
Abstract
The embryonal tumor with abundant neuropil and true rosettes is a rare and highly malignant variant of embryonal brain tumors. It usually affects infants and young children under the age of 4 years and exhibits a very aggressive course with a dismal prognosis. For the 68 cases reported to date the mean age at diagnosis was 25.42 months (range 3–57 months). Survival data are available for 48 children (including our case): the median overall survival is 13.0 months, though 6 (9 %) of the children have had a relative long survival (>30 months). The aggressive combined treatment, involving primary surgical tumor removal, adjuvant polychemotherapy, including high-dose chemotherapy with stem cell transplantation, radiotherapy and radiochemotherapy, might play an important role in the longer survival. We have performed a literature review and we present here a multimodal-treated case of a 2- year-old girl with a long survival, who was reoperated when recurrence occurred. The residual tumor demonstrated a good response to temozolomide radiochemotherapy (craniospinal axis + boost) and followed by maintenance temozolomide. The described complex aggressive treatment option might be considered for future cases of this tumor entity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The embryonal tumor with abundant neuropil and true rosettes (ETANTR) was first described in 2000 by Eberhart et al. [1], who reported on 7 and 2 additional children with this central nervous system (CNS) tumor. Histologically, the presence of undifferentiated neuroepithelial cells, broad bands of well-differentiated neuropil islands, and ependymoblastic rosettes are characteristic for this tumor entity [2]. C19MC gene amplification is also characteristic for this tumor entity and predicts an unfavorable clinical course [3, 4]. The 2007 WHO classification of CNS tumors categorized embryonal neoplasms in three groups: medulloblastoma, atypical teratoid/rhabdoid tumor and CNS primitive neuroectodermal tumor (PNET). The highly aggressive ETANTR is briefly mentioned in connection with the description of CNS PNETs, and should probably be regarded as a separate entity [5–8]. Approximately 70 cases of this rare and malignant pediatric embryonal brain tumor have been reported to date [1, 2, 9–30]. Usually very young children are affected and a slight female predominance is observed [1]. We report here on a 2-year-old girl who presented with ETANTR of the left cerebellum and the left occipital lobe, which later recurred. This is the first report of long term survival of a patient with this kind of tumor even after recurrence.
Case report
A previously healthy 2-year-old girl was admitted to the local hospital with a history of a few weeks of headache, nausea, vomiting, tic, unsteady gait, and disturbances in coordination. Magnetic resonance images (MRI) of the brain revealed a contrast-enhancing lesion 6 cm in diameter in the left cerebellum and left occipital lobe. No signs of spinal metastasis were detected. The girl was operated in May 2010 and gross total resection (GTR) of the tumor was performed. Histopathological examination of the tumor sample revealed highly cell- and vessel-dens blue-celled tumor tissue with necroses and hemorrhages. The hyperchromatic tumor cells with mainly round nuclei and high nucleus/cytoplasm ratio formed clusters and were poorly differentiated. The mitotic ratio was high: 25 mitoses per 1 high-power field. Numerous multilayer perivascular, and ependymal true rosettes and broad bands of neoplastic neuropil islands could be observed. Immunohistochemical analysis indicated CD99, vimentin, synaptophysin, glial fibrillary acidic protein, epithelial membrane antigen, neurofilament, BAF47 and integrase interactor-1 positivity. The proliferation marker (MIB-1) showed focally high labelling index (up to 60 %), and ETANTR was diagnosed (Fig. 1). Detailed molecular profiling of the tumor revealed two genetic variations, involving the I391 M of the PIK3CA which is a known polymorphism, and N1118D and the APC gene, previously described as germline variants associated with an increased risk of familial adenomatosus polyposis [31] and frequently mutated (29.2 %) in brain tumors [32]. The girl was then treated according to the Medulloblastoma 2008 high-risk protocol (2 cycles of vincristine 1.5 mg/m2, cyclophosphamide 2 × 1 mg/m2, and etoposide 4 × 100 mg/m2; 2 cycles of vincristine 1.5 mg/m2, carboplatin 2 × 200 mg/m2, and etoposide 4 × 100 mg/m2; 2 cycles of vincristine 1.5 mg/m2, cisplatin 2 × 40 mg/m2, and etoposide 4 × 100 mg/m2; and weekly adjusted intrathecal triplet altogether 7 times). In November 2010 she underwent autologous stem cell transplantation (SCT) with busulfan, thiotepa and etoposide. The patient became symptom-free, and her cognitive and physical development were normal. A control MRI scan in December 2012 (31 months after the operation) revealed a 50 × 45 × 30 mm cystic lesion with a 12 × 33 × 5 mm contrast-enhancing mass inside it in the left occipital lobe, and a 30 × 17 × 18 mm intensive, inhomogeneous, contrast-enhancing lesion in the occipital part of the tentorium (Fig. 2). The spine was free of disease. The girl was reoperated and subtotal resection (STR) was performed. Thereafter, she received craniospinal irradiation (CSI), with 32 Gy in 1.6 Gy daily fractions (fr) followed by chemo-radiotherapy (CRT) comprising tumor bed (tb) boost with 24 Gy in 1.6 Gy/fr and a residual tumor boost with 6 Gy in 1.5 Gy/fr supplemented with 75 mg/m2 temozolomide daily. No anesthesia was necessary during radiotherapy. She has subsequently continued on 150 mg/m2 temozolomide without dose escalation to 200 mg/m2 due to haematological side effects until September 2014. The control MRI in August 2014 (4 years after the initial diagnosis and 21 months after the recurrence) revealed a residual 24 × 16 × 24-mm cystic lesion without any solid, contrast-enhancing mass (Fig. 3). The girl is currently disease-free, has no neurological symptoms or cognitive deficits and has been admitted to a bilingual primary school.

Hematoxilin-eosin staining showing embryonal tumor with the characteristic abundant neuropil and the prescence of multilayered rosettes (a). The proliferation marker (MIB-1, KI67) reveals high labelling index, focally up to 60 % (b). There is synaptophysin immunoreactivity both in the neuropil and in the rosettes (c). Immunostaining shows p53 positivity (d)

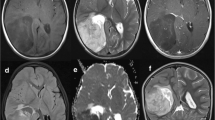
T2 weighted MRI shows the tumor recurrancy in the left occipital lobe (a) and in the postoperative cystic lesion in the left cerebellum and in the tentorium (b)

T2 weighted MRI shows the residual left occipital cystic lesion after the second surgery and RCT
Discussion
Our literature review indicated 69 reported cases [1, 2, 9–30] including our case since the first description of ETANTR by Eberhart et al. in 2000 [1]. The available clinical data demonstrate, that these tumors affect exclusively children, usually under the age of 4 years in different localizations throughout the CNS [9]. The prognosis is regarded as extremely poor, though there has been one case with an event-free survival of up to 7 years after GTR and CRT: CSI with 36 Gy and a boost to 55.8 Gy supplemented with carboplatin and vincristine, and chemotherapy (ChT) over a period of 6 months with cisplatin, vincristine and cyclophosphamide [10]. There have been only 7 reports, including our case, involving beneficial adjuvant treatment after tumor recurrence (Table 1). In these 7 cases the mean age at the time of the diagnosis was 24.9 months (range 16–36 months), with a male: female ratio of 3:4. At a median follow-up of 15 months 3 children died, and 4 were alive. Each of this 7 children underwent primary operation: STR in 4/7 and GTR in 3/7 cases. Thereafter 4 children received combined ChT (vincristine, etoposide, cyclophosphamide, carboplatin, ifosfamide, thiotepa and methotrexate), 2 received intrathecal ChT (cytarabine and methotrexate) and autologous SCT, and 1 of these 2 children was treated by radiotherapy (RT) to the tb with 54 Gy. Tumor recurrence was detected after a mean of 11.6 ± 8.60 months, 6 children were reoperated (4 STR and 2 GTR) while 1 child received only combined ChT and RT. 5 of the reoperated 6 children received combined ChT, 2 of them SCT, 1 proton CSI and our case CSI with 32 Gy and tb CRT with 30 Gy and 75 mg/m2 temozolomide daily.
Alexiou et al. [9] published a literature review of the available case reports and concluded, that patients who underwent STR or GTR had a significant survival benefit in comparison with patients on whom only biopsy could be performed (14 vs. 6 months, p = 0.006) but there were no major differences between the STR and GTR groups. In our analysis of the 7 recurring cases tumor recurrence was somewhat faster among the STR patients than among the GTR group (10.3 ± 0.85 vs. 14.0 ± 8.08 months, p = 0.589) with no significant difference in overall survival (OS) neither (26.3 ± 3.25 vs. 24.7 ± 9.93 months, p = 0.153). 6/7 children were reoperated and interestingly we found that in cases of STR (4/6) as second surgery the mean OS was longer than was in cases of GTR (2/6): 40.5 ± 7.36 vs. 12.5 ± 0.50 months, but because of the small number of patients we must not conclude that STR would be more beneficial than GTR as second surgery.
The majority of the children 6/7 received adjuvant combined high-dose ChT before and/or after the tumor recurrence. In the 3/7 children [1, 10, 11] who did not receive any adjuvant therapy, the tumor recurred faster than in the children treated with ChT (9.00 ± 2.51 vs. 14.00 ± 5.35 months).
4/7 Children received sequential autologous SCT after high-dose ChT [1, 10, 12] and had a better mean OS (31.25 ± 8.89 vs. 15.67 ± 1.09 months).
Focal RT with 54 Gy as adjuvant treatment was administered together with ChT and SCT in only 1 case [12]. This girl was 33 months old at the time of the diagnosis (the second oldest among the 7 cases) and was probably able to cooperate in RT with anesthesia. Three children (including our case) received adjuvant RT after tumor recurrence: 50.4 Gy to the tb [13]; CSI with protons (the dose is not available) [14]; and our case with 32 Gy CSI and a 30 Gy tb boost in 1.6 Gy fr-s with concomitant 75 mg/m2 temozolomide daily. The children who received RT (4/7) had a longer mean OS than that of the non-irradiated children (31.5 ± 8.76 vs. 24.0 ± 4.90 months). The review by Alexiou et al. [9] found that the children who were irradiated had a significant survival benefit relative to non-irradiated children (16 vs. 11 months, p = 0.029).
For the 7 cases treated at least surgically after tumor recurrence we found a quite high mean OS (33.7 ± 6.68 months) which is rather unusual in this tumor entity. Alexiou et al. [9] reported an OS of 13.1 months. Our literature search revealed 6 (including our case) unusually long survivors (at least 30 months after diagnosis) with ETANTR (Table 2). The mean age at diagnosis was 31.2 months (range 7–48 months), with a male: female ratio of 3:3. At the time of the report each of the 6 children was free of disease. They were all operated: 2/6 STR and 4/6 GTR. 1 child who did not receive adjuvant therapy [1] was reoperated 11 months after the first surgery because of tumor recurrence and then received combined ChT followed by SCT. The other 5 children received high-dose ChT in various combinations. RT was applied in 4 cases: hyperfractionated CSI with 36 Gy and a 30 Gy tb boost followed by 8 cycles of carboplatin, vincristine and lomustine [15]; CSI with 36 Gy and a 19.8 Gy tb boost supplemented with concurrent carboplatin and vincristine followed by cisplatin, vincristine, and cyclophosphamide [10] (to date he is the longest survivor); and in our case, CSI with 32 Gy and a 30 Gy tb CRT supplemented with concomitant temozolomide, and thereafter 150 mg/m2 mono temozolomide monthly up to September 2014. The similarities of these cases are that all 6 children were operated after diagnosis, at least STR was performed and combined high-dose ChT was administered for a certain amount of time.
RT is often problematic in the cases of such young children. To avoid major late side-effects (e.g. neurocognitive deficit, intellectual loss, hearing and visual impairment, endocrine dysfunction, asymmetry of the bony and muscular structures, or second malignancies), it is recommended to use the most conformal technique available and delay it as long as possible [33]. In contrast CSI RT is an important part of the treatment protocol of aggressive pediatric medulloblastoma and PNETs [34] and this technique has revealed its benefits in other malignancies with a high tendency to spread along the entire neuroaxis [35]. ETANTR most frequently occurs supratentorially (70.3 %) but has been described at all sites in the CNS [9] and metastatic cases have been reported, too [24]. This malignant feature and the very frequently reported poor outcome underline the need for aggressive multimodal therapy, which should include CSI RT, too. In our case, we delivered CRT for the time of tb irradiation with 30 Gy supplemented with 75 mg/m2 temozolomide after 32 Gy CSI RT and continued only on temozolomide. The CRT and the subsequent ChT were very well tolerated, and the girl is free of disease 52 months after the initial diagnosis. The currently longest survivor [10] received CSI and tb boost CRT with carboplatin and vincristine, continued on ChT with cisplatin, vincristine and cyclophosphamide, and is free of disease 84 months after the diagnosis. These 2 cases appear to suggest that the implementation of CRT may result in long-term disease-free survival. Temozolomide may be better tolerated than carboplatin, vincristine, cisplatin, and cyclophosphamide even by young children and might cause fewer side-effects, but a long-term follow-up is strongly recommended.
In our case report, as well as in many other case reports cited here, neither LIN28 immunostaining nor C19MC amplification analysis were performed. For the appropriate diagnosis these analyses seem to be essential to distinguish between subtypes of embrional tumors and prognosticate favourable and unfavourable clinical course [4, 19, 26–28, 37]. The correct diagnosis is absolutely necessary to choose the best possible treatment. It might be benefitial to perform LIN28 immunostaining and/or C19MC amplification analysis in further suspected cases of ETANTR to provide the exact diagnosis.
Conclusions
ETANTR has been identified as a histologically distinctive CNS embryonal tumor. Since the first description by Eberhart et al. in 2000 [1], we have found 69 reported cases, including our own case. Usually young children under the age of 4 years are affected. ETANTR is associated with a dismal prognosis due to its highly malignant course, but some case reports show that long-term disease-free survival can be achieved through radical tumor resection, ChT, SCT and RT. RT of the entire CSI is strongly recommended because ETANTR can spread via the cerebrospinal fluid. The delivery of CRT can result in a survival benefit, and temozolomide may be less toxic and better tolerated by young children. A long-term follow-up appears necessary and further detailed molecular profiling [36, 37] promote an understanding of the genetic background and molecular mechanism of this rare tumor type and could lead to targeted and more effective therapies.
References
Eberhart C, Brat D, Cohen K, Burgert P (2000) Pediatric neuroblastic brain tumors containing abundant neuropil and true rosettes. Pediatr Dev Pathol 3:346–352. doi:10.1007/s100249910049
Gessi M, Giangaspero F, Lauriola L, Gardiman M, Scheithauer BW, Halliday W, Hawkins C, Rosenblum MK, Burger PC, Eberhart CG (2009) Embryonal tumors with abundant neuropil and true rosettes. a distinctive CNS primitive neuroectodermal tumor. Am J Surg Pathol 33:211–217
Kleinman CL, Gerges N, Papillon-Cavanagh S, Sin-Chan P, Pramatarova A, Quang DAK, Adoue V, Busche S, Caron M, Djambazian H et al (2014) Fusion of TTYH1 with the C19MC microRNA cluster drives expression of a brain-specific DNMT3B isoform in the embryonal brain tumor ETMR. Nat Genet 46:39–47. doi:10.1038/ng.2849
Li M, Lee KF, Lu Y, Clarke I, Shih D, Eberhart C, Collins VP, Meter TV, Picard D, Zhou L et al (2009) Frequent amplification of a chr19q13.41 microRNA polycistron in aggressive primitive neuroectodermal brain tumors. Cancer Cell 16:533–546. doi:10.1016/j.ccr.2009.10.025
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P (2007) The 2007 WHO classification of tumours of the central nervous system (vol 114, pg 97, 2007). Acta Neuropathol 114:547. doi:10.1007/s00401-007-0278-6
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P (2007) The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 114:97–109. doi:10.1007/s00401-007-0243-4
Cenacchi G, Giangaspero F (2004) Emerging tumor entities and variants of CNS neoplasms. J Neuropathol Exp Neurol 63:185–192
Korshunov A, Sturm D, Ryzhova M, Hovestadt V, Gessi M, Jones DTW, Remke M, Northcott P, Perry A, Picard D et al (2014) Embryonal tumor with abundant neuropil and true rosettes (ETANTR), ependymoblastoma, and medulloepithelioma share molecular similarity and comprise a single clinicopathological entity. Acta Neuropathol 128:279–289. doi:10.1007/s00401-013-1228-0
Alexiou GA, Stefanaki K, Vartholomatos G, Sfakianos G, Prodromou N, Moschovi M (2013) Embryonal tumor with abundant neuropil and true rosettes: a systematic literature review and report of 2 new cases. J Child Neurol 28:1709–1715. doi:10.1177/0883073812471434
Manjila S, Ray A, Hu Y, Cai DX, Cohen ML, Cohen AR (2011) Embryonal tumors with abundant neuropiland true rosettes: 2 illustrative cases and a review of the literature. Neurosurg Focus 30:E2. doi:10.3171/2010.10.FOCUS10226
Dunham C, Sugo E, Tobias V, Wills E, Perry A (2007) Embryonal tumor with abundant neuropil and true rosettes (ETANTR): report of a case with prominent neurocytic differentiation. J Neurooncol 84:91–98. doi:10.1007/s11060-007-9346-y
Woehrer A, Slavc I, Peyrl A, Czech T, Dorfer C, Prayer D, Stary S, Streubel B, Ryzhova M, Korshunov A et al (2011) Embryonal tumor with abundant neuropil and true rosettes (ETANTR) with loss of morphological but retained genetic key features during progression. Acta Neuropathol 122:787–790. doi:10.1007/s00401-011-0903-2
Al-Hussaini M, Abuirmeileh N, Swaidan M, Al-Jumaily U, Rajjal H, Musharbash A, Hashem S, Sultan I (2011) Embryonal tumor with abundant neuropil and true rosettes: a report of three cases of a rare tumor, with an unusual case showing rhabdomyoblastic and melanocytic differentiation. Neuropathology 31:620–625. doi:10.1111/j.1440-1789.2011.01213.x
Hervey-Jumper SL, Altshuler DB, Wang AC, He X, Maher CO, Robertson PL, Garton HJL, Fan X, Muraszko KM, Camelo-Piragua S (2014) The role of CD133 + cells in a recurrent embryonal tumor with abundant neuropil and true rosettes (ETANTR). Brain Pathol 24:45–51. doi:10.1111/bpa.12079
La Spina M, Pizzolitto S, Skrap M, Nocerino A, Russo G, Di Cataldo A, Perilongo G (2006) Embryonal tumor with abundant neuropil and true rosettes. A new entity or only variations of a parent neoplasms (PNETs)? This is the dilemma. J Neurooncol 78:317–320. doi:10.1007/s11060-005-9105-x
Adamek D, Sofowora KD, Cwiklinska M, Herman-Sucharska I, Kwiatkowski S (2013) Embryonal tumor with abundant neuropil and true rosettes: an autopsy case-based update and review of the literature. Childs Nerv Syst 29:849–854. doi:10.1007/s00381-013-2037-4
Al-Hussain TO, Dababo MA (2009) Posterior fossa tumor in a 2 year-old girl diagnosis embryonal tumor with abundant neuropil and true rosettes (ETANTR). Brain Pathol 19:343–346. doi:10.1111/j.1750-3639.2009.00279.x
Buccoliero AM, Castiglione F, Degl’Innocenti DR, Franchi A, Paglierani M, Sanzo M, Cetica V, Giunti L, Sardi I, Genitori L et al (2010) Embryonal tumor with abundant neuropil and true rosettes: morphological, immunohistochemical, ultrastructural and molecular study of a case showing features of medulloepithelioma and areas of mesenchymal and epithelial differentiation. Neuropathology 30:84–91. doi:10.1111/j.1440-1789.2009.01040.x
Ceccom J, Bourdeaut F, Loukh N, Rigau V, Milin S, Takin R, Richer W, Uro-Coste E, Couturier J, Bertozzi AI et al (2014) Embryonal tumor with multilayered rosettes: diagnostic tools update and review of the literature. Clin Neuropathol 33:15–22. doi:10.5414/NP300636
Ferri Niguez B, Martinez-Lage JF, Almagro M, Fuster J, Serrano C, Torroba MA, Sola J (2010) Embryonal tumor with abundant neuropil and true rosettes (ETANTR): a new distinctive variety of pediatric PNET: a case based update. Child’s Nerv Syst 26:1003–1008. doi:10.1007/s00381-010-1179-x
Frassanito P, D’Angelo L, Massimi L, Lauriola L, Novello M, Di Rocco C, Tamburrini G (2012) Sudden paraplegia in a case of apparently isolated frontal embryonal tumour with abundant neuropil and true rosettes. Br J Neurosurg 26:284–286. doi:10.3109/02688697.2011.609919
Fuller C, Fouladi M, Gajjar A, Dalton J, Sanford RA, Helton KJ (2006) Chromosome 17 abnormalities in pediatric neuroblastic tumor with abundant neuropil and true rosettes. Am J Clin Pathol 126:277–283. doi:10.1309/TFBX1LWQ93MXQBAW
Judkins AR, Ellison DW (2010) Ependymoblastoma: dear, damned, distracting diagnosis, farewell! Brain Pathol 20:133–139. doi:10.1111/j.1750-3639.2008.00253.x
Kleinschmidt-DeMasters BK, Boylan A, Capocelli K, Boyer PJ, Foreman NK (2011) Multinodular leptomeningeal metastases from ETANTR contain both small blue cell and maturing neuropil elements. Acta Neuropathol 122:783–785. doi:10.1007/s00401-011-0894-z
Neelima R, Easwer HV, Kapilamoorthy TR, Hingwala DR, Radhakrishnan VV (2012) Embryonal tumorwith multilayered rosettes: two case reports with a review of the literature. Neurol India 60:96–99. doi:10.4103/0028-3886.93614
Nobusawa S, Yokoo H, Hirato J, Kakita A, Takahashi H, Sugino T, Tasaki K, Itoh H, Hatori T, Shimoyama Y et al (2012) Analysis of chromosome 19q13.42 amplification in embryonal brain tumors with ependymoblastic multilayered rosettes. Brain Pathol 22:689–697. doi:10.1111/j.1750-3639.2012.00574.x
Pfister S, Remke M, Castoldi M, Bai AHC, Muckenthaler MU, Kulozik A, von Deimling A, Pscherer A, Lichter P, Korshunov A (2009) Novel genomic amplification targeting the microRNA cluster at 19q13.42 in a pediatric embryonal tumor with abundant neuropil and true rosettes. Acta Neuropathol 117:457–464. doi:10.1007/s00401-008-0467-y
Wang Y, Chu S, Xiong J, Cheng H, Chen H, Yao X (2011) Embryonal tumor with abundant neuropil and true rosettes (ETANTR) with a focal amplification at chromosome 19q13.42 locus: further evidence of two new instances in China. Neuropathology 31:639–647. doi:10.1111/j.1440-1789.2011.01215.x
Majumdar K, Batra VV, Tyagi I, Sharma A (2013) Embryonal tumor with abundant neuropil and true rosettes with melanotic (retinal) differentiation. Fetal Pediatr Pathol 32:429–436. doi:10.3109/15513815.2013.799250
Lafay-Cousin L, Hader W, Wei XC, Nordal R, Douglas Strother D, Hawkins C, Chan JA (2013) Postchemotherapy maturation in supratentorial primitive neuroectodermal tumors. Brain Pathol 24:166–172. doi:10.1111/bpa.12089
Nagase H, Miyoshi Y, Horii A, Aoki T, Petersen GM, Vogelstein B, Maher E, Ogawa M, Maruyama M, Utsunomiya J et al (1992) Screening for germ-line mutations in familial adenomatous polyposis patients: 61 new patients and a summary of 150 unrelated patients. Hum Mutat 1:467–473. doi:10.1002/humu.1380010603
Nikuseva-Martic T, Beros V, Pecina-Slaus N, Pecina HI, Bulic-Jakus F (2007) Genetic changes of CDH1, APC, and CTNNB1 found in human brain tumors. Pathol Res Pract 203:779–787
Hoffman KE, Yock TI (2009) Radiation therapy for pediatric central nervous system tumors. J Child Neurol 24:1387–1396. doi:10.1177/0883073809342275
Gerber NU, von Hoff K, Resch A, Ottensmeier H, Kwiecien R, Faldum A, Matuschek C, Hornung D, Bremer M, Benesch M et al (2014) Treatment of children with central nervous system primitive neuroectodermal tumors/pinealoblastomas in the prospective multicentric trial HIT 2000 using hyperfractionated radiation therapy followed by maintenance chemotherapy. Int J Radiat Oncol Biol Phys 89:863–871. doi:10.1016/j.ijrobp.2014.04.017
Mozes P, Szanto E, Tiszlavicz L, Barzo P, Cserhati A, Fodor E, Hideghety K (2014) Clinical course of central neurocytoma with malignant transformation-an indication for craniospinal irradiation. Pathol Oncol Res 20:319–325. doi:10.1007/s12253-013-9697-y
Korshunov A, Remke M, Gessi M, Ryzhova M, Hielscher T, Witt H, Tobias V, Buccoliero AM, Sardi I, Gardiman MP et al (2010) Focal genomic amplification at 19q13.42 comprises a powerful diagnostic marker for embryonal tumors with ependymoblastic rosettes. Acta Neuropathol 120:253–260. doi:10.1007/s00401-010-0688-8
Spence T, Sin-Chan P, Picard D, Barszczyk M, Hoss K, Lu M, Kim SK, Ra YS, Nakamura H, Fangusaro J et al (2014) CNS-PNETs with C19MC amplification and/or LIN28 expression comprise a distinct histogenetic diagnostic and therapeutic entity. Acta Neuropathol 128:291–303. doi:10.1007/s00401-014-1291-1
Acknowledgments
This work was supported by EU-CELLEUROPE (EU315963) project.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors Petra Mozes, Péter Hauser, Tibor Hortobágyi, Gábor Benyó, István Peták, Miklós Garami, Adrienne Cserháti, Katalin Bartyik, László Bognár, Zoltán Nagy, Eszter Turányi, Katalin Hideghéty declare that ther are no conflicts of interest. All author read and approved the final manuscript. HP, BG, GM, BK leaded the oncological treatment and provided the clinical informations. HT, PI, TE did the histopathological and molecular profiling analyses. BL performed the operations and provided the MRI images. HK, NZ were responsible for the radiochemotherapy and provided clinical informations. MP collected the data, did the literature review and wrote the paper.
Rights and permissions
About this article

Cite this article
Mozes, P., Hauser, P., Hortobágyi, T. et al. Evaluation of the good tumor response of embryonal tumor with abundant neuropil and true rosettes (ETANTR). J Neurooncol 126, 99–105 (2016). https://doi.org/10.1007/s11060-015-1938-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-015-1938-3