
Abstract
The success of cancer immunotherapy in patients depends on overcoming immunosuppressive mechanisms in addition to stimulating effective anticancer immune responses. Myeloid-derived suppressor cells (MDSCs) inhibit a spectrum of immune responses, including adaptive immune responses and innate immune responses at the tumor site. MDSCs have been targeted to overcome immunosuppression either by reducing their numbers or downregulating their immunosuppressive activities. Although signal transducer and activator of transcription (STAT) proteins are recognized as signaling and transcription factors induced by cytokines in normal cells, they also have roles in cancer and cancer-related cells, as well as MDSC differentiation and function. In in vitro and in vivo studies, including studies on humans, selective STAT3 inhibitors such as Stattic and S3I-201 have demonstrated potential in regulating MDSC-mediated immunosuppression. Thus, STAT pathways represent a promising target in cancer immunotherapy. Herein, we review the roles of STAT signaling in MDSC biology, and the clinical potential of STAT inhibitors in regulating tumor-associated immunosuppression mediated by MDSCs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
STAT proteins are latent cytoplasmic transcription factors that upon activation by cytokines and growth factors mediate diverse normal and pathological cellular responses in development and immunity (Darnell Jr 1997; Bromberg et al. 2000). In mammals, 7 members of the STAT family have been identified: STAT1, STAT2, STAT3, STAT4, STAT5a, STAT5b, and STAT6. STAT proteins contain an amino-terminal domain, a coiled-coil domain, a DNA-binding domain, an Src homology 2 (SH2) domain, and a transactivation domain. Generally, STAT proteins are phosphorylated and activated by various tyrosine kinases, including Janus kinases (JAKs), receptor tyrosine kinases (RTKs), and non-receptor tyrosine kinases (NRTKs), which promotes STAT dimerization (Levy and Darnell 2002). However, non-phosphorylated STAT proteins also exist as dimers, and both phosphorylated and non-phosphorylated STATs can activate transcription, although of different target genes (Sehgal 2008).
Herein, we focus on the role of STAT signaling in immunosuppression in the tumor microenvironment, which is mediated by several cell types including regulatory T cells (Tregs), MDSCs, and tumor-associated macrophages (Lindau et al. 2013). Owing to its diverse functions, MDSCs are one of the primary cellular mediators of immunosuppression in the tumor environment. Recent studies have demonstrated the importance of STAT signaling in MDSCs and preliminary success with selective STAT inhibitors for the regulation of MDSC function. Hence, we believe that STAT proteins will potentially be an important target for novel cancer immunotherapies.
Roles of STATs in cancer cells
The STAT signaling pathway was originally identified as a signal transduction pathway induced by interferon-α (IFN-α) and interferon-γ (IFN-γ) in normal cells (Darnell Jr and Kerr 1994). However, STATs are also involved in oncogenic signaling, and mediate progression to malignancy (Yu and Jove 2004). Therefore, in addition to mitogen-activated protein kinase (MAPK) (Santarpia et al. 2012) and phosphatidylinositol-3-kinase (PI3 K)–AKT signal transduction pathways (Nitulescu et al. 2016), STAT signal transduction pathways represent promising targets in novel anticancer therapies.
In normal cells, STAT activation is regulated by extracellular ligand binding and it occurs transiently. In contrast, constitutively activated STAT3 is oncogenic (Azare et al. 2007). Activation of STAT3 and STAT5, which prevents apoptosis, has been reported in various human cancer cells (Yu and Jove 2004; Hassel et al. 2008; Liu et al. 2010). Constitutive activation of tyrosine kinase through genetic or epigenetic changes results in unceasing activation of downstream signaling, the STAT pathway, and plays a critical role in oncogenesis. Additionally, constitutive activation of tyrosine kinase signaling, which results in constitutive activation of downstream STAT signaling, is oncogenic (Huang et al. 2002; Levis et al. 2002). In particular, hyperactive STAT3 is frequently detected in myelomas, leukemia, lymphomas, and solid cancers (Al Zaid Siddiquee and Turkson 2008; Sansone and Bromberg 2012); hence a strategy targeting STAT3 may be a wide use approach in cancer therapy. Furthermore, constitutive activation of diverse tyrosine kinase is one of the main mechanism underlying tumor transformation, and diverse tyrosine kinase inhibitors have been developed as cancer therapeutics (Arora and Scholar 2005). Therefore, various types of cancer can be potentially treated with STAT inhibitors, which indirectly and directly target tyrosine kinase and STAT signaling, respectively.
Effects of STAT signaling in the tumor environment
STAT signaling affects not only cancer cells but also the surrounding tumor environment. Constitutive STAT3 activity induces expression of vascular endothelial growth factor (VEGF), a critical angiogenic factor, and promotes invasiveness of cancer cells (Niu et al. 2002). Similarly, STAT3 activation in cancer cells induced by an oncoprotein contributes to VEGF expression and invasiveness of cancer (Wang et al. 2010). STAT3 is also activated in tumor-associated endothelial cells and treatment with STAT3 inhibitors reduces angiogenesis directly (Lee et al. 2015). In addition, STAT5 activation influences endothelial cell migration, invasion, and tube formation (Yang and Friedl 2015). These studies suggest that STAT proteins, and in particular STAT3, are potential targets of anti-angiogenic therapy.
STAT signaling also regulates immune cell responses in the cancer environment. Ablation of STAT3 signaling in hematopoietic cells enhances antitumor responses of T cells and natural killer (NK) cells (Kortylewski et al. 2005). CD8+ T cells expressing constitutively activated STAT6 exhibit defective tumor infiltration (Sasaki et al. 2008). STAT3 activation induced by tumor-derived factors inhibits myeloid cell differentiation and functional dendritic cell (DC) maturation (Nefedova et al. 2004). Constitutive activation of STAT3 in DCs results in the expansion of Tregs (Matsumura et al. 2007). These findings demonstrate the importance of STAT signaling to the antitumor functions of diverse immune cells, and underscore the broad potential of selective STAT inhibitors as immunomodulatory anticancer agents. Collectively, STAT inhibition may be effective for inhibiting the proliferation, growth, and survival of cancer cells and the invasiveness of cancer-associated vascular endothelial cells, and relieving antitumor immunosuppression.
Myeloid-derived suppressor cells (MDSCs)
MDSCs are immature immunosuppressive myeloid cells, including precursors of macrophages, dendritic cells, and granulocytes (Gabrilovich et al. 2007). MDSCs expand and accumulate during pathological conditions such as during infection, inflammation, and cancer (Gabrilovich et al. 2007; Gabrilovich and Nagaraj 2009). MDSCs were originally identified as CD11b+ Gr1+ cells; however, they actually comprise a heterogeneous cell population, which can be generally classified as polymorphonuclear (PMN-MDSC) or monocytic (Mo-MDSC) (Youn et al. 2008; Kim et al. 2012). In tumor-bearing mice, CD11b+Ly-6GlowLy-6Chigh cells are Mo-MDSCs, whereas CD11b+Ly-6GhighLy-6Clow cells are PMN-MDSCs (Kim et al. 2012). In humans, PMN-MDSCs express CD11b+CD14−CD33+CD15+ and/or CD66b+, whereas Mo-MDSCs express CD14+HLA-DRlow (Gabrilovich et al. 2012). S100A9 has also been suggested as a functional marker of MDSCs (Zhao et al. 2012). In addition to PMN-MDSCs and Mo-MDSCs, fibrocytic MDSCs, which are distinguished from conventional fibrocytes, were reported in human umbilical cord blood cell culture systems (Zoso et al. 2014). Notably, B7-H3+ MDSCs were found in the tumor sites of patients with non-small cell lung carcinoma and a murine lung cancer model, and their frequencies were correlated with poor prognosis (Zhang et al. 2015). Other novel functional markers and MDSC subpopulations have also been reported; however, the significance of these putative markers and MDSC subpopulations will require experimental validation showing the loss or gain of MDSC-mediated responses following their selective targeting.
Diverse functions of MDSCs
Eponymously named MDSCs are immune suppressor cells. They are activated by tumor-derived and host-derived factors, including proinflammatory mediators, and inhibit both innate and adaptive immune responses (Ostrand-Rosenberg and Sinha 2009; Gabrilovich et al. 2012). MDSCs mediate immunosuppression through several mechanisms. Arginase-1 (Arg-1)-dependent L-arginine depletion (Rodriguez et al. 2004) and L-cysteine depletion (Srivastava et al. 2010) mediated by MDSCs result in downregulated T cell receptor (TCR) expression and reduced T cell proliferation. MDSCs also generate reactive oxygen species (ROS) and reactive nitrogen species (RNS), which contribute to the inhibition of T cell function. Hydrogen peroxide downregulates TCRζ chain expression and cytokine production of T cells (Schmielau and Finn 2001). Nitrogen oxide (NO)-producing MDSCs impair IL-2 receptor signaling pathways in T cells and inhibit T cell proliferation (Mazzoni et al. 2002). TCR nitration induced by MDSCs affect antigen recognition (Nagaraj et al. 2007). MDSCs limit T cell migration through the downregulation of L-selectin expression in T cells (Hanson et al. 2009) and chemokine nitration (Molon et al. 2011). MDSCs also mediate regulatory T cell activation and expansion (Huang et al. 2006). MDSCs also suppress the activity of other immune cells. They dampen NK cell cytotoxicity and IFN-γ production (Hoechst et al. 2009), and decrease DC-mediated T cell responses (Hu et al. 2011).
Recently, the divergent role of MDSCs in inflammation (Chang et al. 2013) has attracted a lot of attention. Although MDSCs are generally immunosuppressive cells, they are also involved in immune-surveillance as immune effector cells under specific conditions, like acute inflammation (Cuenca et al. 2011). MDSCs, which accumulate in ascites of ovarian carcinoma-bearing mice, are capable of priming cytotoxic T lymphocytes (CTLs) and mediating antitumor immunity (Tomihara et al. 2010). Furthermore, activated natural killer T (NKT) cells convert MDSCs into immunogenic antigen presenting cells (APCs); these MDSCs have the potential to be used as a cell-based vaccine (Ko et al. 2009a; Lee et al. 2012). In addition, injection of attenuated Salmonella into mice induced the conversion of MDSCs into TNF-α producing immune effector cells (Hong et al. 2013b).
Gene expression in MDSCs
Although MDSCs are recognized as major cells of immunosuppression in the tumor environment, the nature and characteristics of MDSC subpopulations and the regulatory factors that modulate their differentiation and functions have not been fully elucidated. Potential therapeutic interventions targeting MDSCs may be based on factors related to their functional markers, accumulation/recruitment to tumor sites, or immunosuppressive activities. These factors have been analyzed in various studies using the following technologies: cDNA microarrays (Kim et al. 2012; Ko et al. 2014), microRNA (miR) microarrays (Hegde et al. 2013; Li et al. 2014), transcriptomics analysis (Fridlender et al. 2012), flow cytometry (Movahedi et al. 2008), or proteomics analysis (Boutte et al. 2011).
Various MDSC phenotypic markers have been identified, the majority of which do not depend on the tumor model used; however, some are still controversial and may be variable in different tumor models (Movahedi et al. 2008; Ko et al. 2009a; Talmadge and Gabrilovich 2013). MDSCs express low levels of CD86 and MHC class II molecules, and are therefore poor antigen presenting cells (Ko et al. 2009a); however, they express significant levels of molecules that suppress T cells, including CD80 (Mencacci et al. 2002) and B7-H1 (Liu et al. 2008). The adhesion molecules, CD11a, CD162, and CD54, are expressed on the surface of both Mo-MDSCs and PMN-MDSCs (Movahedi et al. 2008). IL-4 receptor α (IL-4Rα/CD124), which is reported to mediate the immunosuppressive activities of MDSCs (Kohanbash et al. 2013), is also found on the surface of both MDSC subtypes in mice and humans (Mandruzzato et al. 2009). However, F4/80 and CD115 are Mo-MDSC-specific surface markers (Movahedi et al. 2008), and CD115 defines a specific immunosuppressive MDSC subpopulation (Huang et al. 2006).
As shown by miR microarray analysis, miR-155 and miR-21 are upregulated in MDSCs and are associated with the expansion of functional MDSCs (Li et al. 2014). Expression of miR-494, which targets phosphatase and tensin homolog (PTEN) and AKT signaling pathways, plays a critical role in the accumulation and activity of MDSCs (Liu et al. 2012). In contrast, downregulation of miR-223 in MDSCs is correlated with the differentiation of MDSCs from bone marrow cells (Liu et al. 2011). Overexpression of Ki67, which promotes cell proliferation, in MDSCs facilitates their expansion in tumor-bearing hosts (Kim et al. 2012; Hegde et al. 2013).
The establishment of an immunosuppressive tumor environment depends not only on generating sufficient numbers of MDSCs, but also on recruiting MDSCs to the tumor site. Lack of 5-lipoxygenase, which generates leukotriene B4 and cysteinyl leukotrienes, in MDSCs is associated with their limited recruitment to primary tumor sites and lower expression of Arg-1 (Cheon et al. 2011).
As shown by comparative proteomics analysis, various proteins associated with lipid metabolism, platelet activation, angiogenesis, and tumor invasion are elevated in MDSCs from metastatic tumor models compared to MDSCs from non-metastatic tumor models (Boutte et al. 2011). Proteins that are associated with metastasis and whose expression are regulated by the cAMP-responsive element-binding protein (CREB) transcription factor are also upregulated in MDSCs from malignant tumor-bearing mice compared to in MDSCs from non-metastatic tumor-bearing mice. Consistent with these findings from proteomics analysis, CREB mRNA is upregulated in PMN-MDSCs (Youn et al. 2012). CREB induces the expression of another transcription factor, CCAAT-enhancer-binding protein β (C/EBPβ), which controls downstream M2 macrophage-specific target gene expression (Ruffell et al. 2009). The expression and activity of proteins involved in immunosuppression, including Arg-1 and inducible nitric oxide synthases (iNOS), are decreased in C/EBPβ-deficient MDSCs, suggesting that C/EBPβ is a critical regulator of MDSC function (Marigo et al. 2010).
The proliferation and immunosuppressive functions of MDSCs are regulated by several factors. The calcium binding protein, S100A9, contributes to the proliferation of MDSCs and suppression of T cell responses (Cheng et al. 2008). FK506 binding protein 51 (FKBP51), a member of the immunophilin protein family, is overexpressed in both Mo-MDSCs and PMN-MDSCs, and FKBP51 deficiency in MDSCs attenuates MDSC immunosuppressive activity and reduces Arg-1, iNOS, and NADPH oxidase 2 (NOX2) expression (Kim et al. 2012). Complement factor C5a also contributes to immune suppression via regulation of MDSCs (Markiewski et al. 2008). C5a receptor-deficient MDSCs exhibit defective ROS/RNS production and migration to tumor sites.
Role of STATs in MDSCs
Tumor-derived and host-derived soluble factors affect MDSCs, in terms of expansion, accumulation and suppressive function (Gabrilovich et al. 2012). Inflammation induces MDSC generation by inflammatory mediators, such as IL-1β (Bunt et al. 2006) and PGE2 (Rodriguez et al. 2005). IL-1β induces the production of several cytokines, which can also affect accumulation and immunosuppressive function of MDSCs. In IL-1 receptor-deficient mice, IL-6 partially compensated the proinflammatory function of IL-1β in MDSC induction (Bunt et al. 2007). IL-17 was required for MDSC-mediated immune suppression, and IL-17 receptor-deficient mice exhibited low levels of MDSCs in the tumor sites. Moreover, immune regulation by these MSDCs was defective (He et al. 2010). Proinflammatory S100A8/A9 induced MDSC accumulation and promoted MDSC migration (Sinha et al. 2008). IFN-γ produced by T cells induced MDSCs, which suppressed CD8+ T cells (Gallina et al. 2006).
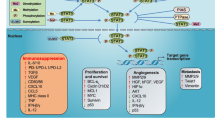
The STAT pathway was originally discovered through the study of IFN-α and IFN-γ-mediated signaling (Darnell Jr and Kerr 1994). IL-6 is one of the most well-known activators of STAT3 (Zhong et al. 1994), and various cytokines, including G-CSF, IL-10, and IL-2, activate other STAT signaling pathways (Yu et al. 2014). Th2 cytokines, IL-4, and IL-13, activate STAT signaling pathways in myeloid cells, which regulate target gene expression (Bhattacharjee et al. 2013). Therefore, it is no wonder that aforementioned MDSC modulating factors are involved in STAT signaling. By activating STAT signaling, these tumor-derived and host-derived soluble ligands modulate MDSC proliferation, survival, and immunosuppressive function (Fig. 1).

Soluble ligands that activate specific STAT signaling in MDSCs. Schematic diagram shows the various soluble factors that activate specific STAT signaling pathways in MDSCs. Receptor engagement with its ligand regulates expansion, differentiation, and/or immunosuppressive functions of MDSCs
As shown in Fig. 1, VEGF, GM-CSF, G-CSF, and IL-6 are involved in MDSC generation. Tumor-derived VEGF, which activates STAT3 and upregulates ROS production in MDSCs, induces MDSCs (Jayaraman et al. 2012). G-CSF contributes to MDSC generation in a STAT3-dependent manner, which is negatively regulated by suppressor of cytokine signaling 3 (SOCS3) (Yu et al. 2015). Another myelopoietic growth factor, GM-CSF activates STAT5 and downregulates interferon regulatory factor-8 (IRF-8) transcription, and induces MDSC accumulation (Waight et al. 2013). IL-6 induces STAT3 phosphorylation, and IL-6 treatment of human PBMCs increases the percentage of MDSCs and upregulates Arg-1 expression in these MDSCs (Chen et al. 2014).
Flt3 ligand (Flt3L), which activates STAT3 signaling, results in the expansion of MDSCs with immunosuppressive functions (Rosborough et al. 2014). Several cytokines also activate these functions. STAT1-activating IFN-γ, STAT3-activating IL-10, and STAT6-activating IL-13 regulate gene expression in MDSCs (Ma et al. 2011). IL-4Rα, which is an important marker of MDSCs, is involved in binding IL-4 and IL-13. IL-4 and IL-13 binding can lead to similar or distinct effects depending on cell type (Kruse et al. 2002; Hallett et al. 2012). In human monocytes, IL-13 binding to its receptor results in JAK2 and TYK2 activation, whereas IL-4 activates JAK1 (Bhattacharjee et al. 2013).
In summary, VEGF, hematopoietic cytokines, such as G-CSF and GM-CSF, Flt3L, and other cytokines that are especially categorized as anti-inflammatory cytokines activate STAT signaling to regulate MDSC proliferation, survival, and activation. Thus, potential anti-cancer therapies targeting STAT signaling may directly affect both cancer cells and tumor-associated immune cells, such as MDSCs.
Regulation of MDSCs in human and mouse cancers via STAT signaling
Although there are several subpopulations of MDSCs in human cancer patients and mouse tumor models, over-activation of STAT signaling is not limited to a specific model (Table 1). In humans, STAT3 signaling is associated with MDSC differentiation and immunosuppressive functions. Immunosuppressive activity of MDSCs in melanoma patients is mediated by STAT3 (Poschke et al. 2010). In MDSCs of patients with head and neck squamous cell carcinoma (HNSCC), phosphorylated STAT3 regulates Arg-1 gene expression by binding to its promoter (Vasquez-Dunddel et al. 2013). In addition to tumor-derived growth factors and cytokines, exosomes can activate STAT3 signaling and modulate the immunosuppressive activities of MDSCs in both mouse and human cancers (Chalmin et al. 2010). Tumor-derived exosomes induce IL-6 production, and autocrine IL-6 signaling triggers STAT3 phosphorylation and activation in MDSCs.
The importance of STAT1 signaling in MDSCs is revealed by mouse knock-out models. Defective STAT1 signaling reduces the immunosuppressive activity of both Mo-MDSCs and PMN-MDSCs (Schouppe et al. 2013; Medina-Echeverz et al. 2014). Under most conditions, STAT1-deficient MDSCs suppress T cell response to a lower extent than normal MDSCs, and this can be understood in the same context with the role of STAT3 in human; however, under specific conditions, STAT1−/− PMN-MDSCs from tumor-bearing mice have higher immunosuppressive activity and longer lifetimes than control MDSCs (Medina-Echeverz et al. 2014).
Thus, STAT signaling pathways, in particular STAT3 signaling, are potential therapeutic targets for modulating MDSC survival, differentiation, and immunosuppressive activities. Several strategies blocking STAT signaling (Table 2) have been attempted to overcome the immunosuppressive activities of MDSCs in the tumor environment.
Targeting STAT pathways for modulating MDSCs
Strategies to overcome tumor-associated immunosuppression mediated by MDSCs can be classified based on their mechanism of action: inhibition of MDSC generation, depletion of MDSCs, differentiation of precursor cells into non-MDSC cells, inhibition of MDSC recruitment to tumor sites, and modulation of immunosuppressive functions of MDSCs (Chang et al. 2013). Numerous host- and tumor-derived factors, such as Flt3L, GM-CSF, and IFN-γ, activate STAT signaling in MDSCs to regulate their accumulation and/or activation.
Several classes of STAT inhibitors, including small molecules, peptides, and oligonucleotides (Furqan et al. 2013), have been reported. Small molecule inhibitors are primarily used to block STAT signaling in preclinical and clinical studies because of their availability, easy application both in vitro and in vivo, and preferable pharmacokinetic properties.
Cucurbitacins are triterpenoids isolated from members of the family, Cucurbitaceae. Several variants of cucurbitacins with improved solubility and efficacy have been developed, including cucurbitacin B and I (JSI-124), which inhibit STAT3 and JAK2 (Alghasham 2013). Treatment with cucurbitacin I induces the differentiation of immature myeloid cells into mature DCs and macrophages and relieves immunosuppression in tumor-bearing mice (Nefedova et al. 2005). Cucurbitacin I also downregulates the expression of NOX in MDSCs and reduces the production of critical ROS (Corzo et al. 2009). It has recently been reported that the oral administration of the JAK2/STAT3 signaling inhibitor, cucurbitacin B, reduces the percentage of Lin-HLA-DR-CD33+ immature myeloid cells in patients with advanced lung cancer and improves anticancer immune responses (Lu et al. 2012). Thus, cucurbitacins can modulate MDSC-mediated immunosuppression either by reducing MDSC load or by inhibiting their activity.
Sunitinib is a multiple receptor tyrosine kinase inhibitor, which is approved for the treatment of several cancers. Its targets include platelet-derived growth factor receptor-α/β (PDGFR-α/β), VEGF receptor-1/2/3 (VEGFR-1/2/3), and c-Kit. Sunitinib inhibits STAT3 phosphorylation, which depends on p-Src but not JAK2 inhibition (Xin et al. 2009). Sunitinib treatment inhibits STAT3-regulated target gene expression, including that of VEGF, and reduces the percentage of MDSCs in the spleen and blood of tumor-bearing mice. Sunitinib treatment also reduces MDSC load in peripheral blood in renal cell carcinoma (RCC) patients, suggesting its potential to target MDSC-mediated immunosuppression (Ko et al. 2009b). The diverse effects of sunitinib likely reflect its diverse targets in tumor cells and immune cells in the tumor environment.
AG490 is a specific JAK2 inhibitor, which also blocks STAT3 activation. However, AG490 also inhibits JAK3, STAT1, STAT3, STAT-5a, STAT-5b, and other tyrosine kinases (Wang et al. 1999). Therefore, the mechanism of action of AG490 should be interpreted with caution. AG490 treatment inhibits STAT3 signaling, relieves MDSC-mediated immunosuppression of T cell responses, and induces the differentiation of MDSCs into immunogenic cells (Poschke et al. 2010).
Stattic, which was identified in a chemical library screen, selectively binds to the SH2 binding domain of STAT3 and inhibits the activation, dimerization and nuclear translocation of STAT3 (Schust et al. 2006). Although its STAT3 selectivity is controversial (Sanseverino et al. 2012), Stattic is widely used as a STAT3 inhibitor (Furqan et al. 2013). Stattic treatment relieves MDSC-mediated immunosuppression and inhibits differentiation into MDSCs. Phosphorylated STAT3 signaling is correlated with Arg-1 expression and Stattic treatment decreases Arg-1 expression in MDSCs. MDSC-mediated immunosuppression of T cells in cancer patients is also reduced by Stattic treatment (Vasquez-Dunddel et al. 2013). The absence of SOCS3, a negative regulator of JAK/STAT signaling, in bone marrow (BM)-derived cells results in the upregulation of STAT3 signaling. Over-activated STAT3 signaling promotes the differentiation of BM-derived cells into MDSCs, whereas Stattic treatment blocks MDSC generation induced by G-CSF (Yu et al. 2015). MDSCs induce cancer stem cells when co-cultured with human pancreatic cancer, which is also prevented by Stattic treatment (Panni et al. 2014).
S3I-201, a small molecule that binds the SH2 domain of STAT3, was identified in an in silico screen of the National Cancer Institute chemical library (Siddiquee et al. 2007). S3I-201 inhibits STAT3-dependent transcription by inhibiting STAT3 homo-dimerization. Unexpectedly, inhibition of STAT3 signaling by S3I-201 results in expansion of MDSCs that depends on Flt3L signaling (Rosborough et al. 2014). However, the immunosuppressive activity of MDSCs is blocked by S3I-201 treatment, indicating its dependence on STAT3 signaling.
In addition to cucurbitacins, several natural compounds, including cryptotanshinone and curcumin, inhibit STAT3 (Furqan et al. 2013). FLL32 is a curcumin analogue with improved pharmacokinetic parameters and potency than curcumin. FLL32 binds the SH2 domain of STAT3 and inhibits STAT3, but not STAT1, phosphorylation/activation and dimerization (Bill et al. 2012). As discussed above, both G-CSF and GM-CSF regulate the generation of MDSCs. FLL32 treatment inhibits G-CSF-induced IRF-8 downregulation, suggesting a critical role of STAT3 in mediating the effects of G-CSF signaling in these cells (Waight et al. 2013). The active ingredient of Herba Epimedii, 3,5,7-trihydroxy-4′-methoxy-8-(3-hydroxy-3-methylbutyl)-flavone (ICT), also reduced STAT3 phosphorylation in MDSCs (Zhou et al. 2011). Treatment with ICT resulted in conversion of MDSCs into DCs and macrophages, and the critical functional mediators, NO and ROS, were downregulated in the MDSCs.
Inhibitors of other STAT homologues were identified by several approaches. Through high-throughput cell-based screening of drugs known to be safe in humans, the antipsychotic drug, pimozide, was identified as a selective STAT5 inhibitor that does not inhibit STAT1 or NF-kB (Nelson et al. 2011). In MDSCs, IRF-8 expression is regulated by GM-CSF via the STAT5 signaling pathway, and inhibition of STAT5 signaling by pimozide increases IRF-8 expression in the presence of GM-CSF (Waight et al. 2013). These results suggest the potential of STAT inhibitors as effective immunotherapeutic anticancer agents.
Conclusion
Cancer immunotherapies can be classified as those that stimulate anticancer immune effectors or those that relieve immunosuppression. The former includes cancer vaccines that comprise stimulatory epitopes or ligands and adjuvants. However, cancer vaccines may not protect patients because of tumor-associated immunosuppressive mechanisms, and both the relief of immunosuppression and activation of immune responses are likely necessary for effective anticancer treatment. As one of the primary cellular effectors of cancer-associated immunosuppression, MDSCs represent an important cellular target of anticancer therapeutics.
STAT signaling is disrupted in various cancers. Various cancer cells are characterized by hyperactive STAT signaling, which contributes to cancer cell survival and proliferation. STAT signaling is also involved in angiogenesis and immune responses at the tumor site. STAT proteins also regulate MDSC survival, differentiation, and immunosuppressive functions. Therefore, in addition to Myd88 (Hong et al. 2013a) and NF-kB signaling (Kim et al. 2012), STAT signaling, in particular STAT3 signaling, is important to MDSC biology.
Various STAT inhibitors have been developed from natural compounds or by screening chemical and drug libraries. Due to the diverse functions of STAT proteins in the tumor environment, STAT inhibitors might be used to regulate cancer cells and the tumor microenvironment, including immune cells. Although most cancer therapies have focused on inducing cancer cell death directly, exploiting the intrinsic potential of the adaptive and innate immune system to target cancer cells provides a powerful means of indirectly removing cancer cells and preventing their recurrence.
Inhibiting STAT signaling may concomitantly decrease the viability of MDSCs and their immunosuppressive functions. In addition, STAT inhibitors have the potential to convert MDSCs into fully mature myeloid cells or non-immune suppressor cells, which may represent a better strategy than simply decreasing MDSC load. Studies on the efficacy of STAT inhibitors as anticancer immunotherapeutic agents are already under way, and together with basic research advances, fulfillment of the clinical promise of STAT inhibitors is on the horizon.
References
Al Zaid Siddiquee K, Turkson J (2008) STAT3 as a target for inducing apoptosis in solid and hematological tumors. Cell Res 18:254–267
Alghasham AA (2013) Cucurbitacins—a promising target for cancer therapy. Int J Health Sci (Qassim) 7:77–89
Arora A, Scholar EM (2005) Role of tyrosine kinase inhibitors in cancer therapy. J Pharmacol Exp Ther 315:971–979
Azare J, Leslie K, Al-Ahmadie H, Gerald W, Weinreb PH, Violette SM, Bromberg J (2007) Constitutively activated Stat3 induces tumorigenesis and enhances cell motility of prostate epithelial cells through integrin beta 6. Mol Cell Biol 27:4444–4453
Bhattacharjee A, Shukla M, Yakubenko VP, Mulya A, Kundu S, Cathcart MK (2013) IL-4 and IL-13 employ discrete signaling pathways for target gene expression in alternatively activated monocytes/macrophages. Free Radic Biol Med 54:1–16
Bill MA, Nicholas C, Mace TA, Etter JP, Li C, Schwartz EB, Fuchs JR, Young GS, Lin L, Lin J, He L, Phelps M, Li PK, Lesinski GB (2012) Structurally modified curcumin analogs inhibit STAT3 phosphorylation and promote apoptosis of human renal cell carcinoma and melanoma cell lines. PLoS One 7:e40724
Boutte AM, Mcdonald WH, Shyr Y, Yang L, Lin PC (2011) Characterization of the MDSC proteome associated with metastatic murine mammary tumors using label-free mass spectrometry and shotgun proteomics. PLoS One 6:e22446
Bromberg J, Darnell Jr JE (2000) The role of STATs in transcriptional control and their impact on cellular function. Oncogene 19:2468–2473
Bunt SK, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S (2006) Inflammation induces myeloid-derived suppressor cells that facilitate tumor progression. J Immunol 176:284–290
Bunt SK, Yang L, Sinha P, Clements VK, Leips J, Ostrand-Rosenberg S (2007) Reduced inflammation in the tumor microenvironment delays the accumulation of myeloid-derived suppressor cells and limits tumor progression. Cancer Res 67:10019–10026
Chalmin F, Ladoire S, Mignot G, Vincent J, Bruchard M, Remy-Martin JP, Boireau W, Rouleau A, Simon B, Lanneau D, De Thonel A, Multhoff G, Hamman A, Martin F, Chauffert B, Solary E, Zitvogel L, Garrido C, Ryffel B, Borg C, Apetoh L, Rebe C, Ghiringhelli F (2010) Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J Clin Invest 120:457–471
Chang SY, Kim YJ, Ko HJ (2013) Potential therapeutic anti-tumor effect of a Salmonella-based vaccine. Hum Vaccin Immunother 9:1654–1660
Chen MF, Kuan FC, Yen TC, Lu MS, Lin PY, Chung YH, Chen WC, Lee KD (2014) IL-6-stimulated CD11b + CD14 + HLA-DR- myeloid-derived suppressor cells, are associated with progression and poor prognosis in squamous cell carcinoma of the esophagus. Oncotarget 5:8716–8728
Cheng P, Corzo CA, Luetteke N, Yu B, Nagaraj S, Bui MM, Ortiz M, Nacken W, Sorg C, Vogl T, Roth J, Gabrilovich DI (2008) Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J Exp Med 205:2235–2249
Cheon EC, Khazaie K, Khan MW, Strouch MJ, Krantz SB, Phillips J, Blatner NR, Hix LM, Zhang M, Dennis KL, Salabat MR, Heiferman M, Grippo PJ, Munshi HG, Gounaris E, Bentrem DJ (2011) Mast cell 5-lipoxygenase activity promotes intestinal polyposis in APCΔ468 mice. Cancer Research 71:1627–1636
Corzo CA, Cotter MJ, Cheng P, Cheng F, Kusmartsev S, Sotomayor E, Padhya T, Mccaffrey TV, Mccaffrey JC, Gabrilovich DI (2009) Mechanism regulating reactive oxygen species in tumor-induced myeloid-derived suppressor cells. J Immunol 182:5693–5701
Cuenca AG, Delano MJ, Kelly-Scumpia KM, Moreno C, Scumpia PO, Laface DM, Heyworth PG, Efron PA, Moldawer LL (2011) A paradoxical role for myeloid-derived suppressor cells in sepsis and trauma. Mol Med 17:281–292
Darnell Jr JE (1997) STATs and gene regulation. Science 277:1630–1635
Darnell Jr JE, Kerr I (1994) Jak-STAT pathways and transcriptional activation in response to IFNs and other extracellular signaling proteins. Science 264:1415–1421
Fridlender ZG, Sun J, Mishalian I, Singhal S, Cheng G, Kapoor V, Horng W, Fridlender G, Bayuh R, Worthen GS, Albelda SM (2012) Transcriptomic analysis comparing tumor-associated neutrophils with granulocytic myeloid-derived suppressor cells and normal neutrophils. PLoS One 7:e31524
Furqan M, Akinleye A, Mukhi N, Mittal V, Chen Y, Liu D (2013) STAT inhibitors for cancer therapy. J Hematol Oncol 6:90
Gabrilovich DI, Nagaraj S (2009) Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 9:162–174
Gabrilovich DI, Bronte V, Chen SH, Colombo MP, Ochoa A, Ostrand-Rosenberg S, Schreiber H (2007) The terminology issue for myeloid-derived suppressor cells. Cancer Res 67:425
Gabrilovich DI, Ostrand-Rosenberg S, Bronte V (2012) Coordinated regulation of myeloid cells by tumours. Nat Rev Immunol 12:253–268
Gallina G, Dolcetti L, Serafini P, De Santo C, Marigo I, Colombo MP, Basso G, Brombacher F, Borrello I, Zanovello P, Bicciato S, Bronte V (2006) Tumors induce a subset of inflammatory monocytes with immunosuppressive activity on CD8+ T cells. J Clin Invest 116:2777–2790
Hallett MA, Venmar KT, Fingleton B (2012) Cytokine stimulation of epithelial cancer cells: the similar and divergent functions of IL-4 and IL-13. Cancer Res 72:6338–6343
Hanson EM, Clements VK, Sinha P, Ilkovitch D, Ostrand-Rosenberg S (2009) Myeloid-derived suppressor cells down-regulate L-selectin expression on CD4+ and CD8 + T cells. J Immunol 183:937–944
Hassel JC, Winnemoller D, Schartl M, Wellbrock C (2008) STAT5 contributes to antiapoptosis in melanoma. Melanoma Res 18:378–385
He D, Li H, Yusuf N, Elmets CA, Li J, Mountz JD, Xu H (2010) IL-17 promotes tumor development through the induction of tumor promoting microenvironments at tumor sites and myeloid-derived suppressor cells. J Immunol 184:2281–2288
Hegde VL, Tomar S, Jackson A, Rao R, Yang X, Singh UP, Singh NP, Nagarkatti PS, Nagarkatti M (2013) Distinct microRNA expression profile and targeted biological pathways in functional myeloid-derived suppressor cells induced by Delta9-tetrahydrocannabinol in vivo: regulation of CCAAT/enhancer-binding protein alpha by microRNA-690. J Biol Chem 288:36810–36826
Hoechst B, Voigtlaender T, Ormandy L, Gamrekelashvili J, Zhao F, Wedemeyer H, Lehner F, Manns MP, Greten TF, Korangy F (2009) Myeloid derived suppressor cells inhibit natural killer cells in patients with hepatocellular carcinoma via the NKp30 receptor. Hepatology 50:799–807
Hong EH, Chang SY, Lee BR, Kim YS, Lee JM, Kang CY, Kweon MN, Ko HJ (2013a) Blockade of Myd88 signaling induces antitumor effects by skewing the immunosuppressive function of myeloid-derived suppressor cells. Int J Cancer 132:2839–2848
Hong EH, Chang SY, Lee BR, Pyun AR, Kim JW, Kweon MN, Ko HJ (2013b) Intratumoral injection of attenuated Salmonella vaccine can induce tumor microenvironmental shift from immune suppressive to immunogenic. Vaccine 31:1377–1384
Hu CE, Gan J, Zhang RD, Cheng YR, Huang GJ (2011) Up-regulated myeloid-derived suppressor cell contributes to hepatocellular carcinoma development by impairing dendritic cell function. Scand J Gastroenterol 46:156–164
Huang M, Dorsey JF, Epling-Burnette PK, Nimmanapalli R, Landowski TH, Mora LB, Niu G, Sinibaldi D, Bai F, Kraker A, Yu H, Moscinski L, Wei S, Djeu J, Dalton WS, Bhalla K, Loughran TP, Wu J, Jove R (2002) Inhibition of Bcr-Abl kinase activity by PD180970 blocks constitutive activation of Stat5 and growth of CML cells. Oncogene 21:8804–8816
Huang B, Pan PY, Li Q, Sato AI, Levy DE, Bromberg J, Divino CM, Chen SH (2006) Gr-1 + CD115 + immature myeloid suppressor cells mediate the development of tumor-induced T regulatory cells and T-cell anergy in tumor-bearing host. Cancer Res 66:1123–1131
Jayaraman P, Parikh F, Lopez-Rivera E, Hailemichael Y, Clark A, Ma G, Cannan D, Ramacher M, Kato M, Overwijk WW, Chen SH, Umansky VY, Sikora AG (2012) Tumor-expressed inducible nitric oxide synthase controls induction of functional myeloid-derived suppressor cells through modulation of vascular endothelial growth factor release. J Immunol 188:5365–5376
Kim YS, Kim YJ, Lee JM, Kim EK, Park YJ, Choe SK, Ko HJ, Kang CY (2012) Functional changes in myeloid-derived suppressor cells (MDSCs) during tumor growth: FKBP51 contributes to the regulation of the immunosuppressive function of MDSCs. J Immunol 188:4226–4234
Ko HJ, Lee JM, Kim YJ, Kim YS, Lee KA, Kang CY (2009a) Immunosuppressive myeloid-derived suppressor cells can be converted into immunogenic APCs with the help of activated NKT cells: an alternative cell-based antitumor vaccine. J Immunol 182:1818–1828
Ko JS, Zea AH, Rini BI, Ireland JL, Elson P, Cohen P, Golshayan A, Rayman PA, Wood L, Garcia J, Dreicer R, Bukowski R, Finke JH (2009b) Sunitinib mediates reversal of myeloid-derived suppressor cell accumulation in renal cell carcinoma patients. Clin Cancer Res 15:2148–2157
Ko J, Rayman PA, Yang Y, Gopalan B, Finke J (2014) Differential gene expression in G-MDSC and neutrophils from renal cell carcinoma patients. J Immunother Cancer 2:P216
Kohanbash G, Mckaveney K, Sakaki M, Ueda R, Mintz AH, Amankulor N, Fujita M, Ohlfest JR, Okada H (2013) GM-CSF promotes the immunosuppressive activity of glioma-infiltrating myeloid cells through interleukin-4 receptor-alpha. Cancer Res 73:6413–6423
Kortylewski M, Kujawski M, Wang T, Wei S, Zhang S, Pilon-Thomas S, Niu G, Kay H, Mule J, Kerr WG, Jove R, Pardoll D, Yu H (2005) Inhibiting Stat3 signaling in the hematopoietic system elicits multicomponent antitumor immunity. Nat Med 11:1314–1321
Kruse S, Braun S, Deichmann KA (2002) Distinct signal transduction processes by IL-4 and IL-13 and influences from the Q551R variant of the human IL-4 receptor alpha chain. Respir Res 3:24
Lee JM, Seo JH, Kim YJ, Kim YS, Ko HJ, Kang CY (2012) The restoration of myeloid-derived suppressor cells as functional antigen-presenting cells by NKT cell help and all-trans-retinoic acid treatment. Int J Cancer 131:741–751
Lee HT, Xue J, Chou PC, Zhou A, Yang P, Conrad CA, Aldape KD, Priebe W, Patterson C, Sawaya R, Xie K, Huang S (2015) Stat3 orchestrates interaction between endothelial and tumor cells and inhibition of Stat3 suppresses brain metastasis of breast cancer cells. Oncotarget 6:10016–10029
Levis M, Allebach J, Tse KF, Zheng R, Baldwin BR, Smith BD, Jones-Bolin S, Ruggeri B, Dionne C, Small D (2002) A FLT3-targeted tyrosine kinase inhibitor is cytotoxic to leukemia cells in vitro and in vivo. Blood 99:3885–3891
Levy DE, Darnell JE (2002) STATs: transcriptional control and biological impact. Nat Rev Mol Cell Biol 3:651–662
Li L, Zhang J, Diao W, Wang D, Wei Y, Zhang CY, Zen K (2014) MicroRNA-155 and MicroRNA-21 promote the expansion of functional myeloid-derived suppressor cells. J Immunol 192:1034–1043
Lindau D, Gielen P, Kroesen M, Wesseling P, Adema GJ (2013) The immunosuppressive tumour network: myeloid-derived suppressor cells, regulatory T cells and natural killer T cells. Immunology 138:105–115
Liu Y, Zeng B, Zhang Z, Zhang Y, Yang R (2008) B7-H1 on myeloid-derived suppressor cells in immune suppression by a mouse model of ovarian cancer. Clin Immunol 129:471–481
Liu Y, Li PK, Li C, Lin J (2010) Inhibition of STAT3 signaling blocks the anti-apoptotic activity of IL-6 in human liver cancer cells. J Biol Chem 285:27429–27439
Liu Q, Zhang M, Jiang X, Zhang Z, Dai L, Min S, Wu X, He Q, Liu J, Zhang Y, Yang R (2011) miR-223 suppresses differentiation of tumor-induced CD11b (+) Gr1 (+) myeloid-derived suppressor cells from bone marrow cells. Int J Cancer 129:2662–2673
Liu Y, Lai L, Chen Q, Song Y, Xu S, Ma F, Wang X, Wang J, Yu H, Cao X, Wang Q (2012) MicroRNA-494 is required for the accumulation and functions of tumor-expanded myeloid-derived suppressor cells via targeting of PTEN. J Immunol 188:5500–5510
Lu P, Yu B, Xu J (2012) Cucurbitacin B regulates immature myeloid cell differentiation and enhances antitumor immunity in patients with lung cancer. Cancer Biother Radiopharm 27:495–503
Ma G, Pan PY, Eisenstein S, Divino CM, Lowell CA, Takai T, Chen SH (2011) Paired immunoglobin-like receptor-B regulates the suppressive function and fate of myeloid-derived suppressor cells. Immunity 34:385–395
Mandruzzato S, Solito S, Falisi E, Francescato S, Chiarion-Sileni V, Mocellin S, Zanon A, Rossi CR, Nitti D, Bronte V, Zanovello P (2009) IL4Rα + myeloid-derived suppressor cell expansion in cancer patients. J Immunol 182:6562–6568
Marigo I, Bosio E, Solito S, Mesa C, Fernandez A, Dolcetti L, Ugel S, Sonda N, Bicciato S, Falisi E, Calabrese F, Basso G, Zanovello P, Cozzi E, Mandruzzato S, Bronte V (2010) Tumor-induced tolerance and immune suppression depend on the C/EBPbeta transcription factor. Immunity 32:790–802
Markiewski MM, Deangelis RA, Benencia F, Ricklin-Lichtsteiner SK, Koutoulaki A, Gerard C, Coukos G, Lambris JD (2008) Modulation of the antitumor immune response by complement. Nat Immunol 9:1225–1235
Matsumura Y, Kobayashi T, Ichiyama K, Yoshida R, Hashimoto M, Takimoto T, Tanaka K, Chinen T, Shichita T, Wyss-Coray T, Sato K, Yoshimura A (2007) Selective expansion of foxp3-positive regulatory T cells and immunosuppression by suppressors of cytokine signaling 3-deficient dendritic cells. J Immunol 179:2170–2179
Mazzoni A, Bronte V, Visintin A, Spitzer JH, Apolloni E, Serafini P, Zanovello P, Segal DM (2002) Myeloid suppressor lines inhibit T cell responses by an NO-dependent mechanism. J Immunol 168:689–695
Medina-Echeverz J, Haile LA, Zhao F, Gamrekelashvili J, Ma C, Metais JY, Dunbar CE, Kapoor V, Manns MP, Korangy F, Greten TF (2014) IFN-gamma regulates survival and function of tumor-induced CD11b + Gr-1high myeloid derived suppressor cells by modulating the anti-apoptotic molecule Bcl2a1. Eur J Immunol 44:2457–2467
Mencacci A, Montagnoli C, Bacci A, Cenci E, Pitzurra L, Spreca A, Kopf M, Sharpe AH, Romani L (2002) CD80 + Gr-1 + myeloid cells inhibit development of antifungal Th1 immunity in mice with candidiasis. J Immunol 169:3180–3190
Molon B, Ugel S, Del Pozzo F, Soldani C, Zilio S, Avella D, De Palma A, Mauri P, Monegal A, Rescigno M, Savino B, Colombo P, Jonjic N, Pecanic S, Lazzarato L, Fruttero R, Gasco A, Bronte V, Viola A (2011) Chemokine nitration prevents intratumoral infiltration of antigen-specific T cells. J Exp Med 208:1949–1962
Movahedi K, Guilliams M, Van Den Bossche J, Van Den Bergh R, Gysemans C, Beschin A, De Baetselier P, Van Ginderachter JA (2008) Identification of discrete tumor-induced myeloid-derived suppressor cell subpopulations with distinct T cell-suppressive activity. Blood 111:4233–4244
Nagaraj S, Gupta K, Pisarev V, Kinarsky L, Sherman S, Kang L, Herber DL, Schneck J, Gabrilovich DI (2007) Altered recognition of antigen is a mechanism of CD8 + T cell tolerance in cancer. Nat Med 13:828–835
Nefedova Y, Huang M, Kusmartsev S, Bhattacharya R, Cheng P, Salup R, Jove R, Gabrilovich D (2004) Hyperactivation of STAT3 is involved in abnormal differentiation of dendritic cells in cancer. J Immunol 172:464–474
Nefedova Y, Nagaraj S, Rosenbauer A, Muro-Cacho C, Sebti SM, Gabrilovich DI (2005) Regulation of dendritic cell differentiation and antitumor immune response in cancer by pharmacologic-selective inhibition of the janus-activated kinase 2/signal transducers and activators of transcription 3 pathway. Cancer Res 65:9525–9535
Nelson EA, Walker SR, Weisberg E, Bar-Natan M, Barrett R, Gashin LB, Terrell S, Klitgaard JL, Santo L, Addorio MR, Ebert BL, Griffin JD, Frank DA (2011) The STAT5 inhibitor pimozide decreases survival of chronic myelogenous leukemia cells resistant to kinase inhibitors. Blood 117:3421–3429
Nitulescu GM, Margina D, Juzenas P, Peng Q, Olaru OT, Saloustros E, Fenga C, Spandidos D, Libra M, Tsatsakis AM (2016) Akt inhibitors in cancer treatment: the long journey from drug discovery to clinical use (Review). Int J Oncol 48:869–885
Niu G, Wright KL, Huang M, Song L, Haura E, Turkson J, Zhang S, Wang T, Sinibaldi D, Coppola D, Heller R, Ellis LM, Karras J, Bromberg J, Pardoll D, Jove R, Yu H (2002) Constitutive Stat3 activity up-regulates VEGF expression and tumor angiogenesis. Oncogene 21:2000–2008
Ostrand-Rosenberg S, Sinha P (2009) Myeloid-derived suppressor cells: linking inflammation and cancer. J Immunol 182:4499–4506
Panni R, Sanford D, Belt B, Mitchem J, Worley L, Goetz B, Mukherjee P, Wang-Gillam A, Link D, Denardo D, Goedegebuure SP, Linehan D (2014) Tumor-induced STAT3 activation in monocytic myeloid-derived suppressor cells enhances stemness and mesenchymal properties in human pancreatic cancer. Cancer Immunol Immunother 63:513–528
Poschke I, Mougiakakos D, Hansson J, Masucci GV, Kiessling R (2010) Immature immunosuppressive CD14 + HLA-DR-/low cells in melanoma patients are Stat3hi and overexpress CD80, CD83, and DC-sign. Cancer Res 70:4335–4345
Rodriguez PC, Quiceno DG, Zabaleta J, Ortiz B, Zea AH, Piazuelo MB, Delgado A, Correa P, Brayer J, Sotomayor EM, Antonia S, Ochoa JB, Ochoa AC (2004) Arginase I production in the tumor microenvironment by mature myeloid cells inhibits T-cell receptor expression and antigen-specific T-cell responses. Cancer Res 64:5839–5849
Rodriguez PC, Hernandez CP, Quiceno D, Dubinett SM, Zabaleta J, Ochoa JB, Gilbert J, Ochoa AC (2005) Arginase I in myeloid suppressor cells is induced by COX-2 in lung carcinoma. J Exp Med 202:931–939
Rosborough BR, Mathews LR, Matta BM, Liu Q, Raich-Regue D, Thomson AW, Turnquist HR (2014) Cutting edge: Flt3 ligand mediates STAT3-independent expansion but STAT3-dependent activation of myeloid-derived suppressor cells. J Immunol 192:3470–3473
Ruffell D, Mourkioti F, Gambardella A, Kirstetter P, Lopez RG, Rosenthal N, Nerlov C (2009) A CREB-C/EBPbeta cascade induces M2 macrophage-specific gene expression and promotes muscle injury repair. Proc Natl Acad Sci U S A 106:17475–17480
Sanseverino I, Purificato C, Gauzzi MC, Gessani S (2012) Revisiting the specificity of small molecule inhibitors: the example of stattic in dendritic cells. Chem Biol 19:1213–1214
Sansone P, Bromberg J (2012) Targeting the interleukin-6/Jak/stat pathway in human malignancies. J Clin Oncol 30:1005–1014
Santarpia L, Lippman SM, El-Naggar AK (2012) Targeting the MAPK-RAS-RAF signaling pathway in cancer therapy. Expert Opin Ther Targets 16:103–119
Sasaki K, Zhao X, Pardee AD, Ueda R, Fujita M, Sehra S, Kaplan MH, Kane LP, Okada H, Storkus WJ (2008) Stat6 signaling suppresses VLA-4 expression by CD8 + T cells and limits their ability to infiltrate tumor lesions in vivo. J Immunol 181:104–108
Schmielau J, Finn OJ (2001) Activated granulocytes and granulocyte-derived hydrogen peroxide are the underlying mechanism of suppression of t-cell function in advanced cancer patients. Cancer Res 61:4756–4760
Schouppe E, Mommer C, Movahedi K, Laoui D, Morias Y, Gysemans C, Luyckx A, De Baetselier P, Van Ginderachter JA (2013) Tumor-induced myeloid-derived suppressor cell subsets exert either inhibitory or stimulatory effects on distinct CD8 + T-cell activation events. Eur J Immunol 43:2930–2942
Schust J, Sperl B, Hollis A, Mayer TU, Berg T (2006) Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem Biol 13:1235–1242
Sehgal PB (2008) Paradigm shifts in the cell biology of STAT signaling. Semin Cell Dev Biol 19:329–340
Siddiquee K, Zhang S, Guida WC, Blaskovich MA, Greedy B, Lawrence HR, Yip ML, Jove R, Mclaughlin MM, Lawrence NJ, Sebti SM, Turkson J (2007) Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc Natl Acad Sci U S A 104:7391–7396
Sinha P, Okoro C, Foell D, Freeze HH, Ostrand–Rosenberg S, Srikrishna G (2008) Proinflammatory S100 proteins regulate the accumulation of myeloid-derived suppressor cells. J Immunol 181:4666–4675
Srivastava MK, Sinha P, Clements VK, Rodriguez P, Ostrand-Rosenberg S (2010) Myeloid-derived suppressor cells inhibit T-cell activation by depleting cystine and cysteine. Cancer Res 70:68–77
Talmadge JE, Gabrilovich DI (2013) History of myeloid-derived suppressor cells. Nat Rev Cancer 13:739–752
Tomihara K, Guo M, Shin T, Sun X, Ludwig SM, Brumlik MJ, Zhang B, Curiel TJ (2010) Antigen-specific immunity and cross-priming by epithelial ovarian carcinoma-induced CD11b (+) Gr-1 (+) cells. J Immunol 184:6151–6160
Vasquez-Dunddel D, Pan F, Zeng Q, Gorbounov M, Albesiano E, Fu J, Blosser RL, Tam AJ, Bruno T, Zhang H, Pardoll D, Kim Y (2013) STAT3 regulates arginase-I in myeloid-derived suppressor cells from cancer patients. J Clin Invest 123:1580–1589
Waight JD, Netherby C, Hensen ML, Miller A, Hu Q, Liu S, Bogner PN, Farren MR, Lee KP, Liu K, Abrams SI (2013) Myeloid-derived suppressor cell development is regulated by a STAT/IRF-8 axis. J Clin Invest 123:4464–4478
Wang LH, Kirken RA, Erwin RA, Yu CR, Farrar WL (1999) JAK3, STAT, and MAPK signaling pathways as novel molecular targets for the tyrphostin AG-490 regulation of IL-2-mediated T cell response. J Immunol 162:3897–3904
Wang Z, Luo F, Li L, Yang L, Hu D, Ma X, Lu Z, Sun L, Cao Y (2010) STAT3 activation induced by Epstein–Barr virus latent membrane protein1 causes vascular endothelial growth factor expression and cellular invasiveness via JAK3 And ERK signaling. Eur J Cancer 46:2996–3006
Xin H, Zhang C, Herrmann A, Du Y, Figlin R, Yu H (2009) Sunitinib inhibition of Stat3 induces renal cell carcinoma tumor cell apoptosis and reduces immunosuppressive cells. Cancer Res 69:2506–2513
Yang X, Friedl A (2015) A positive feedback loop between prolactin and STAT5 promotes angiogenesis. Adv Exp Med Biol 846:265–280
Youn JI, Nagaraj S, Collazo M, Gabrilovich DI (2008) Subsets of myeloid-derived suppressor cells in tumor-bearing mice. J Immunol 181:5791–5802
Youn JI, Collazo M, Shalova IN, Biswas SK, Gabrilovich DI (2012) Characterization of the nature of granulocytic myeloid-derived suppressor cells in tumor-bearing mice. J Leukoc Biol 91:167–181
Yu H, Jove R (2004) The STATs of cancer—new molecular targets come of age. Nat Rev Cancer 4:97–105
Yu H, Lee H, Herrmann A, Buettner R, Jove R (2014) Revisiting STAT3 signalling in cancer: new and unexpected biological functions. Nat Rev Cancer 14:736–746
Yu H, Liu Y, Mcfarland BC, Deshane JS, Hurst DR, Ponnazhagan S, Benveniste EN, Qin H (2015) SOCS3 deficiency in myeloid cells promotes tumor development: involvement of STAT3 activation and myeloid-derived suppressor cells. Cancer Immunol Res 3:727–740
Zhang G, Huang H, Zhu Y, Yu G, Gao X, Xu Y, Liu C, Hou J, Zhang X (2015) A novel subset of B7-H3 + CD14 + HLA-DR-/low myeloid-derived suppressor cells are associated with progression of human NSCLC. Oncoimmunology 4:e977164
Zhao F, Hoechst B, Duffy A, Gamrekelashvili J, Fioravanti S, Manns MP, Greten TF, Korangy F (2012) S100A9 a new marker for monocytic human myeloid-derived suppressor cells. Immunology 136:176–183
Zhong Z, Wen Z, Darnell Jr JE (1994) Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science 264:95–98
Zhou J, Wu J, Chen X, Fortenbery N, Eksioglu E, Kodumudi KN, Pk EB, Dong J, Djeu JY, Wei S (2011) Icariin and its derivative, ICT, exert anti-inflammatory, anti-tumor effects, and modulate myeloid derived suppressive cells (MDSCs) functions. Int Immunopharmacol 11:890–898
Zoso A, Mazza EM, Bicciato S, Mandruzzato S, Bronte V, Serafini P, Inverardi L (2014) Human fibrocytic myeloid-derived suppressor cells express IDO and promote tolerance via Treg-cell expansion. Eur J Immunol 44:3307–3319
Acknowledgments
This work was supported by a National Research Foundation of Korea (NRF) Grant funded by the Korean government (Ministry of Science, ICT & Future Planning) (NRF-2013R1A1A3011482).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflicts of interest.
Rights and permissions
About this article

Cite this article
Ko, HJ., Kim, YJ. Signal transducer and activator of transcription proteins: regulators of myeloid-derived suppressor cell-mediated immunosuppression in cancer. Arch. Pharm. Res. 39, 1597–1608 (2016). https://doi.org/10.1007/s12272-016-0822-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12272-016-0822-9