
Abstract
Ependymoblastoma (EBL) and embryonal tumor with abundant neuropil and true rosettes (ETANTR) are very aggressive embryonal neoplasms characterized by the presence of ependymoblastic multilayered rosettes typically occurring in children below 6 years of age. It has not been established whether these two tumors really comprise distinct entities. Earlier, using array-CGH, we identified a unique focal amplification at 19q13.42 in a case of ETANTR. In the present study, we investigated this locus by fluorescence in situ hybridization in 41 tumors, which had morphologically been diagnosed as EBL or ETANTR. Strikingly, FISH analysis revealed 19q13.42 amplifications in 37/40 samples (93%). Among tumors harboring the amplification, 19 samples were identified as ETANTR and 18 as EBL. The three remaining tumors showed a polysomy of chromosome 19. Analysis of recurrent/metastatic tumors (n = 7) showed that the proportion of nuclei carrying the amplification was increased (up to 80–100% of nuclei) in comparison to the corresponding primary tumors. In conclusion, we have identified a hallmark cytogenetic aberration occurring in virtually all embryonal brain tumors with ependymoblastic rosettes suggesting that ETANTR and EBL comprise a single biological entity. FISH analysis of the 19q13.42 locus is a very promising diagnostic tool to identify a subset of primitive neuroectodermal tumors with distinct morphology, biology, and clinical behavior.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Embryonal tumors represent the largest group of malignant brain tumors in childhood. The 2007 WHO classification of central nervous system tumors recognizes three main histological entities, all of which contain a population of primitive (“embryonal”) cells: (1) medulloblastoma (2) atypical teratoid/rhabdoid tumor (AT/RT), and (3) CNS primitive neuroectodermal tumors (PNETs). Medulloblastoma and AT/RT are well-defined entities in terms of its histopathological features, immunophenotype and genetic profiles. In contrast, the latter entity is less stringently defined and subdivided into CNS neuroblastoma/ganglioneuroblastoma, medulloepithelioma, and ependymoblastoma (EBL) [19].
According to the 2007 WHO classification, EBL is a rare and very aggressive embryonal neoplasm characterized by the presence of true multilayered or “ependymoblastic” rosettes in association with small undifferentiated cells. Because its initial description by Bailey and Cushing [2], it has widely been discussed whether EBL should be regarded a distinct entity of embryonal CNS tumors [5, 6, 20]. Further complicating this classification issue, Eberhart et al. [8] more recently described a yet different pediatric embryonal brain tumor entity. Based on its characteristic histopathological findings, this tumor was designated “embryonal tumor with abundant neuropil and true rosettes (ETANTR)”. The microscopic appearance of this neoplasm includes both ependymoblastic rosettes and patterns of neuronal differentiation, including neurocytes, ganglion cells and neuropil-like background [1, 4, 7–10, 14]. Analyzing a series of tumors exhibiting ependymoblastic rosettes, Judkins and Ellison [11] found that these characteristic morphological structures are most frequently encountered in ETANTR and less frequently in other embryonal tumors including a few cases with mimics of true rosettes, further underlining the difficulties with the diagnosis of EBL.
Molecular analysis of tumors containing true rosettes (both ETANTR and EBL) has thus far provided only limited and heterogeneous findings. Comparative genomic hybridization (CGH) analysis of four EBL revealed that 3 of these tumors showed gain (trisomy) of chromosome 2 [24]. Furthermore, genomic profiling of a small group of ETANTR also showed frequent trisomy of chromosome 2, thus indicating shared genomic imbalances present in both of these entities [7, 9, 10]. Recently, we studied one case of ETANTR using array-CGH and identified a high-level focal amplification within chromosome region 19q13.42 containing a cluster of miRNA coding genes [22]. This aberration has not been detected in any other pediatric brain tumor studied on the same array-CGH platform to date, including 150 medulloblastomas, 122 ependymomas, 12 supratentorial PNETs and 12 AT/RT [12, 13, 21, 23]. Interestingly, however, amplification of 19q13.41 also involving the same miRNA cluster has been described by Li et al. in 11 cases of supratentorial PNET and, most importantly, the authors state that patterns of “ependymal and ependymoblastic differentiation” were observed in most of these cases [16].
Thus, we hypothesized that this focal amplification at 19q13.42 may be specific for tumors with the patterns of “ependymoblastic” differentiation, i.e., for these having multilayered true rosettes in their histological appearance. For these purposes, we performed fluorescence in situ hybridization (FISH) analysis of this candidate locus in 41 cases, which were included here solely by their morphological appearance as diagnosed by 12 distinguished neuropathologists from 8 different centers in 6 countries.
Materials and methods
Tumor samples
Forty-one diagnostic specimens that had histopathologically been diagnosed as either ETANTR or EBL were received for this study from the following sources: cases 1–12 (Burdenko Neurosurgical Institute, Moscow, Russia), cases 13–31 (University of Bonn, Germany), cases 32–34 (Memorial Sloan-Kettering Cancer Center, New York, USA), case 35 (Gade Institute, University of Bergen, Norway), case 36 (South Eastern Area Laboratory Service (SEALS) at Sydney Children’s Hospital, Sydney, Australia), case 37 (Careggi Hospital, Florence, Italy), cases 38,40,41 (Mayo Clinic, Minnesota, USA), and case 39 (Indiana University, Indiana, USA). In addition to 41 primary tumors, we analyzed seven samples obtained after progressive disease (6 local recurrences and one metastasis). Basic clinical characteristics are summarized in Table 1. A fraction of these cases have been previously described [4, 10]. All tumors were subjected to a central histopathological review either by Andrey Korshunov or by Torsten Pietsch.
All cases were routinely formalin-fixed and paraffin-embedded. For diagnostic purposes, routine histopathological examination and immunohistochemical analyses including neuronal and astrocytic differentiation markers, proliferation index and INI1 staining to exclude AT/RT were performed in the different institutions participating in this study.
Fluorescence in situ hybridization
Two-color interphase FISH was performed on de-paraffinized sections using two custom-made fluorochrome-labeled locus-specific probes: FITC-labeled probe 634C1 corresponding to locus 19q13.42 and digoxigenin-labeled probe 2659N19 corresponding to locus 19p13 as a reference.
Pretreatment of slides, hybridization, post-hybridization processing, and signal detection were performed as previously described [17]. Samples showing sufficient FISH efficiency (>90% nuclei with signals) were evaluated by two independent investigators. Signals were scored in at least 200 non-overlapping, intact nuclei. Metaphase FISH for verifying clone mapping position was performed using peripheral blood cell cultures of healthy donors as previously outlined [18].
Specimens were considered amplified for the 19q13.42 locus when more than 10% of tumor cells exhibited either more than eight signals of the corresponding probe with a reference/control ratio >2.0 or innumerable tight clusters of signals of the reference locus probe. Polysomy 19 was defined as >10% of nuclei containing three or more signals for both the target and the reference probes according to the sensitivity of the probe tested in control brain tissue.
Results
Clinical findings
Basic clinical characteristics from the 41 patients included in this study are summarized in Table 1. These tumors occurred predominately in very young children (range 0–6 years, median 2 years, male:female ratio 1.1:1). The predominant tumor location was supratentorial (31 cases) with most frequent origin in the frontal lobe (9 cases). Infratentorial location was less frequent (10 cases) affecting the cerebellum (5), brain stem (3) and midbrain (1). In one case, the tumor was located in the third ventricle.
Clinical follow-up data were available for 16 patients (Table 2). 44% (7/16) of these patients died within the first year after first diagnosis. Median overall survival was 12 months, and median progression-free survival was 6.5 months, which renders these patients a subgroup of PNET with particularly poor outcome (Fig. 1). This may be further illustrated when comparing this cohort with our previously published cohort of classic supratentorial PNETs with neuroblastic differentiation [23] (Fig. 1). Patterns of disease progression were highly variable. Some patients experienced local tumor recurrence, whereas others developed widespread leptomeningeal metastases. One patient additionally showed metastases outside of the CNS. Only a few patients are currently alive without disease progression.

Survival analysis of patients with ETANTR/EBL. Kaplan–Meier survival estimate for PFS (a) and OS (b) from 16 patients with ETANTR/EBL for whom clinical follow-up data was available in comparison with a previously published cohort of classic supratentorial PNETs with neuroblastic differentiation (n = 19) indicating that ETANTR/EBL comprise a subgroup of PNET with particularly poor prognosis
Pathological examination
On light microscopical investigation, all 41 tumors were composed of predominantly poorly differentiated cells with the presence of numerous true multilayered/ependymoblastic rosettes, as described by Mork and Rubinstein [20]. These rosettes were mitotically active and multilayered with well-formed central round or slit-like lumina in the absence of an outer membrane. Mitotic figures were frequently found in the juxtaluminal position. The lumina of the rosettes were either empty or filled with eosinophilic debris. In some rosettes, only a central pinpoint size lumen was discernible.
Tumors were morphologically diagnosed either as ETANTR (20 cases) or EBL (21 cases). ETANTR were defined according to the criteria previously described by Eberhart et al. [8]. These tumors showed a biphasic pattern featuring highly cellular clusters of small cells with round or polygonal nuclei and scanty cytoplasm admixed with large fibrillar and paucicellular neuropil-like areas, sometimes containing neoplastic neurons. Among the clusters and aggregates of small cells, numerous true rosettes were identified. Sometimes, these rosettes were observed abruptly in the neuropil areas and neoplastic neurons were found between the cells composing layers of the rosettes. In one tumor, described previously by Buccoliero et al. [4], “medulloepithelioma-like” areas and patterns of mesenchymal differentiation were additionally found indicating that the morphologic picture may be very diverse in these tumors. EBL was diagnosed based on the exclusive presence of nests and clusters of poorly differentiated cells forming numerous ependymoblastic rosettes while lacking neuropil-like matrix. These rosettes were intermixed with small to medium-sized primitive cells having a high nucleus/cytoplasm ratio and variably developed fibrillary processes. All tumors included in this series showed positive immunostaining for INI1 clearly separating them from AT/RT.
We also analyzed seven recurrent lesions (4 diagnosed as ETANTR and 3 as EBL). Recurrent ETANTRs were characterized by a significant loss of neuropil-like parts, and were predominantly composed of poorly differentiated cellular areas with numerous rosettes (Fig. 2). Extracranial metastases of one ETANTR were mostly composed of clusters of true rosettes different in shape and size, and thin ribbons of small poorly differentiated cells among them. Recurrent EBLs showed histopathological features closely resembling the corresponding primary tumors.

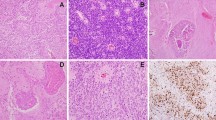
Microscopic appearance of the ETANTR and EBL (H&E ×200). a Representative ETANTR histology: neurocytic paucicellular areas with clusters of multilayered rosettes embedded in abundant neuropil. Single neoplastic neurons were found within the rosettes. b Representative EBL histology: tumor disclosing numerous ependymoblastic rosettes with intermingled clusters of small poorly differentiated cells and absence of neuropil-like areas. c Primary ETANTR (case 7) with single ependymoblastic rosettes and d neuropil-like areas containing ganglioid-like cells. The tumor relapse from the same patient was composed of hypercellular areas with numerous true rosettes but lacking neuropil-like areas. e Primary ETANTR (case 35) with single ependymoblastic rosettes and extensive neurocytic paucicellular areas. f The extracranial metastasis from this tumor was only composed of hypercellular areas and multilayered rosettes with various size and shape
Analysis of the 19q13.42 locus by FISH
One tumor (case 27; diagnosed as EBL) disclosed no signals in tumor nuclei and was considered to be non-informative for further molecular analysis. In the remaining 40 tumors, FISH analysis revealed a high-level focal amplification of the 19q13.42 locus in 37 samples (93%). Among these tumors, 19 had been diagnosed as ETANTR, and 18 as EBL (Fig. 3). The amplification was identified in 30–80% of nuclei in primary tumors. Nuclei with amplification patterns were evenly distributed throughout different components of the tumor including single nuclei as well as tight clusters and collections of tumor cells. By comparison of hematoxylin and eosin (H&E) and FISH sections in the ETANTR cases, nuclei with amplification were found both in neuropil-like and cellular-rich areas. The three remaining tumors (cases 19 and 25; both diagnosed as EBL and case 32 diagnosed as ETANTR) showed polysomy of chromosome 19, i.e. low-level gain of the target locus and the reference locus compatible with an aberration affecting large parts of the chromosome. However, polyploidy could not be completely ruled out in these cases. Analysis of seven recurrent/metastatic tumors revealed that the number of nuclei with amplification was significantly higher in secondary lesions (up to 80–100% of nuclei) in comparison to their primaries. These findings suggest a selective growth advantage for tumor cells harboring the 19q13.42 amplification.

Examples of focal 19q13.42 amplification as detected by FISH analysis. a ETANTR (case 37). b EBL (case 5). c Clusters of numerous nuclei with amplification within the hypercellular part of ETANTR (case 9) and d single nuclei with amplification scattered within the neuropil-like areas of the same tumor. For the FISH analysis, 19q13.42, the locus of common focal amplification was labeled in green, and the reference probe located on 19p13 was labeled in red
Discussion
To date, the distinct nosologic position of some embryonal tumor entities of the CNS including tumors with true multilayered/ependymoblastic rosettes is a subject of intense discussion among neuropathologists [5, 6, 11, 20]. The definition of this tumor entity has a long history and its reclassification as a variant of CNS PNET is an attempt to find consensus for diagnostic assessment. The designation of ETANTR as a yet different embryonal tumor entity has further complicated the search for a diagnostic consensus, although its distinct nosologic position remains unclear. In contrast to EBL, ETANTR shows not only ependymoblastic rosettes, but also a wide spectrum of neuronal differentiation, including neuropil-like areas and neoplastic neurons. Eberhart et al. [8] considered ETANTR to be a unique tumor entity that appears to be distinct from other forms of embryonal CNS tumors, including EBL. Nevertheless, a few cases diagnosed as ETANTR disclosed absence of neuropil-like areas and neuronal differentiation and its distinction from EBL was difficult [10]. Judkins and Ellison [11] came to the conclusion that only ETANTR is a true nosological entity, whereas application of the term “EBL” is both confusing and imprecise. On the other hand, the 2007 WHO classification of CNS tumors contains EBL as a separate entity, whereas ETANTR was only discussed as a possibly unique variant [19]. Thus, the nosologic definition of embryonal tumors with true multilayered rosettes is hampered by divergent histopathological allocations. From the clinical perspective, however, the correct diagnosis of these tumors is of critical importance, because these tumors share a dismal prognosis and typically occur in very young children. Prognosis is by far inferior to other CNS embryonal tumors in childhood including medulloblastoma and cerebral neuroblastoma.
Nowadays, the histopathological classification for various human tumors is frequently complemented by molecular data. For example, the routine application of molecular diagnostics allows separating AT/RTs from other PNETs [3]. Unfortunately, molecular analysis of ETANTR/EBL has been hampered by small investigated tumor cohorts to date. Notably, increased frequencies of chromosome 2 gain were previously observed in both of these tumor entities [7, 9, 10, 22].
In our first study on ETANTR, we performed genome-wide analysis of one tumor by using array-CGH with an average resolution of 0.4 Mb [22]. Among large-scale aberrations, we detected trisomies of chromosomes 2 and 19. Most interestingly, however, we first described a focal high-level amplicon at 19q13.42 spanning 0.89 Mb and covering a cluster of miRNAs (polycistron), which was clearly up-regulated in this tumor. This genetic aberration has not been detected in any of more than 300 pediatric brain tumors studied on the same array-CGH platform to date suggesting that this aberration is specific for ETANTR/EBL. Recently, Li et al. [16] described the same amplification of the miRNA cluster at 19q13.41 using a high-resolution SNP microarray platform in a subset of supratentorial PNETs exhibiting predominantly ependymoblastic and ependymal differentiation.
In the current study, we analyzed the DNA copy number status of this genomic locus at 19q13.42 in a large collection of embryonal tumors with ependymoblastic rosettes diagnosed by a consortium of experienced neuropathologists either as EBL or ETANTR based on their characteristic morphology. These neoplasms predominantly affected infants with preferential location in the cerebral hemispheres. By definition, these tumor entities differ in the presence or absence of extensive neuropil-like areas and other patterns of neuronal differentiation. However, EBL and ETANTR share similar age and sex distribution, tumor localization and dismal prognosis. Moreover, histological analysis of seven recurrent/metastatic ETANTRs revealed that the poorly differentiated component with true rosettes predominated in secondary lesions whereas neuropil-like areas were progressively lost. These findings suggest some similarity between both tumor entities included in this study. FISH analysis disclosed amplification of 19q13.42 locus in 93% of tumors with even distribution between ETANR and EBL. These results demonstrate a very high-level specificity and sensitivity for this genetic aberration in tumors with true multilayered rosettes irrespective of their morphological designation. An increasing number of cells harboring the amplification in recurrent/metastatic tumors suggest that this aberration might serve as a “driver” oncogenic event in these tumors, promoting its clonal expansion, aggressive biological behavior, and treatment resistance. In contrast to all protein-coding genes located in the amplified region, the two miRNA clusters, namely MIR-371–373 and C19MC, were shown to be generally up-regulated in PNETs carrying this specific genomic aberration by us and a group from Toronto [16, 22]. Both of these clusters were previously found to be up-regulated in human embryonic stem cells [15] and, consistent with their function in maintaining stem-cell properties, representatives of the C19MC cluster were shown to drive growth and inhibit differentiation of human neural stem cells [16]. Interestingly, many of the miRNAs targeted by the 19q13.42 amplification share a common seed sequence suggesting that they are cooperatively regulating a defined set of genes [15].
In conclusion, we identified a hallmark cytogenetic aberration occurring in virtually all embryonal brain tumors with ependymoblastic multilayered rosettes indicating that ETANTR and EBL may comprise a single biological entity. FISH analysis of the 19q13.42 locus is a very promising diagnostic tool to identify a subset of primitive neuroectodermal tumors with distinct morphology, biology, and clinical behavior and should be considered a unifying diagnostic marker for these tumors.
References
Al-Hussain TO, Dababo MA (2009) Posterior fossa tumor in a 2 year-old girl. Brain Pathol 19:343–346
Bailey P, Cusing H (1926) A classification of the tumors of the glioma group on a histogenetic basis with a correlated study of prognosis. J.B. Lippincott Co, Philadelphia
Biegel JA, Tan L, Zhang F, Wainwright L, Russo P, Rorke LB (2002) Alterations of the hSNF5/INI1 gene in central nervous system atypical teratoid/rhabdoid tumors and renal and extrarenal rhabdoid tumors. Clin Cancer Res 8:3461–3467
Buccoliero AM, Castiglione F, Degl’innocenti DR, Franchi A, Paglierani M, Sanzo M, Cetica V, Giunti L, Sardi I, Genitori L, Taddei GL (2010) Embryonal tumor with abundant neuropil and true rosettes: morphological, immunohistochemical, ultrastructural and molecular study of a case showing features of medulloepithelioma and areas of mesenchymal and epithelial differentiation. Neuropathology 30:84–91
Cruz-Sanchez FF, Haustein J, Rossi ML, Cervos-Navarro J, Hughes JT (1988) Ependymoblastoma: a histological, immunohistological and ultrastructural study of five cases. Histopathology 12:17–27
Cruz-Sanchez FF, Rossi ML, Hughes JT, Moss TH (1991) Differentiation in embryonal neuroepithelial tumors of the central nervous system. Cancer 67:965–976
Dunham C, Sugo E, Tobias V, Wills E, Perry A (2007) Embryonal tumor with abundant neuropil and true rosettes (ETANTR): report of a case with prominent neurocytic differentiation. J Neurooncol 84:91–98
Eberhart C, Brat D, Cohen K, Burger P (2000) Pediatric neuroblastic brain tumors containing abundant neuropil and true rosettes. Pediatr Dev Pathol 3:346–352
Fuller C, Fouladi M, Gajjar A, Dalton J, Sanford R, Helton K (2006) Chromosome 17 abnormalities in pediatric neuroblastic tumor with abundant neuropil and true rosettes. Am J Clin Pathol 126:277–283
Gessi M, Giangaspero F, Lauriola L, Gardiman M, Scheithauer BW, Halliday W, Hawkins C, Rosenblum MK, Burger PC, Eberhart CG (2009) Embryonal tumors with abundant neuropil and true rosettes: a distinctive CNS primitive neuroectodermal tumor. Am J Surg Pathol 33:211–217
Judkins A, Ellison D (2010) Ependymoblastoma: dear, damned, distracting diagnosis, farewell!*. Brain Pathol 20:133–139
Korshunov A, Remke M, Werft W, Benner A, Ryzhova M, Witt H, Sturm D, Wittmann A, Schöttler A, Felsberg J, Reifenberger G, Rutkowski S, Scheurlen W, Kulozik A, von Deimling A, Lichter P, Pfister S (2010) Adult and pediatric medulloblastomas are genetically distinct and require different algorithms for molecular risk stratification. J Clin Oncol (in press)
Korshunov A, Witt H, Hielscher T, Benner A, Remke M, Ryzhova M, Milde T, Bender S, Wittmann A, Schöttler A, Kulozik A, Witt O, von Deimling A, Lichter P, Pfister S (2010) Molecular staging of intracranial ependymoma in children and adults. J Clin Oncol (in press)
La Spina M, Pizzolitto S, Skrap M, Nocerino A, Russo G, Di Cataldo A, Perilongo G (2006) Embryonal tumor with abundant neuropil and true rosettes. A new entity or only variations of a parent neoplasms (PNETs)? This is the dilemma. J Neurooncol 78:317–320
Laurent L, Chen J, Ulitsky I, Mueller F, Lu C, Shamir R, Fan J, Loring J (2008) Comprehensive MicroRNA profiling reveals a unique human embryonic stem cell signature dominated by a single seed sequence. Stem Cells 26:1506–1516
Li M, Lee KF, Lu Y, Clarke I, Shih D, Eberhart C, Collins VP, Van Meter T, Picard D, Zhou L, Boutros PC, Modena P, Liang ML, Scherer SW, Bouffet E, Rutka JT, Pomeroy SL, Lau CC, Taylor MD, Gajjar A, Dirks PB, Hawkins CE, Huang A (2009) Frequent amplification of a chr19q13.41 microRNA polycistron in aggressive primitive neuroectodermal brain tumors. Cancer Cell 16:533–546
Lichter P, Bentz M, Joos S (1995) Detection of chromosomal aberrations by means of molecular cytogenetics: painting of chromosomes and chromosomal subregions and comparative genomic hybridization. Methods Enzymol 254:334–359
Lichter P, Cremer T, Borden J, Manuelidis L, Ward D (1988) Delineation of individual human chromosomes in metaphase and interphase cells by in situ suppression hybridization using recombinant DNA libraries. Hum Genet 80:224–234
Louis D, Ohgaki H, Wiestler O, Cavenee W, Burger P, Jouvet A, Scheithauer B, Kleihues P (2007) The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol 114:97–109
Mork SJ, Rubinstein LJ (1985) Ependymoblastoma: a reappraisal of a rare embryonal tumor. Cancer 55:1536–1542
Pfister S, Remke M, Benner A, Mendrzyk F, Toedt G, Felsberg J, Wittmann A, Devens F, Joos S, Kulozik A, Reifenberger G, Rutkowski S, Wiestler O, Radlwimmer B, Scheurlen W, Lichter P, Korshunov A (2009) Outcome prediction in pediatric medulloblastoma based on DNA copy-number aberrations of chromosomes 6q, 17q, and the MYC/MYCN loci. J Clin Oncol 27:1627–1636
Pfister S, Remke M, Castoldi M, Bai AH, Muckenthaler MU, Kulozik A, von Deimling A, Pscherer A, Lichter P, Korshunov A (2009) Novel genomic amplification targeting the microRNA cluster at 19q13.42 in a pediatric embryonal tumor with abundant neuropil and true rosettes. Acta Neuropathol 117:457–464
Pfister S, Remke M, Toedt G, Werft W, Benner A, Mendrzyk F, Wittmann A, Devens F, von Hoff K, Rutkowski S, Kulozik A, Radlwimmer B, Scheurlen W, Lichter P, Korshunov A (2007) Supratentorial primitive neuroectodermal tumors of the central nervous system frequently harbor deletions of the CDKN2A locus and other genomic aberrations distinct from medulloblastomas. Genes Chromosomes Cancer 46:839–851
Rickert C, Hasselblatt M (2006) Cytogenetic features of ependymoblastomas. Acta Neuropathol 111:559–562
Acknowledgments
This study was supported by Grants from the Deutsche Kinderkrebsstiftung, and the Tumorzentrum Heidelberg/Mannheim to S.P.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Korshunov, A., Remke, M., Gessi, M. et al. Focal genomic amplification at 19q13.42 comprises a powerful diagnostic marker for embryonal tumors with ependymoblastic rosettes. Acta Neuropathol 120, 253–260 (2010). https://doi.org/10.1007/s00401-010-0688-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-010-0688-8