
Abstract
Genetic variation associated with Picea jezoensis populations of South Korea was investigated using chloroplast (cp), mitochondrial (mt) and nuclear DNA markers. In South Korea, P. jezoensis is distributed across a very restricted area, being found on the summits of three mountains: Mts. Jiri, Dokyu and Gyebang. Examination of five region restriction enzyme combinations for mtDNA and four for cpDNA revealed haplotypes endemic to South Korea. The Gyebang population, the most northerly and most isolated, was genetically distinct from the other populations. Nuclear microsatellite markers indicated, overall, a low level of genetic diversity (H e = 0.406) in South Korea; this could be attributed to genetic drift and/or founder effects associated with historical events. The Wilcoxon sign-rank test did not indicate a recent bottleneck in any of the populations irrespective of the model considered (infinite allele model, two-phased model of mutation, and stepwise mutation model). Microsatellite markers also demonstrated that the Gyebang population was distinct from the others. The results of this study could be used as the basis for conservation guidelines for the management of this species in South Korea.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Picea jezoensis (Sieb. et Zucc.) Carr. is a common and widespread tree species in the cold temperate and boreal forests of east Russia, northeast China, Japan and Korea. P. jezoensis has three varieties based on geographical distribution: P. jezoensis var. jezoensis; P. jezoensis var. hondoensis; and P. jezoensis var. koreana (synonymous with P. jezoensis var. komarovii). Picea jezoensis var. jezoensis is distributed across northern Korea, northeast China, Sakhalin, Kamchatka, the Kuriles and Hokkaido Island in Japan. Picea jezoensis var. hondoensis is restricted to high elevations in the central region of Honshu Island, Japan. Picea jezoensis var. koreana is found on the Korean peninsula and in the southern part of China (Yamazaki 1995). In this study we do not refer to a named variety, since no P. jezoensis variety has been associated with the species in South Korea (Lee 1982, 1985). In South Korea, P. jezoensis has a very limited distribution, growing only on three mountain summits: those of Mts. Jiri, Dokyu and Gyebang. Because of its restricted distribution and ecological value, P. jezoensis is of great conservation importance in South Korea.
Genetic diversity is necessary to facilitate evolutionary responses to environmental change. As such, the World Conservation Union (IUCN) classifies genetic variation as one of the key aspects of biodiversity to be conserved. In addition, information pertaining to genetic variation is essential when planning both in situ and ex situ gene conservation strategies.
In the Pinaceae, the mitochondrial genome is inherited maternally and the chloroplast genome is inherited paternally (Neale and Sederoff 1989; Sutton et al. 1991; Hipkins et al. 1994; Chen et al. 2002). The DNA in these organelles can be used to investigate genetic differentiation among populations. Maternally inherited mitochondrial DNA (mtDNA) is more useful for determining genetic differentiation than is biparentally inherited nuclear-rDNA or paternally inherited chloroplast DNA (cpDNA) (Neale and Sederoff 1988). However, nuclear microsatellites are suitable for investigating genetic diversity since they are co-dominant markers and have relatively high polymorphism even in populations with low allozyme polymorphism (Hughes and Queller 1993; Sugaya et al. 2003).
In the present study, using chloroplast (cp), mitochondrial (mt) and nuclear DNA markers, we investigated the genetic variation in the extant P. jezoensis populations in South Korea. The results of this study could be used to inform the gene conservation program of this species in South Korea.
Materials and methods
Study sites, plant materials and total DNA extraction
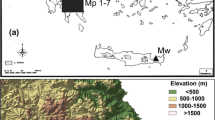
The locations of the four natural populations of P. jezoensis that we studied are shown in Fig. 1. Of the four populations, Gyebang is the smallest and the most northerly; it is over 200 km away from the other three populations. The Jiri-Chunwang and Jiri-Banya populations are located at the most southerly extent of the range of P. jezoensis, and occupy two separate peaks of the Jiri Mountains. These two populations are 10 km apart. The Dokyu population is located between the Gyebang and Jiri populations, but is closer to the latter.

The location of the studied populations of Picea jezoensis. Numbers represent each population sampled (1 Jiri-Chunwang, 2 Jiri-Banya, 3 Dokyu, 4 Gyebang)
We collected needles from four populations of P. jezoensis in South Korea between late February and mid May 2007. Within each population, we tried to sample needles from more than 30 trees, separated by a minimum distance of 30 m in order to avoid the risk of sampling closely related individuals. Although, we aimed to sample at least 30 trees, this was not possible in the Gyebang population because of its small size (Table 1).
Total genomic DNA was isolated from 25 mg needle tissue following the protocol described by Qiagen (DNeasy plant mini handbook; Qiagen, Hilden, Germany).
Organelle DNA PCR-RFLP analysis
The five region restriction enzyme combinations for mtDNA, nad1/Hin6I (HhaI), nad1/HinfI, nad1/HpyF3I (DdeI), nad1/Eco32I (EcoRV) and mh02/Tru1I (Tru9I), and the four for cpDNA, trnK/DraI, trnC-trnD/DraI, trnC-trnD/RsaI and trnD-trnT/TaqI, that were used in this study had showed polymorphism in a previous study (Aizawa et al. 2007).
Polymerase chain reactions (PCR) were performed in a volume of 15 μL with final concentrations of 15 ng genomic DNA; 0.1 mM of each of the four dNTPs; 2 mM MgCl2 (ABgene, Epsom, UK); 1×PCR buffer (ABgene); 0.3 U Taq DNA polymerase (ABgene) and 0.2 μM of each primer. The PCR thermal profile was as follows: an initial denaturing step of 5 min at 94°C followed by 35 cycles of 45 s at 94°C (denaturation), 45 s at 53–62°C (annealing), and 2 min 30 s at 72°C (extension) before final elongation at 72°C for 5 min in a PTC-200 thermal cycler (MJ Research, Waltham, MA) or a Tetrad 2 thermal cycler (Bio-Rad, http://www.bio-rad.com/).
To confirm the presence of amplification, 5 μL of the PCR products were separated on a 1% agarose gel stained with ethidium bromide in 1× TBE buffer. After checking the successful amplification of the mtDNA and cpDNA regions, the PCR products were digested separately with the restriction enzymes. These restriction reactions were carried out for 2 h at 37°C (65°C for TaqI and Tru1I) by adding 10 μL PCR product to a mixture containing 1.1 μL 10× enzyme buffer (Fermentas, St. Leon-Rot, Germany) and 3 U of the enzyme. The restriction fragments were separated on a 1% agarose gel containing 0.005% ethidium bromide in 1× TBE buffer at 200 V for 0.5–1 h then visualized and photographed under UV light. The haplotype name and the restriction fragment length of each of the region restriction enzyme combinations followed Aizawa et al. (2007).
Analysis of data obtained from organelle DNA PCR-RFLP analysis
The F ST values (Weir and Cockerham 1984) of all mtDNA and cpDNA loci were calculated using Arlequin (Schneider et al. 1997).
To determine the phylogenetic relationships between the mtDNA haplotypes, a median-joining network was constructed using NETWORK software (http://www.fluxus-engineering.com; Bandelt et al. 1999). Haplotype data for populations from Japan, Russia and China were obtained from Aizawa et al. (2007) and were combined with the data from the current study to construct the haplotype network.
Nuclear DNA microsatellite analysis
We used six microsatellite primer pairs developed in Picea species (SpAGG3, SpAGD1 and SpAGC1, Pfeiffer et al. 1997; UAPgAG150A, UAPgAG105 and UAPgAC/AT6, Hodgetts et al. 2001; see details in Results). PCR reactions were performed in a volume of 10 μL with final concentrations of 5 ng genomic DNA; 0.2 mM of each of the four dNTPs; 2 mM MgCl2 (ABgene); 1× PCR buffer (ABgene); 1 U Taq DNA polymerase (ABgene) and 0.2 μM of each primer. The PCR thermal profile was as follows: an initial denaturing step of 5 min at 94°C followed by 35–40 cycles of 30 s at 94°C (denaturation), 30 s at 60–66°C (annealing), and 30 s at 72°C (extension) before final elongation at 72°C for 5 min in Tetrad 2 thermal cycler (Bio-Rad). The genotypes of each microsatellite marker were determined using ABI PRISM 3130 genetic analyzers (Applied Biosystems, Foster City, CA).
Analysis of data obtained from nuclear DNA microsatellite analysis
For each nuclear microsatellite locus, the total number of alleles detected (A), the observed heterozygosity (H o), the unbiased expected heterozygosity (H e; Nei 1987), F ST (Weir and Cockerham 1984) and the pairwise F ST between each pair of populations were calculated using the FSTAT program (Goudet 2001). The differences in F ST between each pair of populations were tested based on 1,000 permutations.
For each population, we estimated the average number of alleles per locus (N a), the allelic richness, which was calculated using a fixed sample size of 26 individuals (A g[52]; El Mousadik and Petit 1996), the observed heterozygosity (H o) and the unbiased expected heterozygosity (H e; Nei 1983). We also estimated that the unbiased expected heterozygosity (H e; Nei 1983) using four loci, namely SpAGG3, SpAGD1, SpAGC1 and UAPgAC/AT6, to compare the genetic diversity of P. jezoensis in this study with that found by Aizawa et al. (2008b). These parameters were calculated using POPGENE (Yeh and Boyle 1999) and FSTAT software (Goudet 2001).
Population structure was inferred using STRUCTURE 2.1 (Pritchard et al. 2000). This program was designed to identify the optimal value of K, i.e., the number of populations from which the samples originated, and simultaneously to assign individuals to the populations (clusters). We performed 13 independent runs (K = 1–4) with 105 Markov chain monte carlo (MCMC) repetitions after a burn-in period of 104 interactions, using an admixture ancestral model and a model in which allele frequencies were correlated. We calculated the posterior probability for each K value using the estimated log-likelihood of K to select the optimal K.
To investigate the relationship between the census population size and the allelic richness based on a sample size of 26 diploid individuals (A g[52]), we calculated Spearman’s ρ rank correlation coefficients. The census population size of each population was estimated visually. Spearman’s ρ values were computed with the JMP4 software package (SAS Institute, Cary, NC).
We used the BOTTLENECK program (Piry et al. 1999) to determine whether effective population size had been restricted in the recent past. We employed the infinite allele model (IAM), the two-phased model of mutation (TPM) and the stepwise mutation model (SMM); TPM generally provides the best fit for microsatellite data (Piry et al. 1999). Wilcoxon sign-rank tests were also performed.
Nei's D A distance (Nei 1983) was calculated between each pair of populations using the POPULATIONS program (Langella 2007). The genetic relationships between populations were visualized by constructing a neighbor-joining (NJ) tree based on the D A distance. To assess the confidence limits associated with the topology of the NJ tree, bootstrap proportions were estimated on the basis of 1,000 replicates.
Results
Organelle DNA PCR-RFLP variation
Of the five region restriction enzyme combinations for mtDNA that exhibited polymorphism in Asian P. jezoensis populations (according to Aizawa et al. 2007), only one, mh02/Tru1I (Tru9I) exhibited no variation in this study (Table 2a). Combining all PCR-RFLP band patterns detected by each marker allowed the identification of four mtDNA haplotypes (hereafter referred to as ‘mitotypes’) (Fig. 2; Table 2a). While only one mitotype (V) was found in Dokyu, Jiri-Banya and Jiri-Chunwang, three different mitotypes VIII, IX and XII [mitotypes X and XI are already used in P. alcoquiana (Aizawa et al. 2008a)] were found in Gyebang. Thus, only the Gyebang population exhibited intra-population variation on the basis of mtDNA.

Chloroplast (cp) and mitochondrial (mt) haplotypes revealed by organelle DNA polymerase chain reaction-restriction fragment length polymorphism (PCR-RFLP) analysis. Numbers represent each population sampled (1 Jiri-Chunwang, 2 Jiri-Banya, 3 Dokyu, 4 Gyebang)
Similarly, out of four region restriction enzyme combinations for cpDNA, only one, trnC-trnD/RsaI, was monomorphic in this study. By combining all PCR-RFLP band patterns detected by each marker, we identified six cpDNA haplotypes (hereafter referred to as ‘chlorotypes’) (Fig. 2; Table 2b). All of the populations except Gyebang contained chlorotypes F and K. However, chlorotype L was found only in the Jiri-Chunwang population.
The overall genetic differentiation among populations based on mtDNA was remarkably high (F ST = 0.921), but the F ST value for cpDNA was low (F ST = 0.056).
As described above, mtDNA readily conserves genetic differentiation. Therefore, we constructed a network of mitotypes to determine the migration route of P. jezoensis across Korea. The constructed network of mitotypes indicated that endemic mitotypes VIII, IX and XII had diverged from mitotype IV (Fig. 3). Mitotypes IV and V were derived from mitotype III as reported by Aizawa et al. (2007).

Haplotype network of the mitotypes obtained using the median-joining method (Bandelt et al. 1999)
Nuclear DNA microsatellite variation
The total number of alleles detected (A) at each locus ranged from 4 to 24 (Table 3). The observed heterozygosity (H o) ranged from 0.076 to 0.856 (average over all loci was 0.418) and the unbiased expected heterozygosity (H e) ranged from 0.074 to 0.857 (the average over all loci was 0.406). The values of F ST ranged from 0.005 to 0.320 with a mean of 0.102, indicating that most genetic variation (about 90%) occurred within populations. The F ST values for each pair of populations varied between 0.008 and 0.229 (Table 4) with an average of 0.111. All the pairwise F ST values were statistically significant with the exception of that between the Jiri-Chunwang and Jiri-Banya populations.
In the STRUCTURE analysis (Pritchard et al. 2000), a value of K = 2 resulted in the maximum log-likelihood value. This result suggests that there were probably two original populations, from which the 134 individuals in the current four populations were derived. The frequency of cluster one individuals was high in Jiri-Chunwang, Jiri-Banya and Dokyu populations. In contrast, the frequency of cluster two individuals was high in the Gyebang population (Fig. 4).

Genetic relationship between the four populations (including 134 individuals) surveyed in this study, estimated from six microsatellite loci using STRUCTURE (Pritchard et al. 2000)
The intra-population genetic diversity measures are presented in Table 1. The mean number of alleles per locus (N a) varied from 4.2 to 8.0 (average 6.5). The H o and H e values ranged from 0.394 to 0.452 (average 0.418) and from 0.353 to 0.437 (average 0.406), respectively. The allelic richness based on the sample size of 26 diploid individuals (A g[52]) varied between 4.167 and 6.947 (average 5.966). The H e value, estimated using four loci, ranged from 0.492 to 0.522 (average 0.503). In the present study, a statistically significant correlation was found between the census population size and allelic richness (A g[52]) based on the sample size of 26 diploid individuals (Spearman’s ρ = −1.00; P < 0.0001).
The Wilcoxon sign-rank tests indicated that there had been no significant bottlenecks in any of the four populations under any of the models considered (IAM, TPM and SMM).
Nei’s (1983) D A distance ranged from 0.0563 to 0.1964, with a mean of 0.1361 (Table 4). The NJ dendrogram based on the D A distance is presented in Fig. 5. The dendrogram revealed a clear geographic structure among populations: pairs of populations from geographically close locations clustered together. The isolated northern population, Gyebang, was distinct from the other three populations, whereas the two populations from Jiri (i.e., Jiri-Chunwang and Jiri-Banya) were the most similar.

Neighbor-joining (NJ) dendrogram for the four populations of P. jezoensis based on Nei’s D A distance (Nei 1983)
Discussion
Genetic variation within populations
The average expected heterozygosity of P. jezoensis in South Korea using four loci (H e = 0.503; Table 1) was somewhat lower than that recorded for other P. jezoensis populations in Japan, Russia and China, which ranged from 0.41 to 0.83 (Aizawa et al. 2008b). On the basis of the fossil data, Kong and Watts (1993) suggested that spruces in Korea appeared in the Miocene and became the dominant species during the Quaternary. Subsequently, spruce forests retreated northwards, fragmented, and shifted to high elevations (over 1,000 m above sea level) during the Holocene warming (about 6,000 years ago). Accordingly, the level of genetic diversity in the P. jezoensis populations of South Korea may have been reduced because they were the subject of elevated levels of genetic drift. Recent habitat loss and/or a reduction in population size appear to have contributed relatively little to the low genetic variability in the P. jezoensis populations of South Korea. We found no evidence that any of the four populations have been through a recent bottleneck causing a reduction in numbers. Thus, the low level of genetic diversity in the P. jezoensis populations of South Korea may represent a more ancient restriction in population size.
It is widely assumed that a small population size significantly affects population dynamics. In theory, smaller populations are more at risk of genetic drift, which may result in reduced genetic variation. In the present study, the Gyebang population, the smallest census population, exhibited the lowest values of A g[52], H o, and H e.
Level of genetic differentiation among populations
The level of genetic differentiation among the populations of P. jezoensis in South Korea (F ST = 0.102) was higher than that of other anemophilous and widespread species determined on the basis of nuclear microsatellites, e.g., Pinus contorta (0.028; Thomas et al. 1999), Fraxinus excelsior (0.076; Heuertz et al. 2004), Cryptomeria japonica (0.028; Takahashi et al. 2005) and Cercidiphyllum japonicum (0.043; Sato et al. 2006). The somewhat higher estimated F ST value in the present study might be the result of the genetic distinctness of the Gyebang population. The F ST value obtained from mtDNA was notably higher than those based on nuclear microsatellites and cpDNA. This is due to maternal inheritance retaining ancient genetic structures for a long time.
The NJ dendrogram based on nuclear microsatellite variation showed that the Gyebang population was genetically distinct from the other three populations. Even at the individual level, individuals in the Gyebang population were different from those in the other three populations. The organelle DNA variation also showed clear differentiation of the Gyebang population. The Gyebang population contained unique mtDNA haplotypes, including mitotypes VIII, IX and XII, which were not found in the other populations from South Korea. In contrast, chlorotypes F and K were detected in the Dokyu and Jiri populations, but not in the Gyebang population.
Phylogeographical implications
The mitotype network constructed here supports the results obtained by Aizawa et al. (2007), adding weight to their hypothesis that P. jezoensis from Honshu Island, Japan (mitotypes VI and VII) colonized this region from the Asian continent (mitotype III) via the Korean peninsula (mitotype V). The endemic mitotypes VIII, IX and XII in the Gyebang population diverged from mitotype IV. This result implies that there may be an unknown or extinct population with mitotype IV in northeast China or North Korea.
Aizawa et al. (2007) detected four haplotypes, i.e., mitotype V and chlorotypes B, F and K, in P. jezoensis trees from Jiri. Of the four haplotypes, mitotype V and chlorotype K were not observed in any of the other 32 populations sampled from Russia, China and Japan. In addition to these haplotypes, we found mitotypes VIII, IX and XII endemic to Gyebang and chlorotype L endemic to Jiri-Chuwang. Therefore, the residual populations in South Korea are very valuable because of their rarity and the presence of their endemic haplotypes.
Species with low levels of genetic variability are known to be at risk from new threats and to be more vulnerable to extinction. Changing climate conditions, such as global warming, would render the current mountaintop habitat of P. jezoensis unsuitable for this species. Therefore, both the low level of genetic diversity and the unique organelle DNA variation in P. jezoensis in South Korea must be given serious consideration in ex situ and in situ conservation programs. In particular, priority should be given to the Gyebang population since it holds unique organelle DNA variations and is genetically distinct from the other P. jezoensis populations occurring in South Korea.
References
Aizawa M, Yoshimaru H, Saito H, Katsuki T, Kawahara T, Kitamura K, Shi F, Kaji M (2007) Phylogeography of a northeast Asian spruce, Picea jezoensis, inferred from variation observed in organelle DNA markers. Mol Ecol 16:3393–3405
Aizawa M, Yoshimaru H, Katsuki T, Kaji M (2008a) Imprint of post-glacial history in a narrowly distributed endemic spruce, Picea alcoquiana, in central Japan observed in nuclear microsatellites and organelle DNA markers. J Biogeogr 35:1295–1307
Aizawa M, Yoshimaru H, Saito H, Katsuki T, Kawahara T, Kitamura K, Shi F, Savirov R, Kaji M (2008b) Range-wide genetic structure in a north-east Asian spruce (Picea jezoensis) determined in nuclear microsatellite markers. J Biogeogr (in press)
Bandelt HJ, Forster P, Röhl A (1999) Median-joining network for inferring intraspecific phylogenies. Mol Biol Evol 16:37–48
Chen J, Tauer CG, Huang Y (2002) Paternal chloroplast inheritance patterns in pine hybrids detected with trnL-trnF intergenic region polymorphism. Theor Appl Genet 104:1307–1311
El Mousadik A, Petit RJ (1996) High level of genetic differentiation for allelic richness among populations of the argan tree (Argania spinosa L. Skeels) endemic to Morocco. Theor Appl Genet 92:832–839
Goudet J (2001) FSTAT, a program to estimate and test gene diversities and fixation indices (version 2.9.3). Available from http://www2.unil.ch/popgen/softwares/fstat.htm
Heuertz M, Hausman JF, Hardy OJ, Vendramin GG, Frascaria-Lacoste N, Vekemans X (2004) Nuclear microsatellites reveal contrasting patterns of genetic structure between western and southeastern European populations of the common ash (Fraxinus excelsior L.). Evolution 58(5):976–988
Hipkins VD, Krutovskii KV, Strauss SH (1994) Organelle genomes in conifers: structure, evolution, and diversity. For Genet 1:179–189
Hodgetts RB, Aleksiuk MA, Brown A, Clarke C, Macdonald E, Nadeem S, Khasa D (2001) Development of microsatellite markers for white spruce (Picea glauca) and related species. Theor Appl Genet 102:1252–1258
Hughes CR, Queller DC (1993) Detection of highly polymorphic microsatellite loci in a species with little allozyme polymorphism. Mol Ecol 2:131–137
Kong WS, Watts D (1993) Geobotany 19: the plant geography of Korea with an emphasis on the alpine zones. Kluwer, Dordrecht
Langella O (2007) POPULATIONS (version 1.2.30). http://bioinformatics.org/~tryphon/populations/
Lee CB (1982) Illustrated flora of Korea. Hyangmoonsa, Seoul, pp 59–60
Lee CB (1985) Dendrology, 9th edn. Hyangmoonsa, Seoul, p 235
Neale DB, Sederoff RR (1988) Inheritance and evolution of conifer organelle genomes. In: Hanover J, Keathley D (eds) Genetic manipulation of woody plants. Plenum, New York, pp 251–264
Neale DB, Sederoff RR (1989) Paternal inheritance of chloroplast DNA and maternal inheritance of mitochondrial DNA in loblolly pine. Theor Appl Genet 77:212–216
Nei M (1983) Genetic polymorphism and the role of mutation in evolution. In: Nei M, Koehn R (eds) Evolution of genes and proteins. Sinauer, Sunderland, pp 165–190
Nei M (1987) Molecular evolutionary genetics. Columbia University Press, New York
Pfeiffer A, Olivieri AM, Morgante M (1997) Identification and characterization of microsatellites in Norway spruce (Picea abies K.). Genome 40:411–419
Piry S, Luikart G, Cornuet JM (1999) BOTTLENECK: a computer program for detecting recent reduction in the effective population size using a allele frequency data. J Hered 90:502–503
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Sato T, Isagi Y, Sakio H, Osumi K, Goto S (2006) Effect of gene flow on spatial genetic structure in the riparian canopy tree Cercidiphyllum japonicum revealed by microsatellite analysis. Heredity 96:79–84
Schneider SJ, Kueffer M, Roessli D, Excoffier L (1997) Arlequin, Version 2.0: software for population genetic data analysis. Genetic and Biometry Laboratory, University of Geneva, Switzerland. http://lgb.unige.ch/arlequin/
Sugaya T, Yoshimaru H, Takeuchi T, Katsuta M, Fujimoto K, Changtragoon S (2003) Development and polymorphism of simple sequence repeat DNA markers for Bruguiera gymnorrhiza (L.) Lamk. Mol Ecol Note 3:88–90
Sutton BCS, Flanagan DJ, Gawley JR, Newton CH, Lester DT, El-Kassaby YA (1991) Inheritance of chloroplast and mitochondrial DNA in Picea and composition of hybrids from introgression zones. Theor Appl Genet 82:242–248
Takahashi T, Tani N, Taira H, Tsumura Y (2005) Microsatellite markers reveal high allelic variation in natural populations of Cryptomeria japonica near refugial areas of the last glacial period. J Plant Res 118:83–90
Thomas BR, Macdonald SE, Hicks M, Adams DL, Hodgetts RB (1999) Effects of reforestration methods on genetic diversity of lodgepole pine: an assessment using microsatellite and randomly amplified polymorphic DNA markers. Theor Appl Genet 98:793–801
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Yamazaki T (1995) Pinaceae. In: Iwatsuki K, Yamazaki T, Boufford DE, Ohba H (eds) Flora of Japan: vol 1. Pteriodophyta and Gymnospermae. Kodansya, Tokyo, pp 266–277
Yeh FC, Boyle T (1999) POPGENE Version 1.32. http://www.ualberta.ca/~fyeh/
Acknowledgments
This study was supported by the post-doctoral fellowship program for foreign researchers run by the Korea Forest Research Institute. The authors thank S.-D. Hur, J.-J. Lee, J.-H. Song and B.-H. Yang for their help in collecting needles, and the Korea National Park Service for its cooperation in allowing us to collect samples from two national peaks: Dokyu and the Jiri mountains. We also thank M. Aizawa, Y. Tsumura and Y. Tsuda for their kind advice and valuable suggestions.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Moriguchi, Y., Kang, KS., Lee, KY. et al. Genetic variation of Picea jezoensis populations in South Korea revealed by chloroplast, mitochondrial and nuclear DNA markers. J Plant Res 122, 153–160 (2009). https://doi.org/10.1007/s10265-008-0210-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10265-008-0210-8