
Abstract
In this study, 129 wheat genotypes from globally diverse origins were genotyped using DArTseq (SilicoDArT and SNP) markers. After filtering markers for quality-filtering, 14,270 SilicoDArTs and 6484 SNPs were retained and used for genetic diversity, population structure and linkage disequilibrium analyses. The highest number of SilicoDArT and SNP markers mapped on genome A and B compared to genome D. In both marker types, polymorphism information content (PIC) values ranged from 0.1 to 0.5, while > 0.80% of SilicoDArTs and > 0.44% SNPs showed PIC value more than median (0.25%). Un-weighted Neighbor Joining cluster analysis and Bayesian-based model population structure grouped wheat genotypes into three and four clusters, respectively. Principal component analysis and discriminant analysis of principal component results showed highly match with cluster and population structure analysis. Linkage disequilibrium (LD) was more extensive in both marker types, while graphical display of LD decay for both marker types showed that LD declined in the region close to 15 kbp, where r2-values corresponded to r2 = 0.16. Overall, our genetic diversity analysis showed high level of variation in studied wheat genotypes, even though there was no relationship between wheat grouping and origins. This might be attributed to admixture level that occurred during long-term natural selection of wheat genotypes in different parts of the world. Highly diverse wheat genotypes used in this study may possess unique genes and are useful sources in breeding programs to improve grain yield and quality.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Bread wheat (Triticum aestivum L.) is one of the most important staple food cereal crops that cultivated in most of climatic zones worldwide (Schuster et al. 2009). The rapidly growing world population resulted in the increasing consumption of wheat. Therefore, it is necessary to deploy advanced breeding technologies to increase crop production, especially for strategic crops for human feed like as wheat, rice and corn (Ray et al. 2013; Marcussen et al. 2014; Alipour et al. 2017). Bread wheat is an allohexaploid and evolved through hybridization and comprised three different genomes; A (Triticum uratu), B (Aegilops speltoides) and D (Aegilops tauschii) (Zohary et al. 1969; Dvorak and Zhang 1992). Archaeological studies revealed the wheat domestication in a part of the Fertile Crescent area at least 12,000 years ago. Then bread wheat spread by the first farmers to Asia, Europe and Eastward from 8500 to 2300 BP (Greger 2015). During the domestication, farmers have preserved the superior genotypes and keep diversity of wheat genotypes that slowly adapted to local environments, becoming so-called landraces (Zeven 1998; Balfourier et al. 2019). Mostly, landraces are genotypes with high potential to adapt in different environments, and could tolerate biotic and abiotic stresses (Sohail et al. 2015). The evolution of wheat is a complex history of hybridization and gene flow events, which is associated with human migration from Mediterranean region to Southern Europe and North Africa (Baloch et al. 2017; Balfourier et al. 2019). Wheat is the most important cultivated crops in Middle East, North Africa and Mediterranean regions, providing 15% of calories for human diet (Balfourier et al. 2019). The reduction of genetic diversity in wheat, like many crops affected by domestication and also by the post-Mendlian adoption of breeding activities (Morgante and Salamini 2003; Novoselovic et al. 2016). Traditional crop breeding continuously imposed high pressure on wheat germplasm by selection for superior cultivars (Rubenstein et al. 2005) that lead to uniformity in germplsm for certain zone and climate.
This genetic diversity uniformity led to genetic vulnerability of crop species to various biotic and abiotic stresses (van de Wouw et al. 2010; Talebi and Fayyaz 2012; Nielsen et al. 2014). Therefore, narrow genetic base is still major concern for plant breeder to meet this genetic uniformity and reduce the genetic vulnerability by introducing the diverse genotypes into breeding materials from national and international germplasms (Cox 1998; van de Wouw et al. 2010; Novoselovic et al. 2016). Recently studies on worldwide phylogeography and history of wheat diversity revealed that most of Asian modern cultivars relied on European germplasm, while a huge diversity of landraces in Asia denied for their potential use in breeding programs (Alipour et al. 2017; Balfourier et al. 2019). This can be proved by high level of genetic diversity between improved cultivars with landraces originated from Fertile Crescent (Balfourier et al. 2019). Depending on geographical regions, genotypes domesticated in diverse climates and especially landraces have specific background with rich allelic diversity, can be used in different breeding programs (Smale et al. 2002; Talebi and Fayyaz 2012; Zhang et al. 2011a). Therefore, information on diversity level in crop genetic resources can help breeders to develop new cultivars with high yield potential and desirable alleles (Govindaraj et al. 2015). Characterization of genetic diversity in crops can be made by different methods like morphological, biochemical and DNA-based molecular markers (de Vicente et al. 2005). Unlike morphological and biochemical markers are easily influenced by environmental factors, DNA-based molecular markers are precise and environment-independent. Therefore, these markers providing versatile tools with broad applicability and thereby increasingly becoming valuable tools in breeding programs (Laido et al. 2014; Ghaffari et al. 2014; Novoselovic et al. 2016). Different types of molecular markers such as amplified fragment-length polymorphism (AFLP) (Soleimani et al. 2002; Talebi and Fayyaz 2012), simple sequence repeat (SSR) (Marzario et al. 2018) and restriction fragment-length polymorphism (RFLP) have been utilized for genetic diversity and population structure analysis in different wheat gene pools. However, low number of markers, primarily gel based, poor genome coverage, costly and time consuming are the major limitation factors with utilization of these markers (Mwadzingeni et al. 2017; Alam et al. 2018). Over the last decade, microarray based Diversity Array Technology (DArT) markers have been developed and used as a high-throughput genetic tool for genome diversity and population structure analyses, association mapping or linkage map construction in different crops as well as in wheat (Nielsen et al. 2014; Kabbaj et al. 2017; Mwadzingeni et al. 2017; Alam et al. 2018). Recently, a new genotyping platform called DArTseq has been developed including: ‘SilicoDArT and SNP markers’ based on genotyping-by-sequencing to sequence the most informative representations of genomic DNA samples (Kilian et al. 2012). DArTseq technology develops much higher markers with broad genome coverage in compare to the array version of DArT. Therefore, it serves as a cheap alternative tool for genome analysis by whole genome sequencing (Edet et al. 2018). These markers have been developed and utilized for genome diversity and genome-wide association mapping in different plant species (Ndjiondjop et al. 2017; Pailles et al. 2017; Zaitoun et al. 2018) and also in wheat (Monostori et al. 2017; Alam et al. 2018).
Because of the above-mentioned, it is important to study diverse germplasm from both improved cultivars and landraces for monitoring and measuring genetic diversity and population structure. Therefore, the main objectives of this study were to evaluate the genetic diversity in a globally diverse wheat germplasm to providing new information about the level of genetic diversity within wheat genotypes from globally geographical regions using DArTseq and SNP markers generated by DArTseq technology platform.
Materials and methods
Plant materials and DNA extraction
The 129 wheat genotypes, including 40 Iranian wheat genotypes (26 Iranian commercial cultivars and 14 landraces), 10 international cultivars (mostly used in worldwide breeding programs) and 79 accessions from diverse geographical wheat origins (Supplementary Table S1) were employed for genetic diversity, linkage disequilibrium and population structure analysis using both SilicoDArT and SNP markers. Wheat genotypes were grown in Iranian cereal research institute and selected for 6–7 generation by single seed selection to ensure the homozygosity, as we assumed that wheat is a self-pollinated and homozygous plant. The seeds are available upon request from the corresponding author.
For DNA extraction, ten seeds of each genotypes were sown in controlled greenhouse and after 10 days a pooled leaves for each genotypes harvested and kept in liquid nitrogen. DNA was extracted using CTAB protocol (Lassner et al. 1989) with minor modification (Talebi 2008). A DNA concentration was adjusted to 100–150 ng/µl by spectrophotometer using 260 nm wave length.
Genotyping using SilicoDArT and SNP markers
Sample DNA used for DArTseq platform genotyping by Diversity Arrays Technology Pty Ltd (https://www.diversityarrays.com/) for development high density SilicoDArT and SNP markers as described in previous studies (Ren et al. 2015; Ndjiondjop et al. 2017; Monostori et al. 2017). Overall, genotyping using DArTseq platform generated 54,309 SilicoDArT and 40,225 SNP markers. Since it was assumed that improved wheat genotypes used in this study are completely homozygous and landraces were selected from single plant, SNPs showing heterozygous alleles due to the detection of multiple loci were noted as missing and then markers with > 20% missing data were removed (Bellucci et al. 2015; Monostori et al. 2017, Baloch et al. 2017). All markers filtered using a minimum minor allele frequency (MAF) of 0.1 across the 129 wheat genotypes in TASSEL v.5.2.37 software (Bradbury et al. 2007). Additionally, markers with unknown chromosome position were also removed from the analysis. Overall, 25,066 polymorphic DArTseq markers (14,339 SilicoDArTs and 6484 SNPs) (Supplementary Table S2) remained for further analysis of genetic diversity, population structure and linkage disequilibrium in studied wheat genotyes.
Data analysis
The markers (DArTseq and SNP) were scored using DArTsoft v.7.4.7 as binary data (1 present, 0 absent, for a marker in the genomic DNA of each accession) (Akbari et al. 2006). Several quality parameters for DArTseq and SNP markers including call rate, polymorphic information content (PIC), and reproducibility, were computed by the DArTsoft v.7.4.7 (Cruz et al. 2013; Ndjiondjop et al. 2017). This information was provided by DArT Pty Ltd to produce “SilicoDArT” table and “SNP” table (Supplementary Table S2).
A genetic distance matrix for both marker types were calculated using the identity-by-state (IBS) method implemented in TASSEL v.5.2.37. Cluster analysis was performed on the genetic distance matrix using the Un-weighted Neighbor Joining algorithm (Perrier et al. 2003) implemented in the Dissimilarity Analysis and Representation for windows (DARwin) ver.5.0 software (Perrier and Jacquemoud-Collet 2006). Mantel coefficient correlation test between both marker systems, as well as between markers and geographical origins were implemented in the ape package of statistical software program ‘R’.
Principal coordinate analysis (PCoA) was also used for wheat genotypes grouping, by means of the software package PAST v.3.12 (Hammer et al. 2001). Linkage disequilibrium (LD) for SilicoDArT and SNP markers was implemented in TASSEL v.5.2.37 and graphical LD decay for each marker types were imputed by GAPIT R package (Van Raden 2008; Lipka et al. 2012). For both SilicoDArT and SNP markers, population structure and discriminant analysis of principal component (DAPC) also were calculated. Population structure imputed in STRUCTURE 2.1 based on an admixture model (Evanno et al. 2005), as in model the K-values ranging from 1 to 10 with 3 independent runs and 50,000 Markov chain Monte Carlo iterations.
The results of structure analysis for estimate the optimal value of K using the Delta (K) method (Evanno et al. 2005) were extracted using STRUCTURE HARVESTER web version 0.6.94 (Earl and Vonholdt 2012). Genetic diversity among and between populations, proportion of safflower genotypes membership in each cluster and Wright's F-statistics (FST) among subpopulations were extracted from STRUCTURE 2.1. DAPC analysis for finding the fitting pupation structure of studied wheat genotypes was computed using the adegenet package (Jombart 2008) for R software version 3.2.3 (R Core Team 2014).
Results
Marker polymorphism analysis and genetic relationship between wheat genotypes
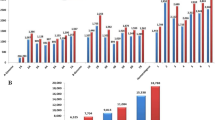
The initial data set consist of 54,309 SilicoDArT and 40,225 SNP markers. These markers are filtered for minor allele frequency lower than 0.1, missing data > 20% and unknown chromosomal position. Finally, a total of 25,066 polymorphic DArTseq1markers (14,339 SilicoDArTs and 6484 SNPs) were assigned to 21 wheat chromosomes and considered for genetic diversity and population structure in 129 wheat genotypes. The highest number of SilicoDArT and SNP markers mapped on genome A and B compared to those mapped on genome D (Fig. 1). The highest and lowest numbers of both marker types were observed for chromosome 2B and 4D, respectively. All SilicoDArT and SNP markers showed > 90% reproducibility. Call rate were > 0.84 and > 0.90 for all SilicoDArT and SNP markers, respectively (Table S2). For both marker types, PIC values ranged from 0.1 to 0.5, while > 0.80% of SilicoDArTs and > 0.44% SNPs showed PIC value more than median (0.25%) (Fig. 2). Kinship coefficients between pairs of genotypes based on SilicoDArTs and SNPs varied from − 0.41 to 2.18 and − 0.42 to 1.98, respectively (Supplementary Table S3).

Genome coverage and distribution of SilicoDArT and SNP markers on different chromosomes of wheat

Frequency distribution of PIC values for SilicoDArT and SNP markers
Wheat genotypes grouped into 3 and 4 clusters according to kinship matrix obtained by SilicoDArT and SNP markers, respectively (Fig. 3). Genetic distance between pairs of genotypes based on SilicoDArTs and SNPs ranged from 0.005 to 0.47 and 0.07 to 0.47, respectively (Supplementary Table S4).

Heatmap plot of kinship matrix displaying relationships of 129 wheat genotypes based on SilicoDArT and SNP markers. The details of members of these groups are presented in Table S3
The Un-weighted Neighbor Joining cluster analysis based on SilicoDArT and SNP markers differentiate the 129 wheat genotypes into 3 and 4 clusters, respectively (Fig. 4). Iranian Landrace and improved cultivars distributed into different clusters. There were no relationships between cluster grouping and origins of wheat genotypes. Most of the genotypes used in this study used as parental lines or have same pedigree background. Therefore, mixture of origins observed in all clusters. Clusters shows relatively same number of genotypes, although cluster III for SilicoDArT and cluster IV in SNPmarkers are larger groups of wheat genotypes. Mantel coefficient test showed positive but non-significant correlation between SilicoDArT and SNPclusters (r = 0.27; P < 0.05). Comparisons among the clusters derived from SilicoDArT and SNPmarkers showed positive significant association between both markers system (r = 0.797; P < 0.0001; Fig. 5) through Mantel test showing a good fit between SilicoDArT and SNPmarker data sets. The results showed non-significant correlation between both marker types and geographical origins of wheat genotypes.

The nieghbor-joining cluster analysis using SilicoDArT (a) and SNP markers (b) for grouping 129 wheat genotypes

Mantel correlation test between SilicoDArT and SNP markers
Population structure and discriminate analysis of principal coordinate (DAPC)
A population structure of 129 wheat genotypes based on Bayesian model in STRUCTURE, showed population structure at K = 3 and K = 4 for SilicoDArT and SNP markers, respectively (Fig. 6). Sub-populations based on SilicoDArT markers contained 33% (POP1), 34.9% (POP2) and 32% (POP3) of wheat genotypes and the genetic divergence within each population (expected heterozygosity) varied from 0.27 (POP1) to 0.35 (POP3) (Table 1). Genetic diversity among the populations based on net nucleotide distance revealed a higher distance between POP2 and POP3 compared to the genetic distance between POP1 and POP2 (Table 1). Four sub-populations based on SNP markers showed relatively low genetic divergence among sub-populations (from 0.28 for POP4 to 0.33 for POP3), while higher divergence between sub-populations was observed (Table 1). Mean fixation index of sub-populations based on SilicoDArT and SNP markers ranged from 0.018 to 0.404 and 0.205 to 0.399, respectively (Table 1).

Determination of the optimal value of K and population structure of 129 wheat genotypes using SilicoDArT and SNP markers
Principal component analysis (PCoA) based on SilicoDArT and SNP markers revealed three and four distinct groups of genotypes, respectively, and two principal components displayed 72.18% (SilicoDArT) and 81.11% (SNP) of total variation (Fig. 7).

Principal coordinate analysis of 129 wheat genotypes based on SilicoDArT and SNP markers
Discriminant analysis of principal component (DAPC) also was employed to fine the fitting population structure based on SilicoDArT and SNP markers. The lowest BIC value was obtained at K = 3 and K = 4 for SilicoDArT and SNP markers, respectively. Therefore, two and three discrimination function were detected which these components explained 37.74 and 25.12 for SilicoDArTs and 26.39, 21.47 and 19.13 for SNP markers (Fig. 8). Results from the DAPC analysis was match with population structure analysis.

The percentage of cumulative variance for the retained PCA eigen vectors and Scatter plot from the DAPC analysis for 129 wheat genotypes used to determine the optimal k number of clusters using SilicoDArT and SNP markers
Linkage disequilibrium analysis
Distribution of LD within chromosomes based on SilicoDArT and SNP markers for each of three wheat genomes are summarized in Table 2. LD was extensive for both marker types, as in the entire population 120,226 (16.9%) and 16,483 (8%) intra-chromosomal pairs showed significant level (P < 0.001) of LD for SilicoDArT and SNP markers, respectively (Table 2). LD analysis for different genomes showed high numbers of significant pairs for genome-B in compare to genome-A and genome-D. Lower marker density and lower number of significant pairs of LD was observed in genome-D compared to genome-A and B, while higher rate of linked marker pairs and higher number of pairs in complete LD (r2 = 1) were found in genome-D compared to genome-A and B. Mean r2-value in both marker types was similar, while genome-D showed higher mean r2-value for both marker types with respect to genome-A and B. The mean critical r2-value for both marker types was similar, although the relatively higher value observed for genome-D (Table 2). Graphical display of LD decay for both marker types, showed that the LD declined in the region close to 15 kb, where r2-values corresponded to r2 = 0.16 (Fig. 9).

Linkage disequilibrium (LD) measured r2 plotted vs. the physical map (bp) between pairs of SilicoDArT and SNP markers in a panel of 129 wheat genotypes located on genome A, B, D and whole chromosome region
Discussion
Characterization of genetic diversity in crop species is prerequisite and important for efficient germplasm conservation for utilization in different breeding programs (Laido et al. 2014). During the second half of twentieth century, due to extensive use of high-yielding semi-dwarf wheat cultivars in most of national and international breeding programs, the genetic diversity was narrowed down (Rufo et al. 2019). Previous studies demonstrated that the durum and bread wheat domesticated in Fertile Crescent and last decades breeding activities of wheat germplasms lead to narrowing their genetic diversity and consequently vulnerability to abiotic and biotic stresses (Zeven 2000; Baloch et al. 2017). Landraces have been considered as a valuable source of favorable genes that can be used introduce into improved cultivars (Lopes et al. 2015; Rufo et al. 2019). But, more studies estimated high genetic distances between landraces and modern cultivars; it clearly shows low usage of landraces in wheat breeding programs (Soriano et al. 2016). Therefore, the monitoring the genetic diversity of landraces with modern cultivars assist breeders to select appropriate parental lines with desirable alleles for crossing and monitoring best progenies with high rate of allelic frequencies and transgressive segregation (Christiansen et al. 2002; Nielsen et al. 2014) for integration into target crossing schemes (Kabbaj et al. 2017). We tried here to explore the genetic diversity of selected modern cultivars and landraces from diverse global geographical origins; however, a large proportion of the landraces were from Iran. These landraces are mainly grown under rain-fed conditions in Iran and selected based on their adoption to various abiotic and biotic stresses (Fayaz et al. 2019). Despite extensive wheat breeding programs worldwide as well as in Iran, it appears most of the modern high-yielding wheat cultivars are vulnerable to environmental stresses. Therefore, continuous screening and characterization of wheat genotypes possessing favorable genes is required to integrate these new sources into breeding gene pool (Mehrabi et al. 2015; Alipour et al. 2017; Fayaz et al. 2019). Recently, Diversity Arrays Technology provided a DArTseq platform as robust molecular markers (SilicoDArT and SNP) for genetic diversity and genome mapping in many crops (Pailles et al. 2017; Mogga et al. 2018; Ndjiodjop et al. 2017; Alam et al. 2018) as well as in wheat (Monostori et al. 2017; Ovenden et al. 2017; Baloch et al. 2017). In this study we highlight the efficiency of SilicoDArT and SNPmarkers for genome diversity analysis in an originally diverse wheat collection.
In this study a total 54,309 SilicoDArT and 40,225 SNPmarkers initially were developed, of which 14,339 SilicoDArT and 6484 SNPmarkers showed highly polymorphism with known chromosomal position and further used for genetic diversity and population structure analyses. SilicoDArT and SNPmarkers used showed an average PIC value of 0.40 and 0.38, respectively. More than 50% of markers showed PIC value higher than average, which suggests the sufficient efficiency of these markers that has been reported previously (Zhang et al. 2011a; Monostori et al. 2017; Baloch et al. 2017). Physical distribution of mapped markers with known positions among A, B and D genome showed that most markers located on B genome followed by A and D genome, providing higher marker density compared to some previous DArTseq genotyping in wheat (Bellucci et al. 2015; Monostori et al. 2017). This finding was similar to previous reports on DArTseq and SNP markers distribution through wheat genomes (Mwadzingeni et al. 2017; Baloch et al. 2017; Alipour et al. 2017).
Average inter-chromosomal LD decay in both marker types was relatively similar, which is in agreement with previous reports using DArTseq platform in wheat (Bellucci et al. 2015; Monostori et al. 2017). Wheat gemplasm studies here, showed long distances for marker pairs in LD plot (Fig. 7), which may be attributed to genetic admixture apart from the genetics or physical distances that has been reported previously (Monostori et al. 2017). LD decay of genome-A, B and D were relatively similar to the LD decay of the total population, although higher rate of linked marker pairs and LD of markers for genome-D were observed. This finding is in agreement with previous studies in wheat (Chen et al. 2012; Nielsen et al. 2014), which was attributed to higher selection pressure on genome D and different selection evolutionary pressures on these marker regions in different wheat genomes (Nielsen et al. 2014). Genetic distance between pairs of genotypes based on both marker types showed relatively highly genetic variability (0.37) among wheat genotypes. Cluster analysis, principal component and model-based population structure analyses revealed three and four distinct groups of wheat genotypes using SilicoDArT and SNP markers, respectively. Wheat genotypes grouping were the same for most of genotypes by both marker systems, which was confirmed by the Mantel test to check association between both markers systems. DArTseq markers (SilicoDArTs and SNPs) are based on genotyping by sequencing (GBS) and providing lots of markers with high genome coverage. There was non-significant correlation between distance matrixes of both marker types with geographical origins of wheat genotypes, which is in agreement with previous DArTseq markers studies on durum wheat (Baloch et al. 2017) and snake melon (Omari et al. 2018). Both SilicoDArT and SNP markers have not separated landraces according to their corresponding geographical origin, and is not surprising as Iranian modern cultivars and landraces grouped into separate clusters. This suggests that Iranian breeders mostly used exotic genetic materials in wheat breeding programs. But there were somewhat congruent between cluster groups with ecological/climate adaptation of wheat genotypes used in this study. Cluster III generated by SNP markers and Cluster II by SilicoDArT markers mainly included wheat genotypes originated from Middle-East (Iran, Afghanistan, Turkey), Kazakhstan, Russia, Romania and Finland. This close relationship may be anticipated, as we know that western Iran, eastern Turkey, northern Syria and Africa and part of Siberian continent are the main centers of diversity and domestication of wheat (Kilian et al. 2007). So, it is possible that these genotypes have been utilized in regional breeding programs and it does admixture of these genotypes with local varieties. In other hand, the evolutionary processes of wheat domestication during thousands years in widespread area may influenced by mutation, migration, selection by farmer and genetic drift (Vikram et al. 2016; Baloch et al. 2017; Kabbaj et al. 2017). Therefore, breeding activities with different strategies in different regions led to significant impact on global wheat genetic structure (Zhang et al. 2011b).
Admixture level in different wheat germplasm observed previously may be reflected to long term natural selection and differentiations that occurred during initial of wheat spread from Fertile Crescent into other parts of the world (Balfourier et al. 2019). In the other hand, single individual or a few genotypes is not sufficient to describe the whole diversity (Golicz et al. 2016). Therefore, selected genotypes in this study might not be representative of the accurate worldwide wheat diversity. Most of genotypes used in this study have not been studied before and showed highly genetic diversity. Therefore, there is a highly chances to find unique genotypes with useful genes for introducing into breeding programs. These wheat genotypes will be highly valuable sources as a donor parent in crossing programs. Although, high density genotyping data generated in this germplasm can be used as an interesting panel for genome-wide association mapping studies for desirable traits in wheat.
Conclusion
Characterization the genetic diversity in crop germplasm is a prerequisite and key strategy for germplasm conservation in plant breeding programs. In this study, 129 wheat genotypes from diverse origins were genotyped with DArTseq platform (SilicoDArT and SNP markers). Our results showed high polymorphism for both marker types as well as high genetic diversity between wheat genotypes. Population structure and cluster analysis grouped genotypes in distinct clusters, which this grouping was not match with geographical origins and high mixture observed in studied germplasm. This finding will support breeding programs for better utilization and expanding the wheat gene pools by germplasm exchange between diverse origins. This study also demonstrated the efficiency of DArTseq platform genotyping for large scale genome analysis in wheat. Therefore, this studied panel is useful for conduct association mapping studies focusing on seed yield, abiotic and biotic stresses in wheat.
References
Akbari M, Wenzl P, Caig V, Carling J, Xia L, Yang S et al (2006) Diversity arrays technology (DArT) for high-throughput profiling of the hexaploid wheat genome. Theor Appl Genet 113:1409–1420
Alam M, Neal J, O’Connor K, Kilian A, Topp B (2018) Ultra-high-throughput DArTseq-based silicoDArT and SNP markers for genomic studies in macadamia. PLoS ONE 13(8):e0203465
Alipour H, Bihamta MR, Mohammadi V, Peyghambari SA, Bai G, Zhang G (2017) Genotyping-by-sequencing (GBS) revealed molecular genetic diversity of Iranian wheat landraces and cultivars. Front Plant Sci 8:1293
Balfourier F, Bouchet S, Robert S, De Oliveira R, Rimbert H, Kitt J, Choulet F, International Wheat Genome Sequencing Consortium, BreedWheat Consortium, Paux E (2019) Worldwide phylogeography and history of wheat genetic diversity. Sci Adv 5:eaav0536
Baloch FS, Alsaleh A, Shahid MQ, Çiftçi V, Sáenz de Miera L, Aasim M, Nadeem MA, Aktas S, Ozkan H, Hatipoglu R (2017) A whole genome DArTseq and SNP analysis for genetic diversity assessment in Durum wheat from central fertile crescent. PLoS ONE 12(1):e0167821–3486
Bellucci A, Torp AM, Bruun S, Magid J, Andersen SB, Rasmussen SK (2015) Association mapping in Scandinavian winter wheat for yield, plant height, and traits important for second-generation bioethanol production. Front Plant Sci 6:1046
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635
Chen X, Min D, Yasir TA, Hu Y-G (2012) Genetic diversity, population structure and linkage disequilibrium in elite Chinese winter wheat investigated with SSR markers. PLoS ONE 7(9):e44510–2830
Christiansen MJ, Andersen SB, Ortiz R (2002) Diversity changes in an intensively bred wheat germplasm during the 20th century. Mol Breed 9:1–11
Cox TS (1998) Deepening the wheat gene pool. J Crop Prod 1:1–25
Cruz VMV, Kilian A, Dierig DA (2013) Development of DArT marker platforms and genetic diversity assessment of the U.S. collection of the new oilseed crop lesquerella and related species. PLoS ONE 8:e64062
de Vicente MC, Guzmán FA, Engels J, Rao VR (2005) Genetic characterization and its use in decision making for the conservation of crop germplasm. The Role of Biotechnology, Villa Gualino, Turin, Italy, 5–7 March 2005
Dvorak J, Zhang HK (1992) Reconstruction of the phylogeny of the genus Triticum from variation in repeated nucleotide sequences. Theor Appl Genet 84:419–429
Earl DA, Vonholdt BM (2012) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4(2):359–361
Edet OU, Gorafi YSA, Cho SW, Kishii M, Tsujimoto H (2018) Novel molecular marker-assisted strategy for production of wheat–Leymus mollis chromosome addition lines. Sci Rep 8:16117
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Fayaz F, Aghaee M, Talebi R, Azadi A (2019) Genetic diversity and molecular characterization of Iranian durum wheat landraces (Triticum turgidum durum (Desf.) Husn.) using DArT markers. Biochem Genet 57(1):98–116
Ghaffari P, Talebi R, Keshavarz F (2014) Genetic diversity and geographical differentiation of Iranian landrace, cultivars and exotic chickpea lines as revealed by morphological and microsatellite markers. Physiol Mol Biol Plants 20(2):225–233
Golicz AA, Batley J, Edwards D (2016) Towards plant pangenomics. Plant Biotechnol J 14:1099–1105
Govindaraj M, Vetriventhan M, Srinivasan M (2015) Importance of genetic diversity assessment in crop plants and its recent advances: an overview of its analytical perspectives. Genet Res Int 2015:431487
Greger L (2015) How wheat came to Britain. Science 347:6225
Hammer Ø, Harper DAT, Ryan PD (2001) Paleontological statistics software package for education and data analysis. Palaeontol Electron 4:9–18
Jombart T (2008) adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24(11): 1403–1405
Kabbaj H, Sall AT, Al-Abdallat A, Geleta M, Amri A, Filali-Maltouf A, Belkadi B, Ortiz R, Bassi FM (2017) Genetic diversity within a global panel of durum wheat (Triticum durum) landraces and modern germplasm reveals the history of allele’s exchange. Front Plant Sci 8:1277
Kilian B, Özkan H, Walther A, Kohl J, Dagan T, Salamini F, Martin W (2007) Molecular diversity at 18 loci in 321 wild and 92 domesticate lines reveal no reduction of nucleotide diversity during Triticum monococcum (einkorn) domestication: implications for the origin of agriculture. Mol Biol Evol 24:2657–2668
Kilian A, Wenzl P, Huttner E, Carling J, Xia L, Blois H, Caig V, Heller-Uszynsk K, Jaccoud D, Hopper C, Aschenbrenner-Kilian M, Evers M, Peng K, Cayla C, Hok P, Uszynski G (2012) Diversity arrays technology: a generic genome profiling technology on open platforms. Methods Mol Biol 888:67–89
Laido G, Marone D, Russo MA, Colecchia SA, Mastrangelo AM, De Vita P, Papa R (2014) Linkage disequilibrium and genome-wide association mapping in tetraploid wheat (Triticum turgidum L.). PLoS ONE 9(4):e95211
Lassner MW, Peterson P, Yoder JI (1989) Simultaneous amplification of multiple DNA fragments by polymerase chain reaction in the analysis of transgenic plants and their progeny. Plant Mol Biol Rep 7:116–128
Lipka AE, Tian F, Wang Q, Peiffer J, Li M, Bradbury PJ, Gore MA, Buckler ES, Zhang Z (2012) GAPIT: genome association and prediction integrated tool. Bioinformatics 28:2397–2399
Lopes MS, El-Basyoni I, Baenziger PS, Singh S, Royo C, Ozbek K, Aktas H, Ozer E, Ozdemir F, Manickavelu A, Ban T, Vikram P (2015) Exploiting genetic diversity from landraces in wheat breeding for adaptation to climate change. J Exp Bot 66(12):3477–3486
Marcussen T, Sandve SR, Heier L, Spannagl M, Pfeifer M, Jakobsen KS, Wulff BBH, Steuernagel B, Mayer KFX, Olsen O-A, Rogers J, el Dole J, Pozniak C, Eversole K, Feuillet C, Gill B, Friebe B, Lukaszewski AJ, Sourdille P (2014) Ancient hybridizations among the ancestral genomes of bread wheat. Science 345:1250092
Marzario S, Logozzo G, David JL, Zeuli PS, Gioia T (2018) Molecular genotyping (SSR) and agronomic phenotyping for utilization of durum wheat (Triticum durum Desf.) ex-situ collection from southern Italy: a combined approach including pedigreed varieties. Genes 9:465
Mehrabi R, Makhdoomi A, Jafar-Aghaie M (2015) Identification of new sources of resistance to septoria tritici blotch caused by Zymoseptoria tritici. J Phytopathol 163:84–90
Mogga M, Sibiya J, Shimelis H, Lamo J, Yao N (2018) Diversity analysis and genome-wide association studies of grain shape and eating quality traits in rice (Oryza sativa L.) using DArT markers. PLoS One 14(2):e0212078
Monostori I, Szira F, Tondelli A, Arendas T, Gierczik K, Cattivelli L, Galiba G, Vagujfalvi A (2017) Genome-wide association study and genetic diversity analysis on nitrogen use efficiency in a Central European winter wheat (Triticum aestivum L.) collection. PLoS ONE 12(12):e0189265
Morgante M, Salamini F (2003) From plant genomics to breeding practice. Curr Opin Biotechnol 14:214–219
Mwadzingeni L, Shimelis H, Rees DJG, Tsilo TJ (2017) Genome-wide association analysis of agronomic traits in wheat under drought-stressed and non-stressed conditions. PLoS ONE 12(2):e0171692
Ndjiondjop MN, Semagn K, Gouda AC, Kpeki SB, Dro Tia D, Sow M, Goungoulou A, Sie M, Perrier X, Ghesquiere A, Warburton ML (2017) Genetic variation and population structure of Oryza glaberrima and development of a mini-core collection using DArTseq. Front Plant Sci 8:1748
Nielsen NH, Backes G, Stougaard J, Andersen SU, Jahoor A (2014) Genetic diversity and population structure analysis of European hexaploid bread wheat (Triticum aestivum L.) varieties. PLoS ONE 9:e94000
Novoselovic D, Bentley AR, Šimek R, Dvojkovic K, Sorrells ME, Gosman N, Horsnell R, Drezner G, Šatovic Z (2016) Characterizing Croatian wheat germplasm diversity and structure in a European context by DArT markers. Front Plant Sci 7:184
Omari S, Kamenir Y, Benichou JIC, Pariente S, Sela H, Perl-Treves R (2018) Landraces of snake melon, an ancient Middle Eastern crop, reveal extensive morphological and DNA diversity for potential genetic improvement. BMC Genet 19:34
Ovenden B, Milgate A, Wade LJ, Rebetzke GJ, Holland JB (2017) Genome-wide associations for water-soluble carbohydrate concentration and relative maturity in wheat using SNP and DArT marker arrays. G3: Genes Genomes Genet 7(8):2821–2830
Pailles Y, Ho S, Pires IS, Tester M, Negrão S, Schmöckel SM (2017) Genetic diversity and population structure of two tomato species from the Galapagos Islands. Front Plant Sci 8:138
Perrier X, Jacquemoud-Collet JP (2006) DARwin software. https://darwin.cirad.fr/darwin
Perrier X, Flori A, Bonnot F (2003) Data analysis methods. In: Hamon P, Seguin M, Perrier X, Glaszmann JC (eds) Genetic diversity of cultivated tropical plants. Science Publishers, Enfield, pp 43–76
R Core Team (2014) R: a language and environment for statistical computing. R Core Team, Vienna
Ray DK, Mueller ND, West PC, Foley JA (2013) Yield trends are insufficient to double global crop production by 2050. PLoS ONE 8:e66428
Ren R, Ray R, Li P, Xu J, Zhang M, Liu G, Yao X, Kilian A, Yang X (2015) Construction of a high-density DArTseq SNP-based genetic map and identification of genomic regions with segregation distortion in a genetic population derived from a cross between feral and cultivated-type watermelon. Mol Genet Genom 290(2):1457–1470
Rubenstein DK, Heisey P, Shoemaker R, Sullivan J, Frisvold G (2005) Crop genetic resources: an economic appraisal. United States Department of Agriculture (USDA). Economic Information Bulletin No. 2. (www.ers.usda.gov)
Rufo R, Alvaro F, Royo C, Soriano JM (2019) From landraces to improved cultivars: assessment of genetic diversity and population structure of Mediterranean wheat using SNP markers. PLoS ONE 14(7):e0219867
Schuster I, Vieira ESN, da Silva GC, de Assis FF, Marchioro VS (2009) Genetic variability in Brazilian wheat cultivars assessed by microsatellite markers. Genet Mol Biol 32:557–563
Smale M, Reynolds MP, Warburton M, Skovmand B, Trethowan R, Singh RP, Ortiz-Monasterio I, Crossa J (2002) Dimensions of diversity in modernspring bread wheat in developing countries from 1965. Crop Sci 42:1766–1779
Sohail Q, Manickavelu A, Ban T (2015) Genetic diversity analysis of Afghan wheat landraces (Triticum aestivum) using DArT markers. Genet Resour Crop Evol 62(8):1147–1157
Soleimani VD, Baum BR, Johnson DA (2002) AFLP and pedigree-based genetic diversity estimates in modern cultivars of durum wheat [Triticum turgidum L. ssp. Durum (Desf.) Husn.]. Theor Appl Genet 104:350–357
Soriano JM, Villegas D, Aranzana MJ, Garcıa del MoralL F, Royo C (2016) Genetic structure of modern durum wheat cultivars and Mediterranean landraces matches with their agronomic performance. PLoS ONE 11(8):e0160983
Talebi R (2008) An alternative strategy in rapid DNA extraction protocol for high throughput RAPD analysis in chickpea and its wild related species. J Appl Biol Sci 2(3):121–124
Talebi R, Fayyaz F (2012) Quantitative evaluation of genetic diversity in Iranian modern cultivars of wheat (Triticum aestivum L.) using morphological and amplified fragment length polymorphism (AFLP) markers. Biharean Biol 6:14–18
Van de Wouw M, van Hintum T, Kik C, van Treuren R, Visser B (2010) Genetic diversity trends in twentieth century crop cultivars: a meta-analysis. Theor Appl Genet 120:1241–1252
Van Raden PM (2008) Efficient methods to compute genomic predictions. J Dairy Sci 91(4):414–4423
Vikram P, Franco J, Burgueño-Ferreira J, Li H, Sehgal D, Pierre CS, Ortiz C, Sneller C, Tattaris M, Guzman C, Sansaloni CP, Ellis M, Fuentes-Davila G, Reynolds M, Sonder K, Singh P, Payne T, Wenzl P, Sharma A, Bains NS, Singh GP, Crossa J, Singh S (2016) Unlocking the genetic diversity of Creole wheats. Sci Rep 6:23092
Zaitoun SYA, Jamous RM, Shtaya MJ, Mallah OB, Eid IS, Ali-Shtayeh MS (2018) Characterizing Palestinian snake melon (Cucumis melo var.flexuosus) germplasm diversity and structure using SNP and DArTseq markers. BMC Plant Biol 18:246
Zeven AC (1998) Landraces: a review of definitions and classification. Euphytica 104:127–139
Zeven AC (2000) Traditional maintenance breeding of landraces: 1. Data by crop. Euphytica 116:65–85
Zhang LY, Liu DC, Guo XL, Yang WL, Sun JZ, Wang DW, Sourdille P, Zhang AM (2011a) Investigation of genetic diversity and population structure of common wheat cultivars in northern China using DArT markers. BMC Genet 12:42
Zhang Z, Belcram H, Gornicki P, Charles M, Just J, Huneau C, Magdelenat G, Couloux A, Samain S, Gill BS, Rasmussen JB, Barbe V, Faris JD, Chalhoub B (2011b) Duplication and partitioning in evolution and function of homoeologous Q; loci governing domestication characters in polyploid wheat. Proc Natl Acad Sci USA 108:18737–18742
Zohary D, Harlan JH, Vardi A (1969) The wild diploid progenitors of wheat and their breeding value. Euphytica 18:58–65
Acknowledgements
The study was conducted as a part of PhD thesis of first author and supported by Islamic Azad University, Sanandaj Branch, Iran. We are grateful to Iranian Cereal Research Institute for providing wheat genotypes used in this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Mahboubi, M., Mehrabi, R., Naji, A.M. et al. Whole-genome diversity, population structure and linkage disequilibrium analysis of globally diverse wheat genotypes using genotyping-by-sequencing DArTseq platform. 3 Biotech 10, 48 (2020). https://doi.org/10.1007/s13205-019-2014-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s13205-019-2014-z