Marine Geol Quat Geol 12:45–56
Tong GB, Chen Y, Wu XH, Li ZH, Yang ZJ, Wang SB, Cao JD (1999) Pleistocene environmental megaevolution and indicated by the sporopollen floras in China. J Geophys Res 5:11–21
Tran TTT (2011) Spatial distribution and historical dynamics of threatened conifers of the Dalat Plateau. Master thesis. University of Missouri, Columbia
Traula H (1968) The phytogeographic evolution of the genus Ginkgo L. Bot Notiser 120:409–422
Vendramin GG, Anzidei M, Madaghiele A, Sperisen C, Bucci G (2000) Chloroplast microsatellite analysis reveals the presence of population subdivision in Norway spruce (Picea abies K.). Genome 43:68–78
Wachowiak W, Prus-Glowacki W (2008) Hybridisation processes in sympatric populations of pines Pinus sylvestris L., P. mugo Turra and P. uliginosa Neumann. Plant Syst Evol 271:29–40
Wang JP, Hsu KC, Chiang TY (2000) Mitochondrial DNA phylogeography of Acrossocheilus paradoxus (Cyprinidae) in Taiwan. Mol Ecol 9:1483–1494
Wang JF, Pan YZ, Gong X, Chiang YC, Kuroda C (2011) Chloroplast DNA variation and phylogeography of Ligularia tongolensis (Asteraceae), a species endemic to the Hengduan Mountains region of China. J Syst Evol 49:108–119
Wang J, Abbott RJ, Peng YL, Du FK, Liu JQ (2012) Species delimitation and biogeography of two fir species (Abies) in central China: cytoplasmic DNA variation. Heredity 107:362–370
Wang JF, Gong X, Chiang YC, Kuroda C (2013) Phylogenetic patterns and disjunct distribution in Ligularia hodgsonii Hook. (Asteraceae). J Biogeogr 40:1741–1754
Watterson GA (1975) On the number of segregating sites in genetical models without recombination. Theor Popul Biol 7:256–276
Wiens JJ, Penkrot TA (2002) Delimiting species using DNA and morphological variation and discordant species limits in spiny lizards (Sceloporus). Syst Biol 51:69–91
Wolfe KH, Li WH, Sharp PM (1987) Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc Natl Acad Sci USA 84:9054–9058
Xu JX, Ferguson DK, Li CS, Wang YF (2008) Late Miocene vegetation and climate of the Lühe region in Yunnan, southwestern China. Rev Palaeobot Palynol 48:36–59
Zhou QX (2001) Studies on systematics of the Taxaceae. Dissertation, Kunming Institute of Botany, Kunming, China
Zhou B, Li G, Yao M, Fang L (1994) An analysis on the periglacial geomorphy and paleogeographic environment during the Quaternary of the Mount Jiuhuashan. J Anhui Normal Univ Natur Sci 17:48–53
Zhou YF, Abbott RJ, Jiang ZY, Du FK, Milne RI, Liu JQ (2010) Gene flow and species delimitation: a case study of two pine species with overlapping distributions in southeast China. Evolution 64:2342–2352
Zou J, Sun Y, Li L, Wang G, Yue W, Lu Z, Wang Q, Liu J (2013) Population genetic evidence for speciation pattern and gene flow between Picea wilsonii, P. morrisonicola and P. neoveitchii. Ann Bot 112:1829–1844
Acknowledgments
We thank Chao-Yi Deng, Guo-Sheng He, Yun-Fei Deng and Hua-Gu Ye for their help with material sampling. We also thank Joanna Schultz (Lewis-Clark State College, ID) and two reviewers for their helpful comments. This study was supported financially by National Basic Research Program of China (973 Program) (Grant No. 2014CB954100) National Natural Science Foundation of China (No. 30170070), and the National Science Council of Taiwan (NSC 96-2628-B-020 -001 -MY2 and NSC 102-2628-B-110 -001 -MY3).
Author information
Authors and Affiliations
Corresponding authors
Additional information
X.-J. Ge and K.-H. Hung contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
ESM 1
(DOC 77 kb)
Rights and permissions
About this article

Cite this article
Ge, XJ., Hung, KH., Ko, YZ. et al. Genetic Divergence and Biogeographical Patterns in Amentotaxus argotaenia Species Complex. Plant Mol Biol Rep 33, 264–280 (2015). https://doi.org/10.1007/s11105-014-0742-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11105-014-0742-0