
Abstract
Diversity arrays technology (DArT) and simple sequence repeat (SSR) markers were applied to investigate population structure, extent of linkage disequilibrium and genetic diversity (kinship) on a genome-wide level in European barley (Hordeum vulgare L.) cultivars. A set of 183 varieties could be clearly distinguished into spring and winter types and was classified into five subgroups based on 253 DArT or 22 SSR markers. Despite the fact, that the same number of groups was revealed by both marker types, it could be shown that this grouping was more distinct for the SSRs than the DArTs, when assigned to a Q-matrix by STRUCTURE. This was supported by the findings from principal coordinate analysis, where the SSRs showed a better resolution according to seasonal habit and row number than the DArTs. A considerable influence on the rate of significant associations with malting and kernel quality parameters was revealed by different marker types in this genome-wide association study using general and mixed linear models considering population structure. Fewer spurious associations were observed when population structure was based on SSR rather than on DArT markers. We therefore conclude that it is advisable to use independent marker datasets for calculating population structure and for performing the association analysis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Genome-wide association studies (GWAS) are a novel tool in crop genetics for identifying significant marker-trait correlations. In contrast to conventional bi-parental segregation-based mapping, which exploits allelic differences between two parental lines only, whole-genome association scans use the complete genetic variation across a wide spectrum of germplasm. This implies that many traits will vary in a GWAS, and can thus be addressed, whereas in a bi-parental population only those traits that vary between the parents can be mapped. Other advantages are the finer mapping resolution compared to classical mapping in bi-parental populations (Remington et al. 2001) and the direct use of existing genetic variation in diverse genotype collections instead of the need to create bi-parental crosses and time-consuming development of segregating populations. However, the statistical tools required to perform the analysis are more complex (Falush et al. 2003), since false-positive or false-negative associations between a marker and a trait can occur due to population structure. Such structure can be caused by artificial or natural selection, genetic drift or the species-dependent mating system (Flint-Garcia et al. 2003). In crops, domestication and breeding processes also add to the population structure. In cultivated barley, the main population structure is based on the distinction between spring and winter gene pool. Whole-genome association scans were performed earlier on barley. These studies used diversity arrays technology (DArT; Pswarayi et al. 2008; Zhang et al. 2009; Comadran et al. 2009) or single nucleotide polymorphism (SNP) markers genotyped by the Illumina method (Comadran et al. 2011). In association mapping, the probability of getting type I and type II errors is higher compared to bi-parental quantitative trait locus (QTL) analysis. Type I error, or false positives, can arise from unaccounted subdivisions in the sample as a result of population structure (Pritchard et al. 2000). If the presence of related subgroups in the sample set is not included explicitly in the model, they could create covariances among individuals and generate bias in the estimates of allele effects (Kennedy et al. 1992). An increased type II error rate, or reduced power in association analysis, has at least three causes: (1) lower correlation between markers and genes due to the decay of linkage disequilibrium leading to an underestimation of true associations; (2) unbalanced design derived from the presence of alleles at different frequencies; and (3) multiple-testing problems. Therefore, the association mapping approach has a limited application for detection of rare variants or alleles that are variable between populations but almost fixed within subpopulations. Yu and Buckler (2006) proposed to use a set of random markers to estimate population structure (Q), which is incorporated in the general linear model (GLM) in order to reveal significant associations. Considering population structure and kinship allows improved control of both type I and type II error rates, as described by Yu et al. (2006). Another issue that creates type I errors is the fact that in multiple tests false positives will appear by chance; Benjamini and Hochberg (1995) proposed ways of correcting for this effect, where the gain in statistical power is more substantial compared to the Bonferroni–Holm procedure (Holm 1979).
The number and marker type used for investigating population structure has an effect on the rate of significant associations that can be identified. This was shown in simulation studies for one human candidate gene locus by Pritchard and Rosenberg (1999), and recently by van Inghelandt et al. (2010) for the outcrossing species maize. In this regard, nothing is known so far for barley. In order to find the true associations, the false discovery rate was revealed here by incorporating the different matrices of population structure (Q-matrices) that were generated on the basis of the two different random marker types, namely simple sequence repeats (SSRs), and DArTs and comparing their impact on the identification of marker–trait associations (MTA) in barley breeding material.
In contrast to the detection of SNPs in candidate genes, the DArT can detect and type DNA variation at several hundred genomic loci in parallel without the need of sequence information (Wenzl et al. 2004, 2006). The polymorphisms detected in the DArT analysis include SNPs, insertions-deletions (InDels) and heritable methylation changes (Jaccoud et al. 2001). While the DArT provided by Triticarte Pty. Ltd. (Canberra, Australia) is a biallelic marker system and enables whole-genome profiling without sequence information, SSRs comprise a codominant multiallelic marker system. Both marker systems are mainly based on genomic sequences.
The aims of this study were (1) to determine population structure patterns and kinship in the set of 183 European barley cultivars with two random marker types (DArT and SSR), and (2) to compare the influence of the resulting population structure and kinship on the rate of significant associations with linear models.
Materials and methods
Germplasm selection and phenotypic data
In total, 183 European cultivars released for commercial use in the period from 1985 to 2007 were studied. Seeds were obtained directly from the breeders, or from the gene bank of the IPK in Gatersleben. The set of barley cultivars investigated here consisted of 92 two-row spring, and 91 (59 two-rowed and 32 six-rowed) winter types, mostly of German origin. Phenotypic data are accessible in the MetaBrew database (Weise et al. 2009). The data on the four kernel and malting traits considered here for association studies have already been described by Matthies et al. (2009a, b).
Marker analysis and estimation of intra-chromosomal linkage disequilibrium
Genomic DNA was extracted from bulked young leaves of six plantlets according to a modified protocol of Plaschke et al. (1995). For DArT analysis, diluted DNA samples were sent to Triticarte Pty Ltd. (Canberra, Australia; http://www.triticarte.com.au), a whole-genome profiling service laboratory using the Barley PstI (BstNI) vers. 2.0 array which comprises 2,304 clones known to be polymorphic in a wide range of barley cultivars (Wenzl et al. 2004, 2007). The reproducibility of the genotyping was verified by analysing some cultivars in duplicate or in triplicate. In total, 1,915 DArT markers were investigated of which 1,088 were mapped. The patterns of 22 SSRs resulting in 23 loci (Varshney et al. 2007) were analysed by PCR-amplification detection with ALF-express (Automated Laser Fluorescent Sequencer from GE Healthcare, formerly Amersham-Pharmacia, Sweden) according to Malysheva-Otto et al. (2006). All marker data were managed in an in-house database.
The polymorphism information content (PIC) values were calculated for each DArT and SSR marker set using the formula \( {\text{PIC}} = 1 - \sum \left( {P_{i} } \right)^{2} \), where P i is the proportion of the population carrying the ith allele (Botstein et al. 1980; Smith et al. 2000), with the software PowerMarker vers. 3.25 by Liu and Muse (2005).
To estimate the quality of the marker data, the data resolution (DR) values of the DArT and SSR datasets were calculated according to the method described by van Hintum (2007). The Jaccard distance (Jaccard 1908; Sneath 1957) was used for the binary DArT datasets and the Nei–Li distance (Nei and Li 1979) for the multi-allelic SSR dataset.
Furthermore, principal coordinates (PCoA) were calculated from each marker set with TASSEL vers. 2.1 (Bradbury et al. 2007) by applying the covariance matrix and Manhattan’s distance and plotted for all 183 cultivars.
Genome-wide intra-chromosomal linkage disequilibrium (LD) amongst all accessions was studied by using all mapped DArTs after removing 5% minor alleles. LD was determined by the estimation of squared allele frequency distributions (r 2) among all loci according to Hill and Robertson (1968) by the software TASSEL vers. 2.1. Statistical significance (p value) of the observed LD was estimated by Monte-Carlo approximation of Fisher’s exact test (Weir 1996), with 1,000 permutations for unlinked loci and for loci on the same chromosome (unlinked r 2 and linked r 2), respectively. When plotting the linked r 2 against map distance over all chromosomes, the second-degree LOESS curve (Cleveland 1979) was drawn using the statistical program SPSS vers. 16. The critical r 2 value, as an evidence of linkage, was derived from the distribution of the unlinked r 2. A square root transformation was performed with all unlinked r 2 estimates to approximate a normally distributed random variable. The population-specific critical value of r 2, beyond which LD was likely to be caused by genetic linkage, was derived from the parametric 95th percentile of this distribution. Map positions of DArT loci (Wenzl et al. 2006) were used to calculate averages of intra-chromosomal LD.
Population structure, kinship and association analysis
To reveal spurious associations, the genetic structure among all 183 cultivars was investigated either with 22 SSR markers or a representative set of 253 DArT markers (Electronic Supplementary Material Table 1). This subset was selected from the total set of 1,088 mapped markers on the basis of their distance, one marker approximately every 5 cM. All 862 DarTs, after considering 5% minor allele frequency (MAF), were randomly distributed across the genome. Only chromosome 4H showed less marker coverage. The population structure was determined with the STRUCTURE software vers. 2.2 (Pritchard et al. 2000; Falush et al. 2003), using the admixture option with uncorrelated allele frequencies. This model-based procedure probabilistically assigns accessions to an assumed number (k) of different subgroups. In order to ensure consistent estimates, STRUCTURE was run with five iterations independently, with k ranging from 1 to 20 in each run, setting a burn-in period of 100,000 and burn-in length of 100,000 Markov Chain Monte Carlo iterations. For each value of k, STRUCTURE produces a Q-matrix that lists the estimated membership coefficients for each accession in each subgroup.
To decide on the appropriate number of clusters (k), the estimated normal logarithm of the probability of fit, provided in the STRUCTURE output, was plotted against k (Electronic Supplementary Material Fig. 1). This value reaches a plateau when the number of groups that best describe the population substructure has been achieved (Pritchard et al. 2000). The mean of the five iterations of each k showing the maximum likelihood was applied to assign all genotypes with a membership probability to a certain subgroup surpassing the threshold p < 0.50 for each of the two marker types. The assignment to groups that resulted from STRUCTURE was studied for each marker type by comparing the consistency of the assignments over runs, and by calculating the frequencies of accessions according to their seasonality (spring and winter) and row number (2r and 6r) according to their affiliation to different Q-groups (Electronic Supplementary Material Table 2).
Based on either 22 SSRs or 253 DarTs, the kinship (K-) matrix was determined with SPAGeDi (Hardy and Vekemans 2002) using the coefficient of Ritland (1996). Negative values between individuals were set to 0, which indicates their lower relation compared to random individuals. This K-matrix was used in the mixed linear model (MLM) to define the degree of covariance between pairs of individuals. Four different MLMs were calculated using the Q-matrices from STRUCTURE and the kinship matrix derived from SPAGeDi created by the two marker types:
Once every accession was assigned to one of the groups for both marker types, the association analysis was performed. The general and mixed linear model (GLM, MLM, Searle 1987) was applied in order to reveal significant associations using the TASSEL software vers. 2.1. Population structure is incorporated into the model by using covariates that indicate the relative contribution in each genotype. If population structure percentages sum to 100% for each genotype, one of the populations should be excluded from the analysis in order to obtain valid F-tests of the population covariates. Four kernel and malting traits were selected for the GWAS: thousand-grain weight (TGW), glume fineness, extract and friability. Genotyping of the 183 cultivars was performed using all 1,088 mapped DArT markers. After removal of those markers with an allele frequency below 5%, 862 DArTs were considered in the GWAS. All terms in the model are considered to be fixed. Multiple testing corrections were performed by applying the Bonferroni–Holm procedure (Holm 1979).
Results and discussion
Genotyping of the barley varieties
In this barley collection, the population was examined with SSRs and DArTs. Multiallelic SSRs are highly polymorphic markers and represent an excellent molecular marker system for population studies. DArT markers have shown to be repeatable high-throughput multi-locus dominant biallelic markers for whole-genome profiling of barley (Wenzl et al. 2004, 2006). In total, 1,915 DArT markers were generated by Triticarte but only the 1,088 mapped ones were used for the GWAS. A subset of 253 equally spaced DArTs was used for the determination of the population structure. The average PIC was 0.53 for the 22 SSRs, 0.28 for the 253 selected DArTs and 0.24 for the 1,088 mapped DArTs, respectively, which is similar to the findings of Zhang et al. (2009) in Canadian barley accessions. Out of these, 862 DArT markers (77.5%) were sufficiently polymorphic in the investigated set of 183 barley cultivars and were used for GWAS. The remaining 22.5% were either monomorphic or possessed a MAF of <5% and were therefore excluded from further analysis (Electronic Supplementary Material Table 1).
Data resolution of DArT and SSR markers
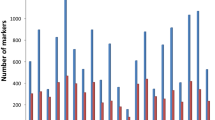
The data resolution (DR) is an indicator of the extent to which the markers are able to describe genetic structure. The DR of the 1,088 DArT markers using the Jaccard distance was 0.938, the reduced set of 253 markers had a DR of 0.832, implying that if the dataset is randomly split in half, and the pairwise similarities of the accessions are calculated on the basis of each half, these similarities would have an average correlation of 0.938 and 0.832, respectively (van Hintum 2007). The complete set of 22 SSR markers had a DR of 0.421 using the Nei–Li distance. This implies that a DR of 0.5 could be reached with 72 DArT markers of the complete set, 51 DArT markers of the reduced set or 32 SSR markers, respectively (Fig. 1).

Data resolution (DR) curves of the three data sets: the 22 SSR markers, the subset of 253 DArT markers selected every 5 cM, and all 1,088 mapped DArT markers. The DR of all 1,088 markers in this set is 0.938
These results would imply that the population structure as described by the 253 DArT markers is more stable, and more repeatable, than that characterized by SSR markers. However, the structures described by the different marker types are not necessarily the same, as they are the result of different population genetic effects. The degree of polymorphism as measured from highly multiallelic SSRs provides more allelic information than that from biallelic DArTs. As a result, SSR markers serve as a better estimator for the population structure.
The advantage of the cost-effective DArT markers is their ideal suitability for high-throughput genome-wide association analysis (Zhang et al. 2009). On the other hand, they suffer to a certain degree of clustering (Wenzl et al. 2006).
Population structure and linkage disequilibrium in European barley
It could be shown that the barley population investigated is highly structured, mainly according to seasonal habit but to a lesser extent according to row number. Either 22 genomic SSRs, representing all seven chromosomes and yielding in total 206 alleles, or a subset of 253 selected random DArT markers representing 506 alleles was applied. The total population of 183 European cultivars could be clearly distinguished into the two main groups according to seasonal habit and further divided into five subgroups by STRUCTURE analysis with both marker types (Fig. 2). Two subgroups were found for the 92 two-rowed spring cultivars, and three for the 91 winter varieties. When looking at the assignments of all analysed cultivars to the different groups in the Q5-Matrix, clear differences were obtained depending on the marker type investigated. Taking a threshold of <50% of membership probability to a certain subgroup revealed by STRUCTURE, only 12 of all 183 analysed cultivars characterised with SSR could not be assigned clearly to one group, in contrast to 19 cultivars with the DArTs (Table 1). A detailed overview of the grouping of all cultivars studied and their origin is given in Electronic Supplementary Material Table 2. For most association mapping methods, genotypes are not assigned to subgroups, but the matrices from STRUCTURE comprising the membership probabilities are used as cofactors (Yu et al. 2006). Therefore, it is expected that the differences in absolute membership probabilities between SSRs and DArTs must have an influence on the results of association mapping approaches as investigated here in barley. This has been studied for SSRs and SNPs by van Inghelandt et al. (2010) in maize.

Population structure of the total set of 183 European cultivars studied with two marker types (22 SSRs and 253 DArTs) illustrated by bar plots. Subclustering in five subgroups for Q5-SSR (a) and Q5-DArT (b)
Grouping of this set of genotypes with SSRs was more distinct than with DArTs (Table 1 and Fig. 2). The population structure matrix obtained with the SSRs also reflects the assignment of the cultivars according to seasonal habit and row number more clearly than the DArTs and is in accordance with the principal coordinate analysis (PCoA), shown in Fig. 3. Notably, only a few genotypes were assigned to the fourth and fifth group of the Q-matrix with DarTs, in contrast to the SSRs. Furthermore, the frequencies were different due to the grouping algorithm. Most of the spring cultivars were assigned to Q1 with the SSRs but to Q2 with the DArTs. The majority of the two-rowed winter accessions clustered in Q1 when analysed with the DArTs, whereas they split in two subgroups (Q3 and Q5) as revealed by SSRs. All six-rowed winter varieties were mostly assigned to Q2 with the SSR and Q3 with the DArT markers (Table 1). No clear grouping was found for 12 genotypes with the SSR and for 19 genotypes with the DArT markers. From these, two cultivars (‘Baccara’ und ‘Maris-Otter’) could not be affiliated clearly to one of the groups either with SSRs or DArTs (Electronic Supplementary Material Table 2). Due to their multiallelic state, SSR alleles show a more diverse pattern in the investigated germplasm (Fig. 3). With both marker types, a clear differentiation could be obtained into three main clusters according to seasonal habit and row number (2r-spring, 2r-winter, 6r-winter). A higher amount of genetic variation is explained by the first two principal axes with the 253 DArTs (32.1%) compared to 26.4% with the SSRs (Fig. 3).

Principal coordinate analysis (PCoA) of all 183 European barley cultivars characterized by 22 SSRs (a) or 253 genome-wide mapped DArT markers, selected every 5 cM (b). The percentage of variance explained by each axis is given. Different plot symbols and colours are indicate the three subpopulations of 2-rowed spring as well as 2- and 6-rowed winter barley cultivars
Linkage disequilibrium statistics (r 2, p values) were calculated for each pair of intra-chromosomal DArT markers and presented in a heat plot (Electronic Supplementary Material Fig. 2). The extent of intra-chromosomal LD was estimated relative to the LD observed among unlinked markers from different chromosomes and a significance threshold for r 2 of 0.21 was determined. Consistently, a low genome-wide intrachromosomal LD was found which serves as a good prerequisite for performing GWAS. There is no intersection of the LOESS curve fit to the critical r 2 of 0.21 (Electronic Supplementary Material Fig. 2).
The LD decay extends for less than 10–15 cM, and therefore GWAS is possible. This rapid intra-chromosomal LD decay within the first few centimorgans is in accordance with other studies in barley cultivars (Waugh et al. 2009; Rostoks et al. 2006; Comadran et al. 2011) and indicates that most of the markers are not tightly linked to each other. The key to association mapping is the LD between functional loci and physically linked markers. The decay of LD over physical distance in a population determines the density of marker coverage needed to perform an association analysis. For example, if LD decays rapidly, a higher marker density is required to capture markers located close enough to functional sites (Yu and Buckler 2006). In other words, fast LD decay results in a fine resolution of loci. The extent and distribution of LD were visualised by plotting intra-chromosomal r 2 values (significant at p < 0.001) against the genetic distance in centimorgans shown in Electronic Supplementary Material Fig. 3. Unlinked r 2 estimates were square-root transformed to approximate a normally distributed random variable and the parametric 95th percentile of that distribution was taken as a critical value of r 2 (0.21), beyond which LD is probably caused by genetic linkage (Breseghello and Sorrells 2006). All linked pairwise marker estimates of r 2 smaller than 0.21 were probably due to genetic linkage and higher than 0.21 were due to population structure. This is another strong indicator for a highly structured population. The frequency of physically linked pairs versus non-physically linked pairs according to higher genetic distance follows a logarithmic function (Electronic Supplementary Material Fig. 3). Long-range genome-wide LD decay is often caused by population structure and/or epistasis which can be addressed by incorporating population structure or kinship information as cofactors in the model and indicates the amount of putative false positives (Yu et al. 2006; Comadran et al. 2009). The remaining significant LD is caused by genetic linkage and residual population structure effects. The observed extent of LD was strongly affected by population structure. This was also noticed by Rostoks et al. (2006).
Effects of population structure on association results
The effects of population structure employing either 22 multiallelic SSRs or 253 biallelic DArTs on the rate of significant associations was assessed. The information obtained about population structure (Q5-Matrix) from both marker types was incorporated in the GLM in order to elucidate significant associations for important malting and kernel quality parameters.
When performing GWAS including the Q-matrix from structure with TASSEL vers. 2.1, it should be noted that this software does not include those genotypes with a group assignment below 50% in the calculation process (Table 1).
Examples of genome-wide associations with random DArTs are shown here for four kernel and malting quality parameters in barley: glume fineness, TGW, extract and friability. Cultivars with fine glumes and high TGW are preferred for the malting process. Genotypes delivering a high extract and good friability values are desired. The friability parameter describes the effects of germination factors, the modification process during malting and also the homogeneity of the sample. The extract represents all water-soluble substances in the fine coarse meal.
The marker positions are given on the barley integrated map (Wenzl et al. 2006) and 862 mapped DArTs considering 5% MAF were used as genomic marker data in the association analysis by applying the GLM with regard to population structure. The results from the GWAS were compared for each trait considering two different Q-matrices calculated with two different marker systems (Q5-SSR, and Q5-DArT) obtained from STRUCTURE. There is an effect on the rate of significant MTAs when employing either SSR or DArT for analysing population structure (Figs. 4, 5). Considering the cumulative p values, the association model including five subgroups for the total population determined by SSRs results in a lower rate of significant MTAs and seems to be more specific for correcting population structure effects (Fig. 4). However, assuming the same number of subpopulations, a difference could be observed in the number of significant associations depending on the marker type in the GLM (Fig. 5 and Electronic Supplementary Material Table 3). This is also true for all four traits considered here applying the MLM (Electronic Supplementary Material Table 4 and Figs. 4 and 5).

Comparison of association results by applying the GLM, when different marker types were used for revealing population structure: a GLM = Q5_SSR, and b GLM = Q5_DArT. The following traits were considered: glume fineness, thousand-grain weight (TGW), extract and friability. The cumulative distribution of the observed p values is shown

Genome-wide association studies of 183 barley cultivars considering the GLM for four traits: a glume fineness, b thousand-grain weight, c extract, and d friability. Population structure was taken into account by using the Q5 matrix calculated either with SSR or DArT markers. The calculated p values were converted into −log10 p. The significance thresholds p < 0.05 and p < 0.001 are indicated by dashed lines. The location of mapped genes for row number (vrs1, vrs5) is shown
Assessing population structure and kinship using the subset of 253 random DArT markers each leads to more spurious associations, with a higher significance (MLM_1) followed by MLM_3 with the combination of Q5-DArT with K-SSR (Electronic Supplementary Material Fig. 4a, c) for all four traits. This is strikingly obvious for the GWAS regarding malt extract but less clear for TGW (Electronic Supplementary Material Fig. 5a, b). Yu et al. (2006, 2009) also stated a specific impact of the kinship estimation with molecular markers on the model fitting for different quantitative traits. In particular, TGW in barley is structure-dependent, since the grouping is predominantly determined by seasonal habit and row type. These parameters are highly correlated with this trait. Linear models accounting for relatedness (K) have a better fit even when a small number of background markers were used in estimating kinship (Yu et al. 2009), which can also be confirmed here for barley. Furthermore, the Q also provides more explanation. The robustness of population structure estimates from random background markers has been studied previously (Pritchard et al. 2000) and validated (Camus-Kulandaivelu et al. 2007). The robustness of kinship estimates with varied numbers of background markers provides further insight into the application of the mixed-model approach in the context of association mapping. There is also a clear marker effect. The SSRs give a better estimation of the kinship than DArTs, resulting in less spurious associations (Electronic Supplementary Material Fig. 5).
Even though a lower number of markers are applied (22 SSRs compared to 253 DArTs), it can be concluded that the population structure based on 206 SSR alleles results in a better differentiation than the 506 DArT alleles. This is also supported by the findings according to DR. Fewer SSR markers than DArTs are needed to obtain the same DR (Fig. 1). Another possible explanation may be that the SSR data used for population structure represent an independent marker set, while DArTs are also used for defining the genotype in the association algorithm. When tracing the population structure with DArTs instead of SSRs, this biallelic marker type was used for both, as genotype and for population structure. To our knowledge, this effect has not been investigated so far. We assume that the biallelic state of DArT markers provides less information than multiallelic SSRs. Microsatellites are also “older” in an evolutionary context, being mostly located in untranscribed regions. Accordingly, the SSRs were probably less exposed to genetic selection pressure than the DArTs. These markers were generated mostly from expressed sequence tags based on microarray hybridisations (Wenzl et al. 2004). SSR analysis provided a higher resolution and allowed a better discrimination between genotypes (Russell et al. 2000, 2004).
Approaches which appropriately control type I errors should approximate to a uniform distribution of the p values. This is the case when the GLM with a population structure of five subgroups derived from SSR data was applied (Fig. 4a) in contrast to Q5_DArTs which resulted in more spurious associations and false positives (Fig. 4b). SSRs are less conserved and are more informative when used for population structure than DArTs. Notably, this effect is not so clear for the quantitative yield component TGW (Figs. 4, 5b). The significance of DArT markers when using different marker systems for determination of population structure after association performed with all 183 cultivars assuming GLM for four traits is depicted in Fig. 5. No coincidences of highly significant MTAs for all four traits with known genes coding for row number in barley (vrs1, vrs5) were observed. Their mapping positions (Wenzl et al. 2006; Pourkheirandish and Komatsuda 2007; Ayoub et al. 2002) are indicated on the whole-genome scan (Fig. 5).
Except for TGW, far more significant MTAs were found for glume fineness, extract and friability with the GLM when considering the population structure matrix derived with the DArTs. In contrast, the lower rate of MTAs for these three traits when taking the Q5-SSR into account seems to be more specific. Association mapping is a method for detection of gene effects based on LD that complements QTL analysis in the development of tools for molecular plant breeding. Significantly associated genomic regions were linked to known QTLs, available from the website http://www.graingenes.org and summarised in Table 2, for the yield component TGW and for the malting quality parameter extract in Table 3. No known reference QTLs colocalising with significant MTAs for TGW were found on chromosome 6H and for extract on 3H and 7H. Eight QTL regions for TGW did coincide with significant MTAs (Table 2). Schmalenbach et al. (2009) identified six QTLs in a backcross study with introgressions lines of H. spontaneum on 2H, 4H, and 6H for this trait. Beattie et al. (2010) found also a highly significant relationship for bPb-0351 with malt extract in their GWAS with DArTs in malting barley (Table 3). Such comparisons were not feasible for glume fineness and friability.
Differences in the rate and chromosomal position of significant hot spots could be shown for each trait. Disregarding TGW, many highly significant associations with a negative log10 p >3.0 were found for the GLM with Q5-DArT, which are not reflected in the GLM with Q5-SSR (Fig. 5). Therefore, we conclude that the use of an independent dataset for assessing population structure is preferable for determining reliable associations compared to using the same data for determining population structure and associations. Nevertheless, the choice of the appropriate number of subgroups also has an impact on the quality of the association result. It is always recommended to compare different association models in order to sort out the false positives. The following two factors were important to consider in order to obtain reliable association results and to avoid false positives or false negatives: correction for population structure is necessary in this type of analysis, especially in structured populations such as the set of barley cultivars investigated here. Furthermore, the number and kind of markers used to determine population structure for the association study has an influence on the result. We found here that even a higher number of DArTs is less meaningful and leads to more spurious significant association results than a lower number of SSRs. A comparison of multiallelic SSRs and biallelic SNPs was recently performed by van Inghelandt et al. (2010), in which they suggest the use of between seven and eleven times as many SNPs than SSRs for analysing population structure and genetic diversity in maize breeding material. To our knowledge, no comparative studies investigating the accuracy and discrimination power of random markers such as SSRs and DArTs in barley have been undertaken to date. The same or even a higher number of SSRs for revealing population structure was used in barley (Stracke et al. 2009; Haseneyer et al. 2009) or in maize (van Inghelandt et al. 2010). Up to now, there have been no specific studies available in barley adressing the optimal number and type of markers which should be used for determination of population structure. The population type and size might have also an impact on the structure of the estimated set of genotypes. The effect of population structure on the association results depends in particular on the number of ancestral groups and on the trait analysed (Mezmouk et al. 2011). We can observe a clear marker effect, when we assume the same number of groups revealed by different kind of markers.
This is the first study to reveal the impact of the marker type on the association result. Hamblin et al. (2007) compared the usefulness of 89 SSR and 847 SNP markers in maize and found a better performance of even a lower number of SSRs when assessing relatedness and genetic diversity. Similar findings are obtained here in terms of population structure, where SSRs led to less false-positive results than DArTs.
In this study, we were able to demonstrate the impact of marker type used in STRUCTURE and the resulting Q-matrix on the number of significant associations when considering the GLM. This same phenomenon was also evident when applying the kinship with different kind of markers in the MLM considering Q and K.
Conclusions and outlook
The marker type (SSR or DArT) used for analysing population structure and kinship has a strong influence on the number and significance of the GWAS detected when applying either the GLM or MLM. It could be shown that our barley population was highly structured, mainly by seasonal habit, and the marker system used has a strong effect on the association results. Multiallelic markers such as SSRs present a more effective tool for assessing accurately the population structure than biallelic DArT markers.
We propose that independent marker sets should be used to assess population structure and to reveal significant marker–trait associations in GWAS. Furthermore, correction for population structure in a set of barley accessions is needed in order to avoid false positives. These are important prerequisites in order to perform meaningful association studies and to provide breeders with well-defined marker–trait associations, which is fundamental for marker-assisted selection in barley breeding for traits such as enhanced kernel and malting quality.
References
Ayoub M, Symons SJ, Edney MJ, Mather DE (2002) QTLs affecting kernel size and shape in a two-rowed by six-rowed barley cross. Theor Appl Genet 105:237–247
Barr AR, Jeffries SP, Broughton S, Chalmers KJ, Kretschmer JM, Boyd WJR, Collins HM, Roumeliotis S, Logue SJ, Coventry SJ, Moody DB, Read BJ, Poulsen D, Lance RCM, Platz GJ, Park RF, Panozzo JF, Karakousis A, Lim P, Verbyla AP, Eckermann PJ (2003a) Mapping and QTL analysis of the barley population Alexis × Sloop. Aust J Agric Res 54:1117–1123
Barr AR, Karakousis A, Lance RCM, Logue SJ, Manning S, Chalmers KJ, Kretschmer JM, Boyd WJR, Collins HM, Roumeliotis S, Coventry SJ, Moody DB, Read BJ, Poulsen D, Li CD, Platz GJ, Inkerman PA, Panozzo JF, Cullis BR, Smith AB, Lim P, Langridge P (2003b) Mapping and QTL analysis of the barley population Chebec × Harrington. Aust J Agric Res 54:1125–1130
Beattie AD, Edney MJ, Scoles GJ, Rossnagel BG (2010) Association mapping of malting quality data from Western Canadian two-row barley cooperative trials. Crop Sci 50:1649–1663
Benjamini Y, Hochberg Y (1995) Controlling the false discovery rate: a practical and powerful approach to multiple testing. J R Stat Soc B 57:289–300
Bezant J, Laurie D, Pratchett N, Chojecki J, Kearsey M (1997) Mapping QTL controlling yield and yield components in a spring barley (Hordeum vulgare L.) cross using marker regression. Mol Breed 3:29–38
Botstein D, White RL, Skolnick M, Davis RW (1980) Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am J Hum Genet 32:314–331
Bradbury PJ, Zhang Z, Kroon DE, Casstevens RM, Ramdoss Y, Buckler ES (2007) TASSEL software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635
Breseghello F, Sorrells ME (2006) Association mapping of kernel size and milling quality in wheat (Triticum aestivum L.) cultivars. Genetics 172:1165–1177
Camus-Kulandaivelu L, Veyrieras J, Gouesnard B, Charcosset A, Manicacci D (2007) Evaluating the reliability of structure outputs case of relatedness between individuals. Crop Sci 47:887–890
Cleveland WS (1979) Robust locally weighted regression and smoothing scatterplots. J Am Stat Assoc 74:829–836
Comadran J, Thomas WTB, van Eeuwijk FA, Ceccarelli S, Grando S, Stanca AM, Pecchioni N, Akar T, Al-Yassin A, Benbelkacem A, Ouabbou H, Bort J, Romagosa I, Hackett CA, Russell JR (2009) Patterns of genetic diversity and linkage disequilibrium in a highly structured Hordeum vulgare association-mapping population for the Mediterranean basin. Theor Appl Genet 119:175–187
Comadran J, Ramsay L, MacKenzie K, Hayes P, Close TJ, Muehlbauer G, Stein N, Waugh R (2011) Patterns of polymorphism and linkage disequilibrium in cultivated barley. Theor Appl Genet. doi:10.1007/s00122-010-1466-7
Falush D, Stephens M, Pritchard JK (2003) Inference of population structure: extensions to linked loci and correlated allele frequencies. Genetics 164:1567–1587
Flint-Garcia SA, Thornsberry JM, Buckler ES IV (2003) Structure of linkage disequilibrium in plants. Annu Rev Plant Biol 54:357–374
Hamblin MT, Warburton ML, Buckler ES (2007) Empirical comparison of simple sequence repeats and single nucleotide polymorphisms in assessment of maize diversity and relatedness. PLoS One 2(12):e1367
Han F, Romagosa I, Ullrich SE, Jones BL, Hayes PM, Wesenberg DM (1997a) Molecular marker-assisted selection for malting quality traits in barley. Mol Breed 3:427–437
Han F, Ullrich SE, Kleinhofs A, Jones BL, Hayes PM, Wesenberg DM (1997b) Fine structure mapping of the barley chromosome-1 centromere region containing malting-quality QTLs. Theor Appl Genet 95:903–910
Hardy OJ, Vekemans X (2002) SPAGeDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes 2:618–620
Haseneyer G, Stracke S, Paul C, Einfeldt C, Broda A, Piepho HP, Graner A, Geiger HH (2009) Population structure and phenotypic variation of a spring barley world collection set up for association studies. Plant Breed 129(3):271–279
Hayes PM, Liu BH, Knapp SJ, Chen F, Jones B, Blake T, Franckowiak J, Rasmusson D, Sorrells M, Ulrich SE, Wesenberg D, Kleinhofs A (1993) Quantitative trait locus effects and environmental interaction in a sample of North American barley germplasm. Theor Appl Genet 87:392–401
Herz MP (2000) Kartierung quantitativer vererbter Eigenschaften einschließlich Brauqualität und Resistenz gegen Krankheiten mit molekularen Markern in Gerste. PhD-Thesis. Technische Universität München (TUM), Lehrstuhl für Pflanzenzüchtung
Hill WG, Robertson A (1968) Linkage disequilibrium in finite populations. Theor Appl Genet 38:226–231
Holm S (1979) A simple sequentially rejective multiple test procedure. Scand J Stat 6(2):65–70
Jaccard P (1908) Nouvelles recherches sur la distribution florale. Bull Soc Vaud Sci Nat 44:223–270
Jaccoud D, Peng K, Feinstein D, Kilian A (2001) Diversity arrays: a solid state technology for sequence information independent genotyping. Nucleic Acids Res 29:e25
Kennedy BW, Quinton M, Vanarendonk JAM (1992) Estimation of effects of single genes on quantitative traits. J Anim Sci 70:2000–2012
Kjaer B, Jensen J (1996) Quantitative trait loci for grain yield and yield components in a cross between a six-rowed and a two-rowed barley. Euphytica 90:39–48
Krumnacker K (2009) Untersuchung der funktionellen Assoziation von Kandidatengenen in Zusammenhang mit der Malzqualität der Gerste durch Transkriptomkartierung. PhD-Thesis. Technische Universität München (TUM), Lehrstuhl für Pflanzenzüchtung
Larson SR, Habernicht DK, Blake TK, Adamson M (1997) Backcross gains for six-rowed grain and malt qualities with introgression of a feed barley yield QTL. J Am Soc Brew Chem 55:52–57
Li JZ, Huang XQ, Heinrichs F, Ganal MW, Röder MS (2005) Analysis of QTLs for yield, yield components, and malting quality in a BC3-DH population of spring barley. Theor Appl Genet 110:356–363
Liu K, Muse SV (2005) PowerMarker: integrated analysis environment for genetic marker data. Bioinformatics 21(9):2128–2129
Malysheva-Otto LV, Ganal MW, Röder MS (2006) Analysis of molecular diversity, population structure and linkage disequilibrium in worldwide cultivated barley germplasm (Hordeum vulgare L.). BMC Genet 7:6
Marquez-Cedillo LA, Hayes PM, Jones BL, Kleinhofs A, Legge WG, Rossnagel BG, Sato K, Ullric E, Wesenberg DM (2000) QTL analysis of malting quality in barley based on doubled-haploid progeny of two elite North American varieties representing different germplasm pools. Theor Appl Genet 101:173–184
Marquez-Cedillo LA, Hayes PM, Kleinhofs A, Legge WG, Rossnagel BG, Sato K, Ullrich SE, Wesenberg DM (2001) QTL analysis of agronomic traits in barley based on the doubled haploid progeny of two elite North American varieties representing different germplasm groups. Theor Appl Genet 103:625–637
Mather DE, Tinker NA, LaBerge DE, Edney M, Jones BL, Rossnagel BG, Legge WG, Briggs KG, Irvine RB, Falk DE, Kasha KJ (1997) Regions of the genome that affect grain and malt quality in a North American two-row barley cross. Crop Sci 37:544–554
Matthies IE, Weise S, Röder MS (2009a) Association of haplotype diversity in the α-amylase gene amy1 with malting quality parameters in barley. Mol Breed 23:139–152
Matthies IE, Weise S, Förster J, Röder MS (2009b) Association mapping and marker development of the candidate genes (1→3),(1→4)-β-d-Glucan-4-glucanohydrolase and (1→4)-β-Xylan-endohydrolase 1 for malting quality in barley. Euphytica 170:109–122
Mezmouk S, Dubreuil P, Bosio M, Décousset L, Charcosset A, Praud S, Mangin B (2011) Effect of population structure corrections on the results of association mapping tests in complex maize diversity panels. Theor Appl Genet 122:1149–1160
Nei M, Li WH (1979) Mathematical models for studying genetic variation in terms of restriction endonucleases. Proc Natl Acad Sci USA 76:5269–5273
Oziel A, Hayes PM, Chen FQ, Jones B (1996) Application of quantitative trait locus mapping to the development of winter-habit malting barley. Plant Breed 115:43–51
Plaschke J, Ganal MW, Röder MS (1995) Detection of genetic diversity in closely related bread wheat using microsatellite markers. Theor Appl Genet 91:1001–1007
Pourkheirandish M, Komatsuda T (2007) The importance of barley genetics and domestication in a global perspective. Ann Bot 100:999–1008
Pritchard JK, Rosenberg NA (1999) Use of unlinked genetic markers to detect population stratification in association studies. Am J Hum Genet 65:220–228
Pritchard JK, Stephens M, Donnelly PJ (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Pswarayi A, van Eeuwijk FA, Ceccarelli S, Grando S, Comadran J, Russell JR, Pecchioni N, Tondelli A, Akar T, Al-Yassin A, Benbelkacem A, Ouabbou H, Thomas WTB, Romagosa I (2008) Changes in allele frequencies in landraces, old and modern barley cultivars of marker loci close to QTL for grain yield under high and low input conditions. Euphytica 163:435–447
Remington DL, Thornsberry JM, Matsuoka Y, Wilson LM, Whitt SR, Doebley J, Kresovich S, Goodman MM, Buckler ES (2001) Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc Natl Acad Sci USA 98:11479–11484
Ritland K (1996) Estimators for pairwise relatedness and individual inbreeding coefficients. Genet Res 67:175–186
Rostoks N, Ramsay L, MacKenzie K, Cardle L, Bhat PR, Roose ML, Svensson JT, Stein N, Varshney RK, Marshall DF, Graner A, Close TJ, Waugh R (2006) Recent history of artificial outcrossing facilitates whole-genome association mapping in elite inbred crop varieties. Proc Natl Acad Sci USA 103:18656–18661
Russell JR, Ellis RP, Thomas WTB, Waugh R, Provan J, Booth A, Fuller J, Lawrence P, Young G, Powell W (2000) A retrospective analysis of spring barley germplasm development from ‘foundation genotypes’ to currently successful cultivars. Mol Breed 6:553–568
Russell J, Booth A, Fuller J, Harrower B, Hedley P, Machray G, Powell W (2004) A comparison of sequence-based polymorphism and haplotype content in transcribed and anonymous regions of the barley genome. Genome 47:389–398
Schmalenbach I, Léon J, Pillen K (2009) Identifcation and verification of QTLs for agronomic traits using wild barley introgression lines. Theor Appl Genet 118:483–497
Searle SR (1987) Linear models for unbalanced data. Wiley, New York
Smith JSC, Kresovich S, Hopkins MS, Mitchell SE, Dean RE, Woodman WL, Lee M, Porter K (2000) Genetic diversity among elite Sorghum inbred lines assessed with simple sequence repeats. Crop Sci 40:226–232
Sneath PHA (1957) Some thoughts on bacterial classification. J Gen Microbiol 17:184–200
Stracke S, Haseneyer G, Veyrieras JB, Geiger HH, Sauer S, Graner A, Piepho HP (2009) Association mapping reveals gene action and interactions in the determination of flowering time in barley. Theor Appl Genet 118(2):259–273
Szücs P, Blake VC, Bhat PR, Chao S, Close TJ, Cuesta-Marcos A, Muehlbauer GJ, Ramsay L, Waugh R, Hayes PM (2009) An integrated resource for barley linkage map and malting quality QTL alignment. Plant Genome 2:134–140
Tinker NA, Mather DE, Rossnagel BG, Kasha KJ, Kleinhofs A, Hayes PM, Falk DE, Ferguson T, Shugar LP, Legge WG, Irvine RB, Choo TM, Briggs KG, Ullrich SE, Franckowiak JD, Blake TK, Graf RJ, Dofing SM, Saghai Maroof MA, Scoles GJ, Hoffman D, Dahleen LS, Kilian A, Chen F, Biyashev RM, Kudrna DA, Steffenson BJ (1996) Regions of the genome that affect agronomic performance in two-row barley. Crop Sci 36:1053–1062
Ullrich SE, Han F (1997) Genetic complexity of the malt extract trait in barley suggested by QTL analysis. J Am Soc Brew Chem 55(1):1–4
van Hintum TJL (2007) Data resolution: a jackknife procedure for determining the consistency of molecular marker datasets. Theor Appl Genet 115:343–349
van Inghelandt D, Melchinger AE, Lebreton C, Stich B (2010) Population structure and genetic diversity in a commercial maize breeding program assessed with SSR and SNP markers. Theor Appl Genet 120:1289–1299
Varshney RK, Marcel TC, Ramsay L, Russell J, Röder MS, Stein N, Waugh R, Langridge P, Niks RE, Graner A (2007) A high density barley microsatellite consensus map with 775 SSR loci. Theor Appl Genet 114:1091–1103
Waugh R, Jannink JL, Muehlbauer GL, Ramsay L (2009) The emergence of whole genome association scans in barley. Curr Opin Plant Biol 12:1–5
Weir BS (1996) Genetic data analysis II. Sinauer, Massachusetts
Weise S, Scholz U, Röder MS, Matthies IE (2009) A comprehensive database of malting quality traits in brewing barley. Barley Genet Newsl 39:1–4
Wenzl P, Carling J, Kudrna D, Jaccoud D, Huttner E, Kleinhofs A, Kilian A (2004) Diversity arrays technology (DArT) for whole-genome profiling of barley. Proc Natl Acad Sci USA 101(26):9915–9920
Wenzl P, Li H, Carling J, Zhou M, Raman H, Paul E, Hearnden P, Maier C, Xia L, Caig V, Ovesná J, Cakir M, Poulsen D, Wang J, Raman R, Smith KP, Muehlbauer GJ, Chalmers KJ, Kleinhofs A, Huttner E, Kilian A (2006) A high-density consensus map of barley linking DArT markers to SSR, RFLP and STS loci and agricultural traits. BMC Genomics 7:206
Wenzl P, Raman H, Wang J, Zhou M, Huttner E, Kilian A (2007) A DArT platform for quantitative bulked segregant analysis. BMC Genomics 8:196
Worch S, Kalladan R, Harshavardhan VT, Pietsch C, Korzun V, Kuntze L, Börner A, Wobus U, Röder MS, Sreenivasulu N (2011) Haplotyping, linkage mapping and expression analysis of barley genes regulated by terminal drought stress influencing seed quality. BMC Plant Biol 11:1
Yu J, Buckler ES (2006) Genetic association mapping and genome organization of maize. Curr Opin Biotechnol 17:155–160
Yu J, Pressoir G, Briggs WH, Vroh Bi I, Yamasaki M, Doebley JF, McMullen MD, Gaut BS, Nielsen DM, Holland JB, Kresovich S, Buckler ES (2006) A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat Genet 38:203–208
Yu J, Zhang Z, Zhu C, Tabanao DA, Pressoir G, Tuinstra MR, Kresovich S, Todhunter RJ, Buckler ES (2009) Simulation appraisal of the adequacy of number of background markers for relationship estimation in association mapping. Plant Genome 2(1):63–77
Zhang LY, Marchand S, Tinker NA, Belzile F (2009) Population structure and linkage disequilibrium in barley assessed by DArT markers. Theor Appl Genet 119(1):43–52
Acknowledgments
The authors gratefully acknowledge the excellent technical assistance of Angelika Flieger in performing the SSR studies and Triticarte Pty. Ltd (Canberra, Australia) for the DArT analyses. This work was granted by the BMBF within the GABI program (GENOBAR, Project-No. 0315066C).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Electronic Supplementary Material Table 1
Number of mapped DArT and SSR markers used for the analysis of population structure, linkage disequilibrium (LD), kinship and association studies in a set of 183 barley cultivars and their average PIC values. The number of DArT-markers which were used in association mapping studies after removal of 5% minor allele frequency (MAF) is also shown. (DOC 40 kb)
Electronic Supplementary Material Table 2
Population structure and assignment (>50% probability) of each cultivar to the five groups (Q1 to Q5) revealed by STRUCTURE analysis with both marker types (sheet a). Population structure and assignment (in % probability) of every cultivar to one of the five groups (Q1 to Q5) revealed by STRUCTURE analysis with the 22 SSRs (sheet b). Population structure and assignment (in % probability) of every cultivar to one of the five groups (Q1 to Q5) revealed by STRUCTURE analysis with the 253 DArTs (sheet c). Origin of cultivars and breeders (sheet d). (XLS 120 kb)
Electronic Supplementary Material Table 3
GWAS with the kernel quality parameter glume fineness (sheet a), GWAS with the kernel quality parameter TGW (sheet b), GWAS with the malting quality parameter extract (sheet c), GWAS with the malting quality parameter friability (sheet d) considering the GLM. (XLSX 218 kb)
Electronic Supplementary Material Table 4
Results of GWAS considering the MLM with four different combinations of marker types used for the estimation the Q-matrix and kinship (MLM_1 = Q5-DArT + K-DArT, MLM_2 = Q5-SSR + K-DArT, MLM_3 = Q5-DArT + K-SSR, MLM_4 = Q5-SSR + K-SSR). Two kernel quality parameters, such as glume fineness (sheet a), and TGW (sheet b), and two malting quality parameters like extract (sheet c), and friability (sheet d) were assessed. (XLSX 593 kb)
Electronic Supplementary Material Fig. 1
Fig. 1 Estimated probability of number of subgroups k (goodness of fit computed as lnPr (X|K) for the investigated set of 183 cultivars studied with two marker types (a) 22 SSRs, and (b) 253 DArTs. The ln likelihood L(K) mean values determined by STRUCTURE are plotted against the assumed number of subgroups (k1 to k20). (PPT 153 kb)
Electronic Supplementary Material Fig. 2
Scatterplot showing the distribution of the intrachromosomal LD-decay parameter r2 in 183 European barley cultivars and plotted against the genetic distance in cM. The horizontal line indicates the 95% percentile of the distribution of unlinked r2, which gives the critical value of r2. Second degree LOESS curve fitted to the plot (black bottom line). (PPT 147 kb)
Electronic Supplementary Material Fig. 3
Proportion of marker pairwise r2 intrachromosomal measurements above and below background linkage disequilibrium with a critical r2 of 0.21 plotted as a logarithmic function of the genetic distance (in classes) of the entire set of 183 European barley cultivars investigated with 862 mapped DArT markers considering 5 % MAF. (PDF 10 kb)
Electronic Supplementary Material Fig. 4
Cumulative distribution of the observed p values assessed for different variantsof the MLM_QK with respect to the marker type used for assessing population structure and kinshipinformation (a) MLM_1 = Q5-DArT + K-DArT, (b) MLM_2 = Q5-SSR + K-DArT, (c) MLM_3 = Q5-DArT + K-SSR, (d) MLM_4 = Q5-SSR + K-SSR). Following traits were considered: Glume fineness,thousand grain weight (TGW), extract, and friability. (PPTX 190 kb)
Electronic Supplementary Material Fig. 5
Genomewide association studies of 183 barley cultivars considering the MLM_QK for four traits (a) extract, and (b) thousand grain weight. Population structure (Q) and kinship (K) was taking into account by using the Q5 matrix calculated either with SSR- or DArT markers. This information was used in different combinations on the MLM in order to assess the effect of the marker type on the rate of signifianct association results (MLM_1 = Q5-DArT + K-DArT, MLM_2 = Q5-SSR + K-DArT, MLM_3 = Q5-DArT + K-SSR, MLM_4 = Q5-SSR + K-SSR). The calculated p-values were converted into –log10(p). The significance thresholds p < 0.05, and p < 0.001 are indicated by dashed lines. (PPTX 320 kb)
Rights and permissions
About this article
Cite this article
Matthies, I.E., van Hintum, T., Weise, S. et al. Population structure revealed by different marker types (SSR or DArT) has an impact on the results of genome-wide association mapping in European barley cultivars. Mol Breeding 30, 951–966 (2012). https://doi.org/10.1007/s11032-011-9678-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11032-011-9678-3