
Abstract
Ischemic preconditioning (IPC), comprising exposure to sub-lethal short term ischemic events, has been shown to exert adaptive responses in many organs including the brain, thus guarding against exacerbations of ischemia reperfusion (IR). However, the mechanisms involved in the early phase of such a protection remain elusive; hence, the present study aimed to investigate the modulatory effect of preconditioning against IR induced injury on infarct size, free radicals, inflammatory/anti-inflammatory markers, caspase-3 and heat shock protein (HSP)70 in the rat hippocampus. To this end, male Wistar rats were divided into 3 groups, (1) sham operated (SO) control; (2) IPC, animals were subject to 3 episodes of ischemia (5 min) followed by reperfusion (10 min), afterwards rats underwent ischemia (15 min) followed by reperfusion (60 min); (3) IR animals were subjected to 15 min global ischemia followed by 60 min reperfusion. IR produced cerebral infarction accompanied by an imbalance in the hippocampal redox status, neutrophil infiltration, elevation in tumor necrosis factor (TNF)-α and prostaglandin (PG)E2, besides reduction in interleukin (IL)-10 and nitric oxide (NO) levels. IPC reverted all changes except for PGE2; however, neither HSP70 nor caspase-3 expression was altered following IR or IPC. The current study points thus towards the activation of the antioxidant system, anti-inflammatory pathway, as well as NO in the early phase of preconditioning protection.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Ischemic preconditioning (IPC), brief non-cytotoxic ischemic episodes, renders cells resistant to subsequent lethal events by activating endogenous protective mechanisms [1].Titration is essential to the effectiveness of all preconditioning stimuli. The stimulus must be strong enough to elicit an adaptive response, but not so intense as to cause injury itself or worsen ischemic outcomes [2, 3]. Notably, subtle non injurious preconditioning episodes reduce adhesion molecules and limit recruitment of inflammatory cells thus conferring neuronal protection [4]. Certainly, following preconditioning, limited amounts of free radicals are produced in response to mildly uncoupling oxidative phosphorylation and the decrease in the inner membrane potential, thus activating protective antioxidant systems [5, 6]. Moreover, inflammatory cytokines have been implicated in ischemic tolerance mechanisms. Tumor necrosis factor (TNF)-α has been shown to confer cytoprotection following preconditioning via activation of manganese superoxide dismutase (MnSOD) [7]. Further support to the role of TNF-α in maintaining cytoprotection resides in the abolition of ischemic tolerance by inhibiting TNF-α release [8]. Furthermore, this cytokine [9] as well as reactive oxygen species (ROS) [10] transcriptionally activate cyclo-oxygenase (COX)-2 that is involved in preconditioning protection [11].
Protective adaptive mechanisms derived from preconditioning have been established in a variety of organ systems, including brain [12, 13]. Noteworthy, preconditioning comprises two different windows namely early and late phases. The former is a transient phase owing to post-translational changes of preexisting proteins through signaling pathways [14, 15], while the latter robust and long lasting phase develops over days and is mediated by protective gene expression and new protein synthesis [14]. Several studies aimed at elucidating effects of either pharmacological or mechanical preconditioning on outcomes of ischemia reperfusion (IR) injury [3, 7, 16–18]. However, the complexity of the system confounds the outcomes. Studies so far, have focused on the delayed stage following preconditioning episodes, but data on the early phase remains scarce [3, 16–18]. Accordingly, the current investigation aimed at defining the protective mechanisms involved in the efficacy of short repetitive ischemic episodes, IPC, in guarding against the subsequent deleterious effects of IR injury in the brain.
Materials and Methods
Animals
Adult male Wistar rats (200–250 g) kept under controlled environmental conditions, at a constant humidity (60 ± 10%), temperature (23 ± 2°C), and a light/dark (12 h) cycle. Animals were allowed food and water ad libtium throughout the experimental procedures. Animal handling and experimental protocols were approved by the Research Ethical Committee of the Faculty of Pharmacy, Cairo University (Cairo, Egypt), and comply with the Guide for the Care and Use of Laboratory Animals [19].
Groups and Treatments and Induction of Transient Global Injury
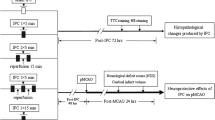
Experimental procedures were subdivided into 4 subsets, where animals were randomly allocated into 3 groups. The first two sets (n = 8 rats per group) were used for biochemical estimations, while the third and forth (n = 4 rats per group) served for infarct size and immunohistochemical assessments, respectively. All rats were anaesthetized with thiopental (50 mg/kg, i.p.) and midline ventral incision was made in neck. Within each subset, animals were assigned (1) sham operated (SO) control, (2) IPC (three episodes of 5 min of global ischemia by bilateral carotid occlusion, followed by 10 min reperfusion before IR exposure, and (3) IR bilateral carotid artery occlusion using small artery clips to induce global cerebral ischemia for 15 min followed by 60 min reperfusion period [20].
Brain Infarct Size
The procedure reported previously in our laboratory was adopted [21]. Briefly, rats were intracardiacally perfused with isotonic saline then sacrificed by spinal dislocation at the end of the reperfusion period. Brain were dissected and two mm coronal brain slices were incubated for 20 min in 1% triphenyltetrazolium chloride (TTC) in 0.2 M Tris buffer (pH 7.4) at 37°C. Infracted cells were either unstained or stained dull yellow, while viable cells stained bright red. In each brain slice, infracted and uninfarcted brain areas were traced using a 100 squares in 1 cm2 transparent plastic grid on both sides and the average infracted/uninfarcted areas were determined. Infarcted areas were expressed as a percentage of total brain area [22, 23].
Tissue Collection
Subsequent to IPC, IR or sham operation, all animals were euthanized and brains were removed immediately on ice cold plates. Both hippocampi were dissected and homogenized immediately in ice-cold saline for all biochemical measurements except for PGE2 (0.1 M phosphate buffer, pH 7.4 containing 1 mM EDTA and 0.1 μM indomethacin) and MPO (100 mM phosphate buffer, pH 6 containing 1% hexadecyltrimethylammonium bromide).
IL-10, TNF-α and PGE2 Estimations
IL-10, TNF-α and PGE2 concentrations were measured using rat ELISA kits purchased from Bender Med Systems (Vienna, Austria), Invitrogen (California, USA) and Cayman Chemical (MI, USA), respectively. All the procedures of the used kits were performed following manufacturers’ instruction manual.
Nitric Oxide Estimation
The method of Miranda et al. [24] was adopted for nitric oxide assay. At 4°C, absolute ethanol was used to deproteinated homogenates for 48 h then centrifuged at 12,000 g for 15 min. Nitrate was reduced to nitrite using vanadium trichloride (0.8% in 1 M HCl). Subsequently, Griess reagent [0.1% N-(1-Naphthyl) ethylenediaminedihydrochloride; 2% sulfanilamide in 5% HCl] was rapidly added and the mixture was incubated for 30 min at 37°C, cooled and the absorbance at 540 nm was measured.
Total Antioxidant Capacity (TAC) Estimation
The method by Koracevic et al. [25] for the assessment of TAC of hippocampi was adopted using commercial kit supplied by Biodiagnostic Co. (Giza, Egypt). Antioxidants eliminate H2O2 in the sample and its residual level is determined by an enzymatic reaction at 505 nm.
Non Protein Thiol (NPSH) Estimation
The method by Beutler et al. [26] for the assessment of NPSH in the hippocampi was utilized. 5-sulfuosalicylic acid (10%, 30 min, 4°C) was used to deproteinate homogenates, which were then centrifuged at 3,000 g for 15 min at 4°C. 5,5′-dithiobis-2-nitrobenzoic acid (1 mM) was added to the supernatant diluted with phosphate buffer (0.3 M, pH 7.7). The optical density was read at 412 nm.
Lipid Peroxides Determination
Lipid peroxides level in the hippocampus was determined by the thiobarbituric acid reaction of Miharaand Uchiyama [27]. Orthophosphoric acid (1%) and thiobarbituric acid (0.6%) were added to hippocampal homogenates, mixtures were boiled for 45 min, and then cooled. The colored product after cooling was extracted by n-butanol and read at 535 and 520 nm and the difference in absorbance was calculated as lipid peroxides level expressed as thiobarbituric acid reactive substances (TBARS).
Myeloperoxidase Activity
Myeloperoxidase (MPO; EC 1.15.1.1) activity (U/g tissue) was estimated as previously described [28]. Briefly, o-dianisidine hydrochloride (0.167%) and H2O2 (0.0005%) in potassium phosphate buffer (50 mM, pH 6) were added to supernatants after 3 freeze/thaw cycles, 10 sec sonication and 15 min centrifugation at 10,000 g for at 4°C. The absorbance kinetics were monitored at 1 min intervals at 460 nm for 4 min.
Immunohistochemistry of Hippocampal Caspase-3 and Heat Shock Protein (HSP)70
Animals of each of the three different manipulations (SO, IPC or IR) were anesthetized with sodium pentobarbital (50 mg/kg, i.p.) and the procedure reported by Abdallah [29] for immunohistochemistry was followed with modification. Briefly, rats were perfused transcardically with 4% paraformaldehyde (PFA) in tris-buffered saline (TBS). Following this, brains were removed post-fixed for 2 h in 4% PFA/TBS, and immersed in 30% sucrose. Paraffin embedded sagittal sections (4 μm) were cut through the entire hippocampus. Subsequently, sections were deparaffinized, rehydrated, and incubated in 0.3% H2O2 for 15 min to block endogenous peroxidase. Non-specific protein binding was blocked for 10 min with normal serum using Universal Quick Kit (Novocastra, Newcastle, UK). For immunohistochemical detection of caspase-3 or HSP70, slides were incubated with primary rat monoclonal antibody (anti-caspase-3 1:100; or anti-HSP70 1:200; Novocastra) for 1 h at room temperature in a humidified chamber. After rinsing twice with TBS, sections were treated with a labeled streptavidin–biotin kit (Novocastra). The sections were then incubated in 3,3′–diaminobenzidine (Novocastra) for 5 min. The sections through the CA3 area were examined under a microscope (×100) for the appearance of a positive brown staining [30].
Statistical Analysis
Data are expressed as mean of 4–8 experiments ± SEM, and statistical comparisons were carried out using one-way analysis of variance (ANOVA) followed by Student–Newman–Keuls multiple comparisons test. All analysis utilized SPSS 16.0 statistical package for Windows (SPSS Inc., Chicago, IL, USA). The minimal level of significance was identified at P < 0.05.
Results
Effect of IPC on Infarct Size Induced by IR Injury
IR induced approximately 60% infarct size compared to control SO rats (Fig. 1). IPC, on the other hand reduced infarct size by 40% compared to IR.

Effect of ischemia preconditioning (IPC) alone or with ischemia reperfusion (IR) on infarct size. Upper panel provides gross inspection from each group, while lower panel depicts infarct area in all test groups. Data represent the means of 4 experiments ± SEM; *; # P < 0.05 compared to sham operated (SO) and ischemic reperfused (IR) groups, respectively, using one-way ANOVA followed by Student–Newman–Keuls Multiple Comparisons Test
Effect of IPC on PGE2 Concentration Induced by IR Injury
The prostanoid PGE2 was increased by transient global ischemia (30%) and was not changed by IPC from SO (control) values (Fig. 2).

Effect of ischemia preconditioning (IPC) alone or with ischemia reperfusion (IR)on prostaglandin (PG)E2 concentration. Data represent the means of 8 experiments ± SEM; *; # P < 0.05 compared to sham operated (SO) and ischemic reperfused (IR) groups, respectively, using one-way ANOVA followed by Student–Newman–Keuls Multiple Comparisons Test
Role of oxidative Stress Mediators in Affording Protection Induced by IPC Against IR Injury
IPC induced an increase in TBARS (36% from SO, Fig. 3a) that was accompanied by a 13% decline in NPSH concentration versus the SO values (Fig. 3b). Meanwhile, IR further intensified lipid peroxidation (68 and 23% from SO and IPC values, respectively). On the other hand, IR evoked a further decline in NPSH (28 and 17%, respectively) from SO and IPC values.

Effect of ischemia preconditioning (IPC) alone or with ischemia reperfusion (IR) on a thiobarbituric acid reactive substances (TBARS) and b non protein thiols (NPSH). Data represent the means of 8 experiments ± SEM; *; # P < 0.05 compared to sham operated (SO) and ischemic reperfused (IR) groups, respectively, using one-way ANOVA followed by Student–Newman–Keuls Multiple Comparisons Test
Effect of IPC on TAC and NO Concentrations Against IR Injury
As seen in Fig. 4a, there was a decline in TAC in the IR group versus the SO compared to IR. Moreover, adaptive preconditioning partially restored NO level to near control value. Moreover, IR reduced it to 53% from vehicle control (Fig. 4b).

Effect of ischemia preconditioning (IPC) alone or with ischemia reperfusion (IR) on total antioxidant capacity (TAC) and nitric oxide (NO) concentration. Data represent the means of 8 experiments ± SEM; *; # P < 0.05 compared to sham operated (SO) and ischemic reperfused (IR) groups,respectively, using one-way ANOVA followed by Student–Newman–Keuls Multiple Comparisons Test
Role of IPC in Modulating Neutrophil Infiltration
IPC prevented neutrophil infiltration as evidenced by the insignificant change in MPO activity compared to the SO animals. Conversely, IR induced almost fivefold increase in MPO activity compared to both SO and IPC (Fig. 5).

Effect of ischemia preconditioning (IPC) alone or with ischemia reperfusion (IR) on myeloperoxidase (MPO) activity. Data represent the means of 8 experiments ± SEM; *; # P < 0.05 compared to sham operated (SO) and ischemic reperfused (IR) groups, respectively, using one-way ANOVA followed by Student–Newman–Keuls Multiple Comparisons Test
Effect of IPC on Proinflammatory TNF-α and Anti-Inflammatory IL-10 Cytokines
In Fig. 6a and b, IR increased TNF-α (58 and 18% from SO and IPC, respectively) and reduced IL-10 concentrations (53 and 48% from SO and IPC values, respectively).

Effect of ischemia preconditioning (IPC) alone or with ischemia reperfusion (IR) on a tumor necrosis (TNF)-α and b interleukin (IL)-10 concentrations. Data represent the means of 8 experiments ± SEM; *; # P < 0.05 compared to sham operated (SO) and ischemic reperfused (IR) groups, respectively, using one-way ANOVA followed by Student–Newman–Keuls Multiple Comparisons Test
Effect of IPC on Caspase-3 and HSP70 Expression in the Hippocampus
Immunhistochemical imaging of the hippocampal CA3 region of caspase-3 (Fig. 7a, b) and HSP70 (Fig. 7c, d) in ischemic reperfused as well as those animals exposed to 3 episodes of ischemic preconditioning showed no stain correspondent to the immunoreactivity of either proteins.

Photomicrographs depicting the immunohistochemical expression of caspase-3 in ischemic preconditioned (IPC) a and ischemic reperfused (IR) animals b as well as heat shock protein (HSP)70 in IPC c and IR d animals in CA3 region of the hippocampus (×100)
Discussion
Ischemic tolerance mechanisms are complex and controversial; however, it appears to involve early [31] and late cellular [32] responses. The present study emphasizes the importance of the early phase protection via transient non-lethal IR episodes against prolonged IR injury evidenced by (1) guarding against sequel of IR injury as evidenced morphologically by decrease of infarction in IPC compared to IR group, (2) enhancement of antioxidant defense systems and reduction in free radical load, (3) amelioration of neutrophil infiltration, as well as (4) decline in TNF-α and an increase in IL-10 concentrations in the hippocampi of preconditioned versus IR animals.
In the brain, the amount and phase of free radical production may provide beneficial or detrimental effects [33, 34]. During the early induction phase of preconditioning, ROS production is required [35] that might be mediated, in part, through PGE2 synthesis via the peroxidative power of COX-2 [36]. A further support to the notion, in the immediate spreading depression (CSD)-induced preconditioning, increased COX-2 protein has been reported as an indicator of early cellular responses [37]. Noteworthy, COX-2 inhibitors [38] have been shown to abolish the protection afforded by IPC, confirming the role of PGs and subsequent controlled release of ROS as crucial defense mechanisms for the development of ensuing ischemic tolerance. Indeed, the use of antioxidants [39] has been shown to reduce IPC induced protection. On one hand, in the current study, IR induced injurious free radical formation manifest as increase TBARS accompanied by a decrease in NPSH and TAC that were partially restored by prior IPC treatment. Such an increase in the former defense systems following preconditioning and within 1 h of reperfusion, imply an enhancement of the antioxidant defense mechanisms. The increase of NPSH may be due to early recruitment or recycling of low molecular weight antioxidant molecules (uric acid and ascorbate) from the periphery [40] or increased glutathione reductase [41]. Moreover, the reduction in neutrophil infiltration, being another source of free radical production, shown in the current investigation after IPC, may afford an additional explanation to the reinstated NPSH/TAC levels and reduced lipid peroxidation. Accordingly, when non-cytotoxic stress, below the threshold of damage, is applied, protective mechanisms prevail, which render cells resilient to further damage. However, when stress overwhelms the unprimed system, damage to the system surmounts, as seen in IR animals in this study, which is in line with our previous finding [42].
On the other hand, in the current study, IR reduced NO that may be attributed to its exhaustion in formation of peroxynitritein the vicinity of superoxide anion formation [42] while IPC, in part reinstated its level. Indeed, NOS [39] inhibitors present another impendence for the protection by IPC. Evidence exists that IPC prevents Na+/K+- ATPase inhibition, thus dissipating membrane potential ensuing inhibition of glutamate release, hence decreasing nNOS activation [14]. This may explain the partial restoration of NO concomitant with the decrease in free radical shown in this study.
Although TNF-α is reported to exert deleterious effects [43], however, an in vitro study by Burkovetskaya et al. [44], an increase in TNF-α as early as early as 10 min in hippocampal slice neurons after a 3 min hypoxic episode, was revealed. Such an increase in this proinflammatory cytokine during preconditioning may stimulate brain parenchymal cells to elicit adaptive responses, hence, ischemic tolerance [7]. Interestingly, the current study reports an increase in TNF-α in IR compared to their control counterpart. IPC partially restored TNF-α to near control values, suggestive of a protective role for this proinflammatory cytokine against exacerbation of IR injury. One plausible explanation for the decreased level of this inflammatory mediator might be the present inhibition of neutrophil infiltration, evidenced by reduction in MPO, that releases TNF-α upon activation during IR episodes [34].
Meanwhile, though IL-10 showed reduction in IPC group compared to SO, this anti-inflammatory cytokine was further decreased in IR animals. Accordingly, the difference in the IL-10 concentration in the IR group compared to IPC in this present study represents an adaptive response to the decrease in TNF-α. The latter effect is in line with the work of Kalpana et al. [45] thus attenuating the production of this proinflkammatory cytokine [46, 47].
Though we report an increase TBRAS, TNF-α as well as PGE2 in ischemic reperfused rats, there was no change in the expression pattern of caspase-3. Reported studies link these mediators to extrinsic and intrinsic death pathways that converge on caspase-3 induction [48, 49]. A plausible explanation for the observed phenomenon could be explained by the finding of other investigators [50, 51] showing increased caspase-3 expression as early as 6 up to 72 h of reperfusion. Noteworthy, the current study displayed no expression of the molecular chaperon, HSP70, following ischemic preconditioning suggesting no involvement in the early preconditioning protection. Indeed, Ge et al. [52] showed that HSP70 is upregulated 24 h following ischemic injury.
Taken all together, ROS and NO production during the early phase of preconditioning afford protection against further damage in cerebral transient global ischemia. Neuroprotection during this phase of preconditioning could be ascribed in part to enhancement of antioxidant defense systems restoring thus pro-oxidant/antioxidant milieu of the hippocampus, as well as a subtotal restoration of inflammatory/anti-inflammatory cytokine concentration. Furthermore, the results of the current investigation suggest that HSP70/caspase-3 are not involved in preconditioning induced neuroprotection nor IR-induced toxicity.
Abbreviations
- NPSH:
-
Nonproteinthiols
- HSP:
-
Heat shock protein
- IL-10:
-
Interleukin10
- IPC:
-
Ischemia preconditioning
- IR:
-
Ischemia reperfusion
- MDA:
-
Malondialdehyde
- MnSOD:
-
Manganese superoxide dismutase
- MPO:
-
Myeloperoxidase
- NO:
-
Nitric oxide
- PGE2 :
-
Prostaglandin E2
- ROS:
-
Reactive oxygen species
- SO:
-
Sham operated
- TAC:
-
Total antioxidant capacity
- TBARS:
-
Thiobarbituric acid reactive substances
- TNF-α:
-
Tumor necrosis factor-alpha
References
Murry CE, Jennings RB, Reimer KA (1986) Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 1986(11/01):1124–1136
Dirnagl U, Becker K, Meisel A (2009) Preconditioning and tolerance against cerebral ischaemia: from experimental strategies to clinical use. Lancet Neurol 2009(03/20):398–412
Stowe AM, Altay T, Freie AB, Gidday JM (2011) Repetitive hypoxia extends endogenous neurovascular protection for stroke. Ann Neurol 2011(03/26):975–985
An P, Xue YX (2009) Effects of preconditioning on tight junction and cell adhesion of cerebral endothelial cells. Brain Res 1272:81–88
Dirnagl U, Meisel A (2008) Endogenous neuroprotection: mitochondria as gateways to cerebral preconditioning? Neuropharmacology 2008(04/12):334–344
Sommer C (2009) Neuronal plasticity after ischemic preconditioning and TIA-like preconditioning ischemic periods. Acta Neuropathol 2008(12/17):511–523
Bhuiyan MI, Kim YJ (2010) Mechanisms and prospects of ischemic tolerance induced by cerebral preconditioning. Int Neurourol J 2011(01/22):203–212
Dirnagl U, Simon RP, Hallenbeck JM (2003) Ischemic tolerance and endogenous neuroprotection. Trends Neurosci 2003(05/15):248–254
Tu XK, Yang WZ, Shi SS, Wang CH, Zhang GL, Ni TR, Chen CM, Wang R, Jia JW, Song QM (2010) Spatio-temporal distribution of inflammatory reaction and expression of TLR2/4 signaling pathway in rat brain following permanent focal cerebral ischemia. Neurochem Res 2010(04/14):1147–1155
Collino M, Aragno M, Mastrocola R, Gallicchio M, Rosa AC, Dianzani C, Danni O, Thiemermann C, Fantozzi R (2006) Modulation of the oxidative stress and inflammatory response by PPAR-gamma agonists in the hippocampus of rats exposed to cerebral ischemia/reperfusion. Eur J Pharmacol 2006(01/03):70–80
Kim EJ, Raval AP, Hirsch N, Perez-Pinzon MA (2010) Ischemic Preconditioning mediates cyclooxygenase-2 expression via nuclear factor-kappa B activation in mixed cortical neuronal cultures. Transl Stroke Res 2010(07/08):40–47
Chen J, Simon R (1997) Ischemic tolerance in the brain. Neurology 1997(02/01):306–311
Rubino A, Yellon DM (2000) Ischaemic preconditioning of the vasculature: an overlooked phenomenon for protecting the heart? Trends Pharmacol Sci 2000(06/06):225–230
Pignataro G, Scorziello A, Di Renzo G, Annunziato L (2009) Post-ischemic brain damage: effect of ischemic preconditioning and postconditioning and identification of potential candidates for stroke therapy. FEBS J 2008(12/18):46–57
Speechly-Dick ME, Mocanu MM, Yellon DM (1994) Protein kinase C. Its role in ischemic preconditioning in the rat. Circ Res 1994(09/01):586–590
Gidday JM (2010) Pharmacologic preconditioning: translating the promise. Transl Stroke Res 2011(01/05):19–30
Hoyte LC, Brooks KJ, Nagel S, Akhtar A, Chen R, Mardiguian S, McAteer MA, Anthony DC, Choudhury RP, Buchan AM, Sibson NR (2010) Molecular magnetic resonance imaging of acute vascular cell adhesion molecule-1 expression in a mouse model of cerebral ischemia. J Cereb Blood Flow Metab 2010(01/21):1178–1187
Liu Y, Xue F, Liu G, Shi X, Liu W, Luo X, Sun X, Kang Z (2011) Helium preconditioning attenuates hypoxia/ischemia-induced injury in the developing brain. Brain Res 2011(01/05):122–129
ILAR (Institute of Laboratory Animal Resources) (1996) Guide for the care and use of laboratory animals. NIH Publication No. 85-23 (revised 1996). National Academy Press, Washington, D.C. Available from www.nap.edu/openbook.php?record_id=5140
Racay P, Chomova M, Tatarkova Z, Kaplan P, Hatok J, Dobrota D (2009) Ischemia-induced mitochondrial apoptosis is significantly attenuated by ischemic preconditioning. Cell Mol Neurobiol 2009(03/14):901–908
Mohamed RA, Agha AM, Nassar NN (2011) SCH58261 the selective adenosine A2A receptor blocker modulates ischemia reperfusion injury following bilateral carotid occlusion: role of inflammatory mediators. Neurochem Res. (In press)
Malik ZA, Singh M, Sharma PL (2011) Neuroprotective effect of Momordica charantia in global cerebral ischemia and reperfusion induced neuronal damage in diabetic mice. J Ethnopharmacol 133:729–734
Rehni AK, Bhateja P, Singh N, Jaggi AS (2008) Implication of mast cell degranulation in ischemic preconditioning-induced prevention of cerebral injury. Fundam Clin Pharmacol 22:179–188
Miranda KM, Espey MG, Wink DA (2001) A rapid, simple spectrophotometric method for simultaneous detection of nitrate and nitrite. Nitric Oxide 2001(02/17):62–71
Koracevic D, Koracevic G, Djordjevic V, Andrejevic S, Cosic V (2001) Method for the measurement of antioxidant activity in human fluids. J Clin Pathol 2001(05/01):356–361
Beutler E, Duron O, Kelly BM (1963) Improved method for the determination of blood glutathione. J Lab Clin Med 1963(05/01):882–888
Mihara M, Uchiyama M (1978) Determination of malonaldehyde precursor in tissues by thiobarbituric acid test. Anal Biochem 1978(05/01):271–278
Krawisz JE, Sharon P, Stenson WF (1984) Quantitative assay for acute intestinal inflammation based on myeloperoxidase activity. Assessment of inflammation in rat and hamster models. Gastroenterology 1984(12/01):1344–1350
Abdallah DM (2010) Anticonvulsant potential of the peroxisome proliferator-activated receptor γ agonist pioglitazone in pentylenetetrazole-induced acute seizures and kindling in mice. Brain Res 1351:246–253
Nassar N, Abdel-Rahman AA (2008) Brainstem phosphorylated extracellular signal-regulated kinase 1/2-nitric-oxide synthase signaling mediates the adenosine A2A-dependent hypotensive action of clonidine in conscious aortic barodenervated rats. J Pharmacol Exp Ther 324:79–85
Kirino T (2002) Ischemic tolerance. J Cereb Blood Flow Metab 2002(11/20):1283–1296
Bolli R (2000) The late phase of preconditioning. Circ Res 87:972–983
Kunz A, Park L, Abe T, Gallo EF, Anrather J, Zhou P, Iadecola C (2007) Neurovascular protection by ischemic tolerance: role of nitric oxide and reactive oxygen species. J Neurosci 27:7083–7093
Wang Q, Tang XN, Yenari MA (2007) The inflammatory response in stroke. J Neuroimmunol 184:53–68
Schaller B, Graf R (2002) Cerebral ischemic preconditioning. An experimental phenomenon or a clinical important entity of stroke prevention? J Neurol 2002(11/07):1503–1511
Im JY, Kim D, Paik SG, Han PL (2006) Cyclooxygenase-2-dependent neuronal death proceeds via superoxide anion generation. Free Radic Biol Med 2006(09/15):960–972
Horiguchi T, Snipes JA, Kis B, Shimizu K, Busija DW (2005) The role of nitric oxide in the development of cortical spreading depression-induced tolerance to transient focal cerebral ischemia in rats. Brain Res 2005(03/23):84–89
Bolli R, Shinmura K, Tang XL, Kodani E, Xuan YT, Guo Y, Dawn B (2002) Discovery of a new function of cyclooxygenase (COX)-2: COX-2 is a cardioprotective protein that alleviates ischemia/reperfusion injury and mediates the late phase of preconditioning. Cardiovasc Res 2002(08/06):506–519
Puisieux F, Deplanque D, Pu Q, Souil E, Bastide M, Bordet R (2000) Differential role of nitric oxide pathway and heat shock protein in preconditioning and lipopolysaccharide-induced brain ischemic tolerance. Eur J Pharmacol 2000(02/25):71–78
Glantz L, Avramovich A, Trembovler V, Gurvitz V, Kohen R, Eidelman LA, Shohami E (2005) Ischemic preconditioning increases antioxidants in the brain and peripheral organs after cerebral ischemia. Exp Neurol 2005(02/09):117–124
Bigdeli MR (2009) Preconditioning with prolonged normobaric hyperoxia induces ischemic tolerance partly by upregulation of antioxidant enzymes in rat brain tissue. Brain Res 2009(01/27):47–54
Watkins LR, Maier SF, Goehler LE (1995) Cytokine-to-brain communication: a review & analysis of alternative mechanisms. Life Sci 57:1011–1026
Abdallah DM, Nassar NN, Abd-El-Salam RM (2011) Glibenclamide ameliorates ischemia-reperfusion injury via modulating oxidative stress and inflammatory mediators in the rat hippocampus. Brain Res 2011(02/15):257–262
Burkovetskaya ME, Levin SG, Godukhin OV (2007) Neuroprotective effects of interleukin-10 and tumor necrosis factor-alpha against hypoxia-induced hyperexcitability in hippocampal slice neurons. Neurosci Lett 2007(03/23):236–240
Kalpana S, Dhananjay S, Anju B, Lilly G, Sai Ram M (2008) Cobalt chloride attenuates hypobaric hypoxia induced vascular leakage in rat brain: molecular mechanisms of action of cobalt chloride. Toxicol Appl Pharmacol 2008(07/19):354–363
Kini H, Pai RR, Kalpana S (2003) Solitary parotid metastasis from columnar cell carcinoma of the thyroid: a diagnostic dilemma. Diagn Cytopathol 2003(02/01):72–75
Qian L, Block ML, Wei SJ, Lin CF, Reece J, Pang H, Wilson B, Hong JS, Flood PM (2006) Interleukin-10 protects lipopolysaccharide-induced neurotoxicity in primary midbrain cultures by inhibiting the function of NADPH oxidase. J Pharmacol Exp Ther 2006(06/30):44–52
Broughton BR, Reutens DC, Sobey CG (2009) Apoptotic mechanisms after cerebral ischemia. Stroke 40:e331–e339
Takadera T, Shiraishi Y, Ohyashiki T (2004) Prostaglandin E2 induced caspase-dependent apoptosis possibly through activation of EP2 receptors in cultured hippocampal neurons. Neurochem Int 2004(10/15):713–719
Liang HW, Qiu SF, Shen J, Sun LN, Wang JY, Bruce IC, Xia Q (2008) Genistein attenuates oxidative stress and neuronal damage following transient global cerebral ischemia in rat hippocampus. Neurosci Lett 2008(05/10):116–120
Teschendorf P, Padosch SA, Spohr F, Albertsmeier M, Schneider A, Vogel P, Choi YH, Bottiger BW, Popp E (2008) Time course of caspase activation in selectively vulnerable brain areas following global cerebral ischemia due to cardiac arrest in rats. Neurosci Lett 2008(10/22):194–199
Ge PF, Luo TF, Zhang JZ, Chen DW, Luan YX, Fu SL (2008) Ischemic preconditioning induces chaperone hsp70 expression and inhibits protein aggregation in the CA1 neurons of rats. Neurosci Bull 2008(10/08):288–296
Conflict of interest
The authors have no conflict of interest to disclose.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Nassar, N.N., Abdelsalam, R.M., Abdel-Rahman, A.A. et al. Possible Involvement of Oxidative Stress and Inflammatory Mediators in the Protective Effects of the Early Preconditioning Window Against Transient Global Ischemia in Rats. Neurochem Res 37, 614–621 (2012). https://doi.org/10.1007/s11064-011-0651-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-011-0651-7