
Abstract
Glioblastoma multiforme (GBM) modulates the immune system to engance its malignant potential. Signal transducer and activator of transcription 3 (STAT3) activation is a regulatory node in modulating the immune microenvironment in several human tumors, including GBM. To investigate whether STAT3 inhibition might enhance anti-tumor responses, we inhibited STAT3 signaling using small interfering RNA against STAT3. We tested the human GBM cell lines U87, U251, and HS683, which are known to constitutively express high levels of phospho-STAT3. STAT3 inhibition resulted in enhanced expression of several pro-inflammatory cytokines and chemokines and supernatants from STAT3-silenced human GBM cell lines increased lipopolysaccharide-induced dendritic cell activation in vitro. We obtained comparable results when STAT3 activity was suppressed with specific small molecule inhibitors. Our results support the hypothesis that activated STAT3 contributes to the immunosuppressive microenvironment in GBM and support previous studies implicating STAT3 as a potential target for immunotherapy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Despite aggressive therapy, patients with glioblastoma multiforme (GBM) continue to have poor prognosis, thus mandating novel approaches to bolster the current therapeutic regimens. Preclinical studies have demonstrated the potential of immunotherapy to provide durable responses in models of GBM; however, these results have not yet been effectively translated to success in clinical trials. One major impediment is the immunosuppressive microenvironment of GBM, which allows the tumor to evade the immune system [1]. GBMs have been shown to downregulate MHC Class II expression, upregulate checkpoint molecules such as B7-H1, and recruit/activate suppressor immune cells such as regulatory T cells (Treg) [2–4].
Signal transducer and activator of transcription 3 (STAT3) is directly involved in the implementation and maintenance of the GBM immunosuppressive microenvironment and plays a central role in many tumors in which STAT3 is consistently activated [5–7]. STAT3 is activated downstream of multiple tyrosine kinase pathways such as epidermal growth factor receptor (EGFR) as well as non-receptor tyrosine kinases such as SRC and ABL, which are constitutively activated in many neoplasms [8, 9]. As a transcription factor, STAT3 mediates immunosuppression in tumors though modulation of cytokine expression and downregulation of MHC Class II and co-stimulatory molecules [10–13]. STAT3 is inducible by several cytokines, including IL-6 and IL-10 [14, 15], and STAT3 mediated expression of these factors drives activation of STAT3 in nearby immune cells (Fig. 1) [9, 16–18]. As a result of this activation loop, STAT3 remains constitutively active in immune cells such as neutrophils, natural killer (NK) cells, dendritic cells (DCs), macrophages, and lymphocytes, profoundly inhibiting their anti-tumor activity. In addition, STAT3 promotes activity of Tregs, which have been shown to inhibit anti-tumor immune responses in a wide variety of cancers [19, 20].

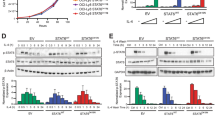
a EMSA demonstrates that STAT3 is constitutively expressed in U251, U87, and HS683. The first lane shows a positive control expressing STAT3 and STAT1, which form homo- and heterodimers. Lane 2 shows anti-STAT3 antibody causing a supershift of the pSTAT3 homodimer. Lanes 3–5 show STAT3 homodimer in HS683, U87, and U251. EMSA is representative of three replications of experiments where the cell lines shown were treated with STAT3 siRNA. b Western Blot shows knockdown of pSTAT3 in HS683, U87 and U251 cell lines after treatment with STAT3 siRNA. Lanes 1,3, and 6 show the untreated cell lines, lanes 2, 4, and 6 show the cell lines treated with control siRNA, and lanes 3, 6, and 9 show knockdown of pSTAT3 in cell lines after treatment with STAT3 siRNA. Lane 10 shows the U937 cell line treated with IL-6 and acts a positive control for pSTAT3 staining in this western blot. This blot is representative of three experimental replications. c EMSA shows that treatment of HS683, U87, and U251 with anti-STAT3 siRNA decreases pSTAT3 levels. Positive control expressing pSTAT3 and pSTAT1 is shown in lane 1. Lane 2 shows that addition of anti-STAT3 antibody causes a supershift in the pSTAT3 homodimer. Lanes 3, 6, 9 show pSTAT3 levels in untreated cell lines. Lanes 4, 7, 10 show pSTAT3 levels when cell lines are treated with control siRNA. Lanes 5, 8, and 11 show pSTAT3 levels decreased when cell lines are treated with anti-STAT3 siRNA delivered by lipofectamine
Inhibition of STAT3 has been investigated as a potential therapeutic approach in preclinical models of GBM. The validity of this approach is bolstered by data which showed that infiltrating cells with activated STAT3 was associated with poorer survival in GBMs [21]. A recent study reported that regulation of STAT3 activation with inhibitor WP1066 was capable of reversing immunosuppression in a murine glioma model, specifically improving microglial activity and survival after treatment [22]. Another group used the small molecule JSI-124 to block STAT3 combined with adoptive cytotoxic T cell therapy in a murine model and reported improved survival and increased T cell response with treatment [23]. A separate group confirmed that effector functions of immune cells are decreased in the presence of glioma cells by evaluating the functional status of STAT3-positive immune cells in co-culture experiments with normal microglial and human glioma cells [24]. These studies suggest that STAT3 activation in GBMs negatively affects immune cells and implicate STAT3 inhibition as a potential therapy; however, small molecule inhibitors tested thus far have not been specific for STAT3.
Here we present our data on the effects of STAT3 inhibition on the immune profile of GBM using STAT3 small interfering RNA (siRNA) and two STAT3-selective small molecule inhibitors, STAT3 Inhibitory Compound (Stattic) and NSC 74859 (S31-201)[25, 26].
Materials and methods
Cell lines
Established human glioma cell lines U251, U87, and HS683 were kindly provided by Dr. Gregory Riggins (Johns Hopkins School of Medicine). U937 is an established monocytic cell line derived from histiocytic lymphoma; this was kindly provided by Dr. Drew Pardoll (Johns Hopkins School of Medicine). These were cultured in DMEM supplemented with 10 % FCS, 100 units/mL penicillin, and 100 μg/mL streptomycin at 37 °C in a humidified atmosphere containing 5 % CO2.
siRNA transfection
Cell lines were transfected with 15 nM STAT3 siRNA oligonucleotide (Santa Cruz Biotechnology, Santa Cruz, CA) using Lipofectamine 2000 (Invitrogen Life Technologies, Grand Island, NY) after the cells reached 90 % confluence. Controls were treated with scrambled siRNA oligonucleotide or Lipofectamine alone, according to the manufacturer’s recommendations. The medium was changed 24 h after treatment. 72 h after treatment, the culture supernatant was collected and cells were harvested by Trypsin-mediated detachment from the flask.
Electrophoresis mobility shift assay (EMSA)
We performed EMSA as previously described [27]. We obtained 5–10 mg of crude nuclear extracts and incubated the extract with the 32P-labeled high-affinity SIE probe derived from the c-fos gene promoter that binds STAT1 and STAT3. We then performed the supershift binding reaction with a rabbit polyclonal antibody specific for STAT3 (Santa Cruz Biotechnology). The Protein-DNA complexes were run on 5 % non-denaturing polyacrylamide gels and analyzed with autoradiography.
Western blot
We performed Western blot with a standard protocol based on commercially available reagents. The pSTAT3 antibody was specific for STAT3 phosphorylated at Y705 (Cell Signaling, Danvers, MA). Antibody was used at a dilution of 1:1,000.
Quantitative real-time PCR (qRT-PCR)
Total RNA was extracted using the RNeasy Mini Kit according to the manufacture’s protocol (Qiagen, Valencia, CA). 1 μg of RNA was used as template for reverse transcription; 100 μL cDNA was synthesized using the SuperScript III First-strand Synthesis SuperMix for qRT-PCR (Invitrogen, Carlsbad, CA). qRT-PCR was performed using the ICycler MyiQ detection system (BioRad, Hercules, CA). Reactions were performed in 25 μL of final volume reaction mixture, containing 5 μL cDNA, 12.5 μL 2X SYBR Green master mix (BioRad), and 1 μmol sense and antisense primer. All oligonucleotide primers for qRT-PCR were synthesized by IDT, Inc. and the sequences are listed in Supplementary Table 1. The same thermal profile conditions were used for all primer sets. Samples were run in triplicate for each primer set. To analyze the relative changes in gene expression of the treatment group versus the untreated control, the 2−ΔΔCT method was used [28]. HS683 cell lines were tested in triplicate a total of three times, and the U87 and U251 cell lines were tested in triplicate once.
DC maturation assay
Human DCs were generated from Buffy coat units (Baxter Healthcare Corporation Fenwal division, Deerfield, IL) or from fresh whole blood, drawn with informed consent from healthy donors into heparinized Vacutainers (Becton–Dickinson Bioscience, San Jose, CA). Peripheral blood mononuclear cells (PBMCs) were isolated by a Ficoll-Hypaque density gradient (Amersham Pharmacia Biotech, Uppsala, Sweden). In accordance to the manufacturer’s protocol, we isolated CD14+ monocytes from the PBMCs through positive selection using a MACS system (Miltenyi Biotech Inc, Auburn, CA).
Subsequently, monocytes were cultured in six-well plates (0.5 × 106 cells/mL) in RPMI-1640 + 10 % FCS supplemented with 1,000 U/mL GM-CSF (R&D Systems, Minneapolis, MN) and 500 U/mL IL-4 (Peprotech, Rocky Hill, NJ). To generate mature DCs, cells were cultured for 6 days and then incubated with 100 ng/mL of lipopolysaccharide (LPS)from Escherichia coli 026:B6 (Sigma, St. Louis MS). Cells were harvested after a total treatment period of 48 h. For DC maturation inhibition experiments, CD14+ monocytes were cultured in standard DC medium supplemented with 25–75 % tumor cell supernatants from the first 24 h after siRNA transfection (see above). Fresh medium supplemented with supernatant was added to the culture on days 2, 4, and 6, at which point LPS was added. On day 8, cultures were harvested. The expression of CD11c, human leukocyte antigen (HLA)-DR, and CD86 on immature and mature DCs was analyzed by flow cytometry. DCs (1 × 104) were labeled (by incubating in 100 μL PBS/5 % FCS/0.1 % sodium azide, staining buffer) with fluorescein isothiocyanate (FITC)- conjugated IgG specific for HLA-DR (Becton–Dickinson Bioscience), phycoerythrin (PE)- conjugated IgG specific for CD86 (eBioscience, San Diego, CA), and allophycocyanin (APC)-conjugated IgG mAb specific for CD11c (Becton–Dickinson Bioscience).
Small molecule inhibitor
STAT Three Inhibitory Compound (Stattic, Calbiochem) was diluted in DMSO following the manufacturer’s protocol. NSC 74859 (S3I-201, Calbiochem) was diluted in DMSO following the manufacturer’s protocol. 5 × 105 adherent cells were seeded in T25 flasks and grown for 24 h before adding 7 mL of medium with 7 μL of 10 mM stock Stattic (final concentration 10 μM), 100 μM stock NSC 74859 (final concentration 100 μM), or DMSO. RNA and nuclear protein were extracted to confirm specific inhibition of STAT3 [27].
Results
STAT3 suppression alters the proinflammatory cytokine and chemokine profile of GBM cell lines
Initially, we tested for phospho-STAT3 (pSTAT3) expression using EMSA of nuclear protein from in vitro culture of untreated GBM cell lines. We found constitutive expression, activating phosphorylation, dimerization, and specific DNA binding of STAT3 in U251, U87, and HS683 (Fig. 1a). This is consistent with the pattern of pSTAT3 overexpression in resected human GBM specimens relative to normal brain and is in agreement with previously published studies [5, 29].
We used siRNA to specifically suppress STAT3 expression in adherent cell lines, as it has been effectively applied in head and neck cancer cell lines [30]. The LipofectAMINE transfection condition effected pSTAT3 suppression in each of the three adherent cell lines, as demonstrated by EMSA and western blot. Compared to controls, treatment with anti-STAT3 siRNA qualitatively reduced the level of DNA binding by pSTAT3 (Fig. 1b, c).
We next evaluated the effects of STAT3 inhibition on expression of the following cytokines and chemokines by qRT-PCR: interferon gamma-inducible protein 10 (IP-10), RANTES, IL-6, IL-8, and tumor necrosis factor alpha (TNFα) (Fig. 2). Compared to controls, HS683 cells treated with anti-STAT3 siRNA expressed higher mRNA levels of IL-6 (p < 0.001), IL-8 (p < 0.001), IP-10 (p = 0.01), and Rantes (p < 0.001). However, although TNFα trended towards an increase in expression, this was not statistically significant (p = 0.07). U251 and U87 cell lines treated with anti-STAT siRNA did not show significantly different mRNA levels of cytokines, however several patterns seen trended towards significance; both cell lines show an increase in IL-8 and IP-10 mRNA expression after treatment. IL-6 mRNA expression increased in U251 but not in U87, TNFa is increased in U251 cells, while Rantes is increased in U87.

STAT3 suppression in HS683 modifies the mRNA expression of cytokines and chemokines as determined by qRT-PCR. Cell lines treated with scrambled siRNA (Control siRNA) and with anti-STAT3 siRNA (STAT3 siRNA) are normalized to the untreated arm (No treatment) using the GAPDH housekeeping gene and the 2−∆∆CT method. Increases in all cytokines except TNF-alpha (TNFα) are statistically significant when compared to either the untreated or Control siRNA arms. IL-6, IP-10, and Rantes also had significant changes when the untreated arm is compared to the Control siRNA arm, but this is much less than the difference between the untreated arm and the anti-STAT3 siRNA arm. Discrepancies between the 95 % confidence interval and the alpha of 0.05 are due to application of the paired t test in p value calculation. Data is the result of three experimental replications with each qRT-PCR run completed in triplicate; error bars are shown, and the statistically significances marked *p < 0.05; **p < 0.01
pSTAT3 inhibition in tumor cells reduces dendritic cell maturation via soluble factors
We next assessed the maturation of DCs exposed to supernatant from each experimental arm of U251 and HS683. Immature DCs express low levels of CD86 and HLA-DR (15 % double-positive, Fig. 3a), while DCs which mature in response to LPS express high levels of CD86 and HLA-DR (72.7 % double positive, Fig. 3b). Supernatants from untreated and Lipofectamine-treated (negative control) U251 and HS683 cell culture did not alter the maturation of DCs (70.5–75.4 % double positive, Fig. 3c, d, f, and g). However, there was a significant increase in the level of expression of CD86 and HLA-DR when DCs were exposed to supernatant from HS683 or U251 cells treated with anti-STAT3 siRNA (92.1, or 92.7 % double positive, Fig. 3e or h). Therefore, blocking STAT3 in the tumor cells stimulates the release of soluble factors that dramatically enhance DC maturation.

Flow cytometry demonstrates DC maturation by HLA-DR and CD86 double positivity. Inhibition of DC maturation by GBM-derived factors is abrogated after STAT3 knockdown. The top row shows immature monocyte-derived DCs (a 15.5 % HLA-DR+/CD86+) and LPS-matured DCs (b 72.7 %). The middle and bottom rows show GBM adherent cell lines U251 and HS683 respectively. DCs were matured in the presence of media from untreated GBM cells (c 74.6 %; f 70.5 %), control (d 72.7 %; g 75.4 %) and STAT3 SiRNA (e 92.7 %; h 92.1 %). DC maturation increases upon the addition of conditioned media (CM) from GBM cells treated with STAT3 SiRNA
Small molecule inhibition of STAT3 reflects the effects of siRNA inhibition
Although siRNA and shRNA are effective in suppressing pSTAT3 in vitro, it has yet to be demonstrated as a feasible therapy in the clinical setting. Stattic and NSC 74859 are two non-peptide small molecule inhibitors which block pSTAT3 dimerization by inhibiting activity at the Src homology 2 (SH2) dimerization domain [25, 26]. Treatment with Stattic and NSC 74859 both decreased the level of pSTAT3 expressed by HS683 cells (Fig. 4a). Treatment with the DMSO diluents served as a negative control and did not qualitatively impact the level of pSTAT3.

a Western blot for pSTAT3 demonstrates a decrease in pSTAT3 when HS683 cells are treated with either NSC (lane 3) or Stattic (lane 6), compared to untreated cells (lanes 1 and 4) and cells treated with DMSO solution (lanes 2 and 5). Lane 7 shows a positive control (GBM55), glioblastoma resected from a patient. b qRT-PCR data is normalized to untreated GBM cell lines with the 2−∆∆CT method and the GAPDH housekeeping gene. Treatment with NSC 74859 (NSC) leads to a statistically insignificant trend towards increased IL-8 (3.74 [2.73–4.74], p = 0.07) and IL-6 (3.61 [1.7–5.52], p = 0.1). It does not significantly change expression of Rantes (1.2 [0.76–1.65], p = 0.5). NSC treatment decreases expression of IP-10 (0.43 [0.2–0.66], p = 0.02) and TNF (0.63 [0.52–0.74], p = 0.009). However, for TNFα (NSC 0.63 [0.52–0.74] vs. DMSO 0.73 [0.46–1.01], p = 0.6), changes in cytokine expression during NSC treatment are not significantly different than with DMSO delivery alone. Discrepancies between the 95 % confidence interval and the alpha of 0.05 is due to application of the paired t test in p value calculation. Data is the result of three experimental replications with each qRT-PCR run completed in triplicate; error bars are shown, and the statistically significances marked *p < 0.05; **p < 0.01. c qRT-PCR illustrates that treatment with Stattic leads to a non-significant increase in IL-6 (2.05 [−0.25–4.34], p = 0.6), IL-8 (3.34 [−0.73–7.4], p = 0.5), and TNFα (2.97 [0.89–5.05], p = 0.3). It also leads to a non-significant decrease in IP-10 (0.44 [0.13–0.75], p = 0.11) and Rantes (0.87 [0.13–1.61], p = 0.8). Discrepancies between the 95 % confidence interval and the alpha of 0.05 is due to application of the paired t test in p value calculation. Data is the result of three experimental replications with each qRT-PCR run completed in triplicate; error bars are shown, and the statistically significances marked *p < 0.05; **p < 0.01
qRT-PCR demonstrated significant change in the expression of inflammatory cytokines and chemokines when HS683 was treated with NSC 74859 or Stattic (Fig. 4b, c). NSC 74859 significantly decreased the expression of IP-10 (p = 0.02) and TNFα (p = 0.009), however NSC non-significantly increased mRNA expression of IL-6 (p = 0.1) and IL-8 (p = 0.07), which is similar to the pattern observed after siRNA treatment. Stattic non-significantly increased expression of IL-6 (p = 0.6), IL-8 (p = 0.5), and TNFα (p = 0.3), which is similar to the pattern observed after siRNA treatment. However, the Stattic arm trended towards decreased expression of IP-10 (p = 0.11) and Rantes (p = 0.8). Some of the effects in cytokine expression after treatment with Stattic and NSC resembled the patterns observed after siRNA; most consistently, IL-6 and IL-8 upregulation. Other cytokines had variable responses to the different methods of STAT3 inhibition and different degrees of STAT3 inhibition, which likely reflects variable sensitivity in pathways downstream of STAT3. Surprisingly, IP-10 was downregulated by both small-molecule inhibitors.
Discussion
We have found that targeting STAT3 (using both siRNA and small molecule inhibitors) changes the expression of paracrine immunologic signaling molecules in adherent human GBM cells. IL-6 and IL-8 were consistently increased by STAT3 blockade, both via siRNA and small molecules. IP-10 consistently increased with STAT3 siRNA but decreased with both small molecule inhibitors. Rantes responded variably to STAT3 siRNA but more subtly than IL-6; IL-8 and was affected by neither small molecule inhibitor small molecule inhibitor. Finally, TNFα presented the most variable response in both siRNA and small molecule treatments. First, IL-6, IL-8 and IP-10 expression consistently increase when STAT3 expression is inhibited. Combined with the IL-6 and IL-8 response to small molecules which interfere at the STAT3 SH2 domain, we infer that IL-6 and IL-8 expression is dependent on tyrosine phosphorylation and dimerization of STAT3, but IP-10 expression may be modulated by other downstream effects of STAT3. Furthermore, the variable response of Rantes and TNFα are not surprising, given additional levels of STAT3 regulation, such as protein inhibitor of activated STAT3 (PIAS3), which act downstream of expression and tyrosine phosphorylation; it is difficult to assess the effect of blocking STAT3 expression or phosphorylation via Rantes and TNFα.
Mechanisms of STAT3 paracrine activity have been well defined in other tumor models. Although we evaluated expression of IP-10, Rantes, IL-8, IL-6, TNFα, and IFNβ, it is possible that other cytokines or chemokines contribute to the increased immune response when pSTAT3 is inhibited. In this study, the relationships between cytokines and STAT3 reflect the published cancer immunology literature on other tumors [9]. It will also be important to evaluate whether pSTAT3 inhibition affects the expression and activity of receptors for the chemokines and cytokines, which has not been evaluated in prior studies. Changes at the receptors could amplify or dampen the functional impact of changes in the signaling molecules.
Despite the non-significant findings of some of our cell lines in the qRT-PCRs, we found consistent functional significance in our DC maturation assay. Exposing DCs to culture medium from untreated U251 and HS683 did not alter the proportion of maturing DCs; however, exposing DCs to the soluble factors in medium from STAT3 siRNA-treated U251 and HS683 cells significantly increased the number of maturing DCs. This increase in DC maturation in response to STAT3 inhibition suggests that the changes in cytokine and chemokines expression are physiologically significant.
While the concept of immunotherapy for cancers is very attractive, current vaccine strategies have yielded marginal benefits [31–33]. Past studies suggested that we can effectively train the immune system to recognize specific peptides, but the immune microenvironment of tumors can inactivate tumor-specific immune cells [34–36]. To neutralize the anti-tumor immune response, cancer cells have replicated the body’s physiologic mechanisms of controlling autoimmunity [37, 38]. Activated STAT3 has been shown to play an important role in mediating the immune microenvironment in other solid tumors and our results support previous studies which have suggested that STAT3 may play an important role in the immune microenvironment of GBM [39–43].
To investigate clinical application of STAT3 inhibition in glioma, we tested Stattic and NSC 74859, which have been shown to affect tumor growth directly as well as through anti-tumor immune response. Although we did not observe qualitative signs of increased cell death during our experiments, it is possible that cell death signals may have effected some of the observed changes. However, this is via STAT3-mediated pathways and cannot be distinguished from the immune response, since we used concentrations of Stattic and NSC 74859 which have been previously shown to be to only induce apoptosis in STAT3-dependent cell lines [25, 26].
Stattic and NSC 74859 are attractive small molecules to apply to the clinics because they effectively inhibited pSTAT3 activity in vitro. However, the therapeutic application of these two small molecules requires additional data and faces potential barriers. First, the in vivo environment involves multidimensional transport of the cytokines and chemokines as well as additional cell types such as normal astrocytes, neurons, vascular endothelial cells, and immune cell populations. Second, these small molecules act by direct interaction with the STAT3 protein, but the intracranial and intra-GBM distribution of these drugs has not been evaluated.
Our data suggest variable STAT3 expression in clinical GBM specimens. This may be due to intratumoral spatial heterogeneity or interpatient heterogeneity. Although there may be populations of GBM cells which may not directly respond to STAT3 inhibition, this is unlikely to alter the clinical effect, since the paracrine pathways of STAT3 activity mean that STAT3 activation in one cell leads to uniform effects on neighboring cells which do not demonstrate STAT3 activation. However, in clinical application, STAT3 inhibition may need to be selectively applied to patients who are screened for STAT3 upregulation, or may need to be applied in combination with other anti-glioma therapies.
Conclusion
Targeting dysregulated STAT3 signaling is an attractive immunotherapy strategy against GBM. Although blocking pSTAT3 has been shown to increase apoptosis and decrease proliferation in tumor cells, this data suggests that it can also increase the anti-tumor immune response against human GBM cells. Stattic and NSC 74859 set the basis for promising future immunotherapies for GBM.
References
Prasad G, Wang H, Hill DL, Zhang R (2004) Recent advances in experimental molecular therapeutics for malignant gliomas. Curr Med Chem Anticancer Agents 4:347–361
Zagzag D, Salnikow K, Chiriboga L, Yee H, Lan L, Ali MA, Garcia R, Demaria S, Newcomb EW (2005) Downregulation of major histocompatibility complex antigens in invading glioma cells: stealth invasion of the brain. Lab Invest 85:328–341
Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, Cachola KE, Murray JC, Tihan T, Jensen MC, Mischel PS, Stokoe D, Pieper RO (2007) Loss of tumor suppressor PTEN function increases B7–H1 expression and immunoresistance in glioma. Nat Med 13:84–88
Akasaki Y, Liu G, Chung NHC, Ehtesham M, Black KL, Yu JS (2004) Induction of a CD4(+) T regulatory type 1 response by cyclooxygenase-2-overexpressing glioma. J Immunol 173:4352–4359
Rahaman SO, Harbor PC, Chernova O, Barnett GH, Vogelbaum MA, Haque SJ (2002) Inhibition of constitutively active Stat3 suppresses proliferation and induces apoptosis in glioblastoma multiforme cells. Oncogene 21:8404–8413
Kortylewski M, Jove R, Yu H (2005) Targeting STAT3 affects melanoma on multiple fronts. Cancer Metastasis Rev 24:315–327
Ma X-T, Wang S, Ye Y-J, Du R-Y, Cui Z-R, Somsouk M (2004) Constitutive activation of Stat3 signaling pathway in human colorectal carcinoma. World J Gastroenterol 10:1569–1573
Yu H, Pardoll D, Jove R (2009) STATs in cancer inflammation and immunity: a leading role for STAT3. Nat Rev Cancer 9:798–809
Wang TH, Niu GL, Kortylewski M, Burdelya L, Shain K, Zhang SM, Bhattacharya R, Gabrilovich D, Heller R, Coppola D, Dalton W, Jove R, Pardoll D, Yu H (2004) Regulation of the innate and adaptive immune responses by Stat-3 signaling in tumor cells. Nat Med 10:48–54
Wang J, Wang X, Hussain S, Zheng Y, Sanjabi S, Ouaaz F, Beg A (2007) Distinct roles of different NF-kappa B subunits in regulating inflammatory and T cell stimulatory gene expression in dendritic cells. J Immunol 178:6777–6788
Yu H, Kortylewski M, Pardoll D (2007) Crosstalk between cancer and immune cells: role of STAT3 in the tumour microenvironment. Nat Rev Immunol 7:41–51
Burdelya L, Kujawski M, Niu GL, Zhong B, Wang TH, Zhang SM, Kortylewski A, Shain K, Kay H, Djeu J, Dalton W, Pardoll D, Wei S, Yu H (2005) Stat3 activity in melanoma cells affects migration of immune effector cells and nitric oxide-mediated antitumor effects. J Immunol 174:3925–3931
Hoentjen F, Sartor RB, Ozaki M, Jobin C (2005) STAT3 regulates NF-kappa B recruitment to the IL-12p40 promoter in dendritic cells. Blood 105:689–696
Herbeuval JP, Lelievre E, Lambert C, Dy M, Genin C (2004) Recruitment of STAT3 for production of IL-10 by colon carcinoma cells induced by macrophage-derived IL-6. J Immunol 172:4630–4636
Zhong Z, Wen ZL, Darnell JE (1994) STAT3—a STAT family member activated by tyrosine phosphorylation in response to epidermal growth-factor and interleukin-6. Science 264:95–98
Ogura H, Murakami M, Okuyama Y, Tsuruoka M, Kitabayashi C, Kanamoto M, Nishihara M, Iwakura Y, Hirano T (2008) Interleukin-17 promotes autoimmunity by triggering a positive-feedback loop via interleukin-6 induction. Immunity 29:628–636
Takeda K, Clausen BE, Kaisho T, Tsujimura T, Terada N, Forster I, Akira S (1999) Enhanced Th1 activity and development of chronic enterocolitis in mice devoid of Stat3 in macrophages and neutrophils. Immunity 10:39–49
Kinjyo I, Inoue H, Hamano S, Fukuyama S, Yoshimura T, Koga K, Takaki H, Himeno K, Takaesu G, Kobayashi T, Yoshimura A (2006) Loss of SOCS3 in T helper cells resulted in reduced immune responses and hyperproduction of interleukin 10 and transforming growth factor-beta 1. J Exp Med 203:1021–1031
Matsumura Y, Kobayashi T, Ichiyama K, Yoshida R, Hashimoto M, Takimoto T, Tanaka K, Chinen T, Shichita T, Wyss-Coray T, Sato K, Yoshimura A (2007) Selective expansion of Foxp3-positive regulatory T cells and immunosuppression by suppressors of cytokine signaling 3-deficient dendritic cells. J Immunol 179:2170–2179
Zou WP (2006) Regulatory T cells, tumour immunity and immunotherapy. Nat Rev Immunol 6:295–307
Mizoguchi M, Betensky RA, Batchelor TT, Bernay DC, Louis DN, Nutt CL (2006) Activation of STAT3, MAPK, and AKT in malignant astrocytic gliomas: correlation with EGFR status, tumor grade, and survival. J Neuropathol Exp Neurol 65:1181–1188
Hussain SF, Kong L-Y, Jordan J, Conrad C, Madden T, Fokt I, Priebe W, Heimberger AB (2007) A novel small molecule inhibitor of signal transducers and activators of transcription 3 reverses immune tolerance in malignant glioma patients. Cancer Res 67:9630–9636
Fujita M, Zhu X, Sasaki K, Ueda R, Low KL, Pollack IF, Okada H (2008) Inhibition of STAT3 promotes the efficacy of adoptive transfer therapy using type-1 CTLs by modulation of the immunological microenvironment in a murine intracranial glioma. J Immunol 180:2089–2098
Kostianovsky AM, Maier LM, Anderson RC, Bruce JN, Anderson DE (2008) Astrocytic regulation of human monocytic/microglial activation. J Immunol 181:5425–5432
Siddiquee K, Zhang S, Guida WC, Blaskovich MA, Greedy B, Lawrence HR, Yip MLR, Jove R, McLaughlin MM, Lawrence NJ, Sebti SM, Turkson J (2007) Selective chemical probe inhibitor of Stat3, identified through structure-based virtual screening, induces antitumor activity. Proc Natl Acad Sci USA 104:7391–7396
Schust J, Sperl B, Hollis A, Mayer TU, Berg T (2006) Stattic: a small-molecule inhibitor of STAT3 activation and dimerization. Chem Biol 13:1235–1242
Yu CL, Meyer DJ, Campbell GS, Larner AC, Cartersu C, Schwartz J, Jove R (1995) Enhanced DNA-binding activity of a Stat3-related protein in cells transformed by the Src oncoprotein. Science 269:81–83
Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2(T)(-Delta Delta C) method. Methods 25:402–408
Liu J, Xu X, Feng X, Zhang B, Wang J (2011) Adenovirus-mediated delivery of bFGF small interfering RNA reduces STAT3 phosphorylation and induces the depolarization of mitochondria and apoptosis in glioma cells U251. J Exp Clin Cancer Res 30:80
Albesiano E, Davis M, See AP, Han JE, Lim M, Pardoll DM, Kim Y (2010) Immunologic consequences of signal transducers and activators of transcription 3 activation in human squamous cell carcinoma. Cancer Res 70:6467–6476
Liau LM, Prins RM, Kiertscher SM, Odesa SK, Kremen TJ, Giovannone AJ, Lin JW, Chute DJ, Mischel PS, Cloughesy TF, Roth MD (2005) Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T-cell responses modulated by the local central nervous system tumor microenvironment. Clin Cancer Res 11:5515–5525
Yamanaka R, Homma J, Yajima N, Tsuchiya N, Sano M, Kobayashi T, Yoshida S, Abe T, Narita M, Takahashi M, Tanaka R (2005) Clinical evaluation of dendritic cell vaccination for patients with recurrent glioma: results of a clinical phase I/II trial. Clin Cancer Res 11:4160–4167
De Vleeschouwer S, Fieuws S, Rutkowski S, Van Calenbergh F, Van Loon J, Goffin J, Sciot R, Wilms G, Demaerel P, Warmuth-Metz M, Soerensen N, Wolff JEA, Wagner S, Kaempgen E, Van Gool SW (2008) Postoperative adjuvant dendritic cell-based immunotherapy in patients with relapsed glioblastoma multiforme. Clin Cancer Res 14:3098–3104
Wheeler CJ, Black KL, Liu G, Mazer M, Mazer M, Zhang X-X, Pepkowitz S, Goldfinger D, Ng H, Irvin D, Yu JS (2008) Vaccination elicits correlated immune and clinical responses in glioblastoma multiforme patients. Cancer Res 68:5955–5964
Yu JS, Liu GT, Ying H, Yong WH, Black KL, Wheeler CJ (2004) Vaccination with tumor lysate-pulsed dendritic cells elicits antigen-specific, cytotoxic T-cells in patients with malignant glioma. Cancer Res 64:4973–4979
Karman J, Ling CY, Sandor M, Fabry Z (2004) Initiation of immune responses in brain is promoted by local dendritic cells. J Immunol 173:2353–2361
Jacobs JFM, Idema AJ, Bol KF, Grotenhuis JA, de Vries IJM, Wesseling P, Adema GJ (2010) Prognostic significance and mechanism of Treg infiltration in human brain tumors. J Neuroimmunol 225:195–199
Jacobs JFM, Idema AJ, Bol KF, Nierkens S, Grauer OM, Wesseling P, Grotenhuis JA, Hoogerbrugge PM, de Vries IJM, Adema GJ (2009) Regulatory T cells and the PD-L1/PD-1 pathway mediate immune suppression in malignant human brain tumors. Neuro Oncol 11:394–402
Garcia R, Yu CL, Hudnall A, Catlett R, Nelson KL, Smithgall T, Fujita DJ, Ethier SP, Jove R (1997) Constitutive activation of Stat3 in fibroblasts transformed by diverse oncoproteins and in breast carcinoma cells. Cell Growth Differ 8:1267–1276
Sato T, Neilson LM, Peck AR, Liu C, Tran TH, Witkiewicz A, Hyslop T, Nevalainen MT, Sauter G, Rui H (2011) Signal transducer and activator of transcription-3 and breast cancer prognosis. Am J Cancer Res 1:347–355
Fernandes A, Hamburger AW, Gerwin BI (1999) Erbb-2 kinase is required for constitutive STAT 3 activation in malignant human lung epithelial cells. Int J Cancer 83:564–570
Grandis J, Drenning S, Chakraborty A, Zhou M, Zeng Q, Pitt A, Tweardy D (1998) Requirement of Stat3 but not Stat1 activation for epidermal growth factor receptor- mediated cell growth in vitro. J Clin Invest 102:1385–1392
Watson CJ, Miller WR (1995) Elevated levels of members of the stat family of transcription factors in breast-carcinoma nuclear extracts. Br J Cancer 71:840–844
Acknowledgments
This research was partially funded by The Johns Hopkins School of Medicine Dean’s Year of Research Program, The W. W. Smith Charitable Trust Medical Research Grant, the Association for Academic Surgery Joel J. Roslyn Faculty Research Award, and the Neurosurgery Research and Education Foundation (NREF). The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. No writing assistance was utilized in the production of this manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Alfred P. See and James E. Han contributed equally to this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
See, A.P., Han, J.E., Phallen, J. et al. The role of STAT3 activation in modulating the immune microenvironment of GBM. J Neurooncol 110, 359–368 (2012). https://doi.org/10.1007/s11060-012-0981-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11060-012-0981-6