
Abstract
Genome-wide association studies have become a wide spread method of quantitative trait locus identification for many crops, including wheat (Triticum aestivum L.). Its benefit over traditional biparental mapping approaches depends on the extent of linkage disequilibrium (LD) in natural populations. We estimated the genetic diversity, population structure, and LD decay rate in a winter wheat association mapping panel (n = 205) and identified markers associated with thousand-kernel weight (TKW) and related traits. The panel was genotyped with a high-density Illumina iSelect 90 K single nucleotide polymorphism assay. PIC values were 0.047–0.375 with a mean of 0.277. Structural analysis suggested the association mapping panel contained four subpopulations. LD decay rates extended to longer genetic distances within the D genome (11.0 cM) relative to the A and B genomes (1.5 and 1.8 cM, respectively). A total of 271 marker-trait associations (MTAs) were identified for TKW and related traits, explaining 5.49–9.86 % of variation in individual traits. Among them, 11 highly significant markers (p < 0.0001), eight stable markers and twelve multi-trait MTAs were detected. Two stable markers, Ku_c9210_105 for KL and BS00023893_51 for TKW, were detected in three environments. These MTAs could be used for developing cleaved amplified polymorphic sequence markers for molecular marker-assisted selection in wheat breeding programs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Wheat (Triticum aestivum L.) is one of the most important crops in the world. Thousand kernel weight (TKW) is one of the three major yield components, and it is not only directly related to wheat grain yield and milling quality, but also affects seedling vigor and growth, which indirectly affects yields (Cui et al. 2011; Ramya et al. 2010). TKW has also been found to be closely associated with kernel size traits, such as kernel length (KL), kernel width (KW), kernel thickness (KT), and kernel diameter ratio (KDR; Sun et al. 2009; Tsilo et al. 2010). Consequently, a better understanding of the relationships between kernel weight and associated traits by analyzing their genetic mechanisms is essential to further increases in wheat grain yields.
Genome-wide association studies (GWAS) and traditional quantitative trait loci (QTL) mapping are the two main approaches to dissecting the genetic bases of complex traits (Risch and Merikangas 1996). To date, numerous traditional QTL studies have been conducted to uncover the genetic basis of TKW and related traits in wheat (Cui et al. 2011, 2015; Huang et al. 2006; Ramya et al. 2010; Sun et al. 2009; Zhang et al. 2014; Zheng et al. 2010). However, the traditional QTL mapping approach often locates genomic regions containing polymorphisms that are limited to the biparental population and with low resolution. GWAS, as a complement to QTL mapping, has rapidly become a promising approach to genetic mapping based on linkage disequilibrium (LD). Association mapping studies have many advantages over traditional QTL mapping, including increased QTL resolution, allele coverage, and potential use of natural germplasm (such as landraces, elite cultivars, and advanced breeding lines) (Buckler and Thornsberry 2002). In wheat, few studies have used association mapping to dissect the genetics of end-use quality traits (Breseghello and Sorrells 2006), yield component traits (Yao et al. 2009), and disease resistance (Tommasini et al. 2007).
Population structure and LD provide strategic information for successful association mapping (Zhang et al. 2010). The presence of population stratification in association mapping panels can result in nonfunctional spurious associations between molecular markers and traits (Flint-Garcia et al. 2003). The prevalence of LD in targeted genomic regions is a prerequisite for association mapping (Al-Maskri et al. 2012). Moreover, LD is population specific, affected by many genetic factors (Flint-Garcia et al. 2003), and highly variable across the genome. The presence of population structure has been widely documented in studies investigating the diversity of elite crop germplasm, especially in self-pollinating cereals (Hao et al. 2011). Therefore, an understanding of the population structure and extent of LD is a prerequisite for meaningful association mapping results.
To achieve high mapping resolution, GWAS require a large number of molecular markers that cover the whole genome at a sufficient resolution (Sajjad et al. 2012; Yao et al. 2009). Because single nucleotide polymorphism (SNP) markers are abundant and evenly distributed across most genomes, they satisfy the large samples and high-density marker requirements of GWAS (Gupta et al. 2008). Coupled with progress in next-generation DNA sequencing, SNP detection and genotyping can be conducted in a high-throughput and cost-effective manner, which has enabled SNP markers to be widely used in GWAS of many animals and plants, including humans (Eric et al. 2011), rice (Huang et al. 2010), and maize (Wilson et al. 2004). However, the use of SNP markers in wheat GWAS is a challenge because of wheat’s complex genomic architecture and incomplete genome sequence (Sukumaran and Yu 2014). Recently, the 90 K iSelect SNP genotyping array permitted a dramatic increase in the numbers of gene-based SNP markers that can be used to construct high-density linkage maps (Wang et al. 2014) and conduct QTL analysis in durum wheat (Colasuonno et al. 2014). GWAS using SNP markers for agronomical traits including TKW has also been applied in wheat (Zanke et al. 2015).
In the present study, we used the currently available 90 K Illumina iSelect SNP array (Wang et al. 2014) to genotype and analyze a panel of 205 diverse Chinese winter wheat lines. Our objectives were to (1) determine the genetic diversity, population structure, and LD extent of the association mapping population using SNP markers and (2) identify molecular markers associated with TKW and related traits in this mapping population. The results of the present study can facilitate marker-assisted breeding and reveal strategic combinations of TKW and related traits in wheat.
Materials and methods
Plant materials
The winter wheat association mapping panel used in this study was comprised of 205 genetically diverse varieties or lines carefully chosen to represent genetic stock used in the regions of China that grow winter wheat. Among them, 203 varieties and lines came from 10 provinces that are major winter wheat production regions in China and the remaining two varieties, used as founder parents, are from other countries (Mexico and France). Of the 205 varieties and lines, there were 77 bred varieties, 55 founder parents, and 73 breeder’s lines. The 73 breeder’s lines were specifically chosen from among varieties used in Shandong provincial regional trials in 2012 (Table S1).
Experimental design and phenotypic trait evaluation
The association mapping panel seeds were planted in Tai’an (116°36′E, 36°57′N) and Dezhou (116°29′E, 37°45′N) for two consecutive years (2013 and 2014), respectively. The experimental design followed a completely randomized block design with two replicates in each environment. All lines were grown in a three-row plot with 2 m long and 25 cm row-to-row distance. Crop production practices followed local procedures. There was no damage attributable to climate, disease, or pests during the growing seasons.
The measurement of KL, KW, KT, and TKW was carried out after the materials were harvested, following natural maturity. One thousand kernels of three independent samples were weighed and the mean was considered the 1000-kernel weight for each replicate of each genotype. The kernel length, width, and thickness were measured using an automatic color seed counter SC-I (Zhejiang Sci-Tech University). KDR was calculated as KDR = KL/KW.
DNA extraction
DNA was extracted from young leaf tissues of each variety using the protocol recommended by Triticarte Pty. Ltd (http://www.triticarte.com.au). DNA quality was checked by electrophoresis on 0.8 % agarose gels, and DNA concentration was determined with a NanoDropND-1000 UV–Vis spectrophotometer (NanoDrop Technologies, Wilmington, USA).
SNP markers and genotyping
The samples were genotyped using the recently developed wheat 90 K Illumina iSelect array comprising 81,587 gene-associated SNPs (Wang et al. 2014). Genotyping was performed at the University of California, Davis, Genome Center following the manufacturer’s recommendations as described by Akhunov et al. (2009). The genotyping assays were carried out on the Illumina iScanReader. Genotypic clusters for each SNP were determined using the polyploid version of GenomeStudio software (Illumina, http://www.illumina.com). The accuracy of SNP clustering was visually checked, and incorrectly clustered SNPs were manually adjusted. The SNPs with a detection rate exceeding 0.8 and a minor allele frequency (MAF) exceeding 0.05 were used for further data analysis.
Data analysis
The basic genetic statistics, including total number of alleles, gene diversity, MAF, and polymorphism information content (PIC), were calculated with Power Marker v 3.25 (Liu and Muse 2005). Phenotypic data analyses were performed using statistical software SPSS version 17.0 (SPSS, Chicago, IL, USA). Phenotypic correlation coefficients between TKW and related traits were calculated separately for each environment. Broad sense heritability was calculated as described by Wyman et al. (1991).
Population structure analysis was performed using the neighbor-joining (NJ) cluster analysis and STRUCTURE v 2.2 software (Pritchard et al. 2000) based on the genotyping data obtained from unlinked SNP markers in the population. In the former method, the genetic distance between genotypes was computed using Rogers (1972) genetic distances with Power Marker v 3.25, and the cluster analysis was carried out using the NJ tree method implemented in MEGA 4.0 (Tamura et al. 2007). In the STRUCTURE analysis, five independent runs were performed with a K-value (the putative number of genetic groups) varying from 1 to 15, with the length of burn-in period and the number of MCMC (Markov chain Monte Carlo) generations after burn-in both set to 100,000 under the ‘admixture model.’ The most likely K-value was determined by the log probability of data [LnP(D)] and an ad hoc statistic ΔK based on the rate of change of LnP (D) between runs using successive K-values as described by Evanno et al. (2005). The maximum membership probability among subgroups was applied to subdivide the accessions into different subgroups.
LD between SNP markers, including pairwise estimates of the squared allele-frequency correlation (r 2) and the significance of each pair of loci, was estimated by TASSEL3.0 (Bradbury et al. 2007). The pair-wise significance was computed from 1,000 permutations. The r 2 among loci was calculated separately for unlinked loci on different chromosomes and for linked loci on the same chromosome. Loci were considered to be in significant LD when p < 0.001. A critical value for r 2, used as evidence of linkage, was derived from the 95th percentile threshold of unlinked loci according to the method described by Breseghello and Sorrells (2006). LD decay scatter plots of syntenic r 2 versus genetic distance (cM) between markers were generated using SPSS 17.0. The smothering second degree LOESS curve was fitted and the point at which the LOESS curve intercepted the critical r 2 was determined to be the average LD decay of the population.
Significant marker-trait associations (MTAs) were identified using a mixed linear model (MLM) in TASSEL3.0, which simultaneously accounted for population structure and kinship. The population structure (summarized by the Q matrix) was inferred by the program STRUCTUTE v 2.2 and the kinship matrix (summarized by the K matrix) was calculated by software TASSEL3.0. The P value determined whether a QTL was associated with the marker or not. The R 2 was used to evaluate the magnitude of the MTA effects. SNPs with p ≤ 0.001 were considered to be significantly associated with individual traits.
Results
Phenotypic data
Phenotypic variation in TKW and related traits within the different environments are shown in Table S2. All of the evaluated traits exhibited wide variation across the association mapping populations in each of the environments. Broad sense heritability of the five traits ranged from 52.78 % for KDR to 72.02 % for KL, indicating that both genetic and environmental factors played roles in the expression of these measured traits. Both absolute values of skewness and kurtosis were less than 1.0, which indicates these traits are typical quantitative traits regulated by multiple loci, and phenotypic data were therefore suitable for GWAS analysis.
To pinpoint the relationships between TKW and related traits, correlation coefficients (r) among these traits were calculated based on environment (Table S3). Specifically, significant positive correlations were observed between TKW and KL, between TKW and KW, and between TKW and KT, whereas significant negative correlations were observed between TKW and KDR across all four environments. The highest correlation coefficients were detected between TKW and KW, while the lowest was between TKW and KDR. The results indicated that KW was most strongly correlated with TKW, followed by KT and KL.
Molecular markers and consensus genetic map
Among 81,587 SNP markers on the 90 K wheat chip, 38,381 SNP markers (47.04 %) were polymorphic in the association mapping population. When SNPs with detection rates less than 0.2 or a MAF less than 0.05 were removed, a total of 32,432 SNPs remained. Of these SNPs, 24,355 SNPs genetically mapped to the wheat consensus linkage map constructed by Wang et al. (2014) and were used for the subsequent study. The distribution of 24,355 SNPs on each chromosome are shown in Table S4. The total length of the map was 3674.16 cM, with a mean genetic distance of 0.15 cM between markers. Chromosome 1B contained the most markers (n = 2390), followed by Chromosome 5B (n = 2187) and 2B (n = 1977), while chromosome 4D had the fewest loci (n = 78). Among the A, B, and D genomes, the B genome contained the most loci (n = 12,321) across a total length of 1150.47 cM, followed by the A genome (n = 9523) with a total length of 1252.51 cM, and the D genome (n = 2511) with a length of 1271.18 cM. SNP density ranged from 0.48 cM−1 for chromosome 4D to 14.00 cM−1 for chromosome 6B.
Genetic diversity
Using 24,355 SNP markers, genetic diversity of the association mapping population was evaluated at the genome level (Table S5). Among the 205 accessions, 48,710 alleles were detected at the 24,355 marker loci, each with two alleles as expected. Approximately 60.33 % of SNPs (14,694/24,355) had an MAF more than 0.2, which were then selected as markers with normal allele frequencies, and 9.63 % of SNPs (2346/24,355) showed almost equal allele frequencies (with MAF close to 0.5) for two alternative alleles (Fig. S1). The SNP markers showed different levels of gene diversity, and values ranged from 0.024 to 0.500 with a mean of 0.337. The average PIC value was 0.277, ranging from 0.047 to 0.375 with a peak distribution between 0.35 and 0.38, and over 69 % of SNPs had a PIC value of 0.20–0.40. Among the A, B, and D genomes, the B genome had the highest PIC value and gene diversity, followed by genomes D and A.
Population structure analysis
A total of 3852 unlinked SNP markers were selected from 24,355 SNPs for population structure analysis. In the NJ cluster analysis, 205 accessions clustered into four groups (Fig. 1). Group 1 comprised 22 varieties (lines) and was dominated by the wheat varieties from Henan province. Group 2 consisted of 23 varieties (lines) and was mainly comprised of varieties from Hebei province. Group 3 was the largest group and included 109 varieties or lines, most of which were from Shandong province. Group 4 included 51 varieties (lines) most from three provinces (Shanxi, Jiangsu, and Ningxia provinces). The clustering analysis based on the genotypic data generally corresponded to known pedigrees, and the lines closely related in the pedigree usually did cluster together. For example, Lin4 (A53), which was derived from Lumai23 (A22), clustered with Lumai23 in group 3. Yannong19 (A32) and Yannong21 (A31), which were both derived from Shan 82-29, clustered together (in group 3). Furthermore, 73 wheat breeder’s lines were scattered across the four groups.

Neighbor-joining (NJ) tree for the association panel based on SNP marker
In the STRUCTURE analysis, the ΔK value was plotted against the number of hypothetical subgroups K, with the highest ΔK observed for K = 4 (Fig. S2). Accordingly, the 205 accessions were segregated into four subpopulations with the maximum membership probability (Fig. S3). Subpopulations 1–4 were respectively comprised of 43, 32, 105, and 24 varieties or lines, which was consistent with the NJ cluster analysis result.
Linkage disequilibrium analysis
Like the above population structure analysis, 3852 unlinked SNP markers were also used to evaluate the LD extent of the association mapping panel (Table S6). Across all 3852 loci, a total of 5784,214 locus pairs (including 144,177 linked locus pairs and 5640,037 unlinked locus pairs) were detected in the mapping population. Of these locus pairs, 355,184 (6.14 %) locus pairs were associated at the p < 0.001 level. Among linked locus pairs, 34,616 (24.01 %) of 144,177 locus pairs were in LD at the p < 0.001 level, with an average r 2 of 0.21 (ranging from 0.043 to1, or complete LD). Higher LD was observed in linked locus pairs than in unlinked locus pairs. Only 320,568 unlinked locus pairs (5.68 % of 5640,037) were in LD at the p < 0.001, and r 2 values varied from 0.044 to 1 with a mean of 0.11. Some differences were observed in the LD extent among the A, B, and D genomes. The B genome contained the largest percentage of significant markers (27.75 %), followed by the A genome (21.44 %) and the D genome (17.86 %).
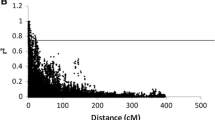
To reveal LD decay distances in the association mapping population, we generated LD decay scatter plots of syntenic r 2 versus genetic distance (cM) and estimated LD decay distances for the whole genome as well as the A, B, and D genomes (Fig. 2). A critical value of r 2, or basal LD, was calculated from inter-chromosomal LD analysis and was estimated to be 0.21 in this study. The point at which the LOESS curve intercepts the critical r 2 was determined as the average LD decay of the population. Based on these criteria, LD decay distance was about 2 cM for the whole genome, and approximately 1.5, 1.8, and 11 cM for the A, B, and D genomes, respectively (Table S6). Extensive variability in the magnitude of r 2 at a given genetic distance was detected, which reflects the wide local variation in the extent of LD extent across the genomes.

Linkage disequilibrium (LD, r 2) decay plot of significant r 2 values and genetic distance (cM) (P < 0.001) of locus pairs on A, B, D and whole genomes in the association panel. The inset provides a more detailed view of the LD decay characteristics over a 20 cM genetic distance; the horizontal line indicates the critical value of r 2
Marker–trait associations
A total of 271 MTAs were detected for TKW and the related traits across four environments. They were distributed across all wheat chromosomes except for 3D, and explained 5.49–9.86 % of the phenotypic variance (Table S7, S8). The highest number of MTA was recorded for KDR (86), followed by KL (65), KT (48), and KW (39), while the fewest MTA were observed for TKW (33). Of these MTAs, eleven markers showed highly significant associations with TKW and related traits (p < 0.0001), eight markers were detected in two or more environments, and twelve MTAs were multi-trait MTAs (Table 1, 2).
Sixty-five MTAs for KL were mapped to 9 of the 21 chromosomes and explained 5.55–9.78 % of the phenotypic variance. Of these MTAs, four MTAs on chromosomes 1B, 2B, and 3A showed highly significant associations with KL (p < 0.0001) explaining 8.04 % to 9.78 % of variation in the trait. Two MTAs (RAC875_c291_696 and RAC875_c291_647) on chromosome 1B were detected in two environments and accounted for 6.83–8.26 % of the phenotypic variance in each environment. One MTA (Ku_c9210_1059) on chromosome 2D showed up in three different environments explaining 6.18–6.34 % of the phenotypic variance, which suggests this MTA could be considered a stable marker for the trait. Moreover, a locus harboring ten MTAs for KL was detected on chromosomes 3A (91 cM), which can therefore be regarded as a strongly associated region for KL.
Thirty-nine MTAs for KW were mapped onto 17 of the 21 chromosomes and explained 5.56–8.18 % of the phenotypic variance. Of them, one MTA (Kukri_rep_c107605_164) on chromosome 6D showed a highly significant association with KW, explaining 8.18 % of trait variation.
Forty-eight MTAs for KT were identified on 12 of the 21 chromosomes, which explained 5.59–7.96 % of the phenotypic variance. Of them, two MTAs on chromosome 4B showed highly significant associations with KT (p < 0.0001) and explained 7.88 % and 7.96 % of trait variation, respectively. One stable marker (BS00110611_51) was detected in two environments on chromosomes 2B and explained 5.86 % and 6.38 % of trait variation in each environment. Furthermore, a strongly associated region harboring six MTAs for KT was noted on chromosome 1D (39 cM).
Eighty-six MTAs for KDR were identified on 17 of the 21 chromosomes, explained 5.49–9.79 % of the phenotypic variance. Of these MTAs, three highly significant association MTAs (p < 0.0001) were detected on chromosomes 1A, 2B, and 4B that explained 9.79, 8.65, and 8.24 % of trait variation, respectively. Three stable markers (Excalibur_c30234_130, RFL_Contig2765_1200, and RFL_Contig3175_749) were detected on chromosome 6A and explained 5.61–5.90 % of the phenotypic variance in each environment. Moreover, the strong association region harboring 21 MTAs for KDR was noted on chromosome 6A (137 cM).
Thirty-three MTAs for TKW were detected on 10 of the 21 chromosomes and accounted for 5.50 % to 9.86 % of the phenotypic variation. One highly significant MTA (p < 0.0001) on chromosome 6D was detected, and explained 7.79 % of the variation in TKW. One stable marker (BS00023893_51) on chromosome 6A was detected in three environments, which accounted for 7.32, 9.86, and 8.13 % of the phenotypic variances in each environment. Moreover, one strong association region harboring six MTAs was noted on chromosome 3A (88 cM).
Multi-trait MTAs for TKW and related traits were detected in this study. Their chromosome positions with their effect values are shown in Table 2. Five MTAs distributed across chromosomes 2B, 2D, 5A, and 5B showed a pleiotropic effect on KL and KDR. Three MTAs that mapped to chromosomes 4B and 7B were pleiotropic loci for KW and KDR. Similarly, TKW and KT had MTA in common on chromosome 1B, TKW and KW had common MTA on chromosome 2A, TKW and KL shared MTA on chromosome 3A, and TKW and KDR had overlapping MTA on chromosome 4B.
Discussion
Genetic diversity
Molecular markers have been used extensively to detect variability in wheat genotypes and to evaluate their genetic diversity (Hao et al. 2011). In this study, 24,355 SNP loci were used to estimate genetic diversity in a mapping panel at the genome level. The mean gene diversity and PIC were 0.340 and 0.277, respectively. The PIC values observed in our mapping population were higher than previous estimates of genetic diversity using SNP markers for a set of US cultivars (PIC = 0.23; Chao et al. 2009). The observed PIC values were similar to those of 287 advanced wheat accessions obtained from CIMMYT, in which the mean genetic diversity value of 0.34 and PIC value of 0.27 were obtained using 3848 SNP markers (Lopes et al. 2014). These results indicate that there was high genetic diversity in our mapping population, which approximately reflected the genetic diversity of Chinese winter wheat collections. Furthermore, over 69 % of the SNPs in our association mapping panel had PIC values of 0.20–0.40, which is regarded as the suitable PIC range for genetic research using molecular markers (Botstein et al. 1980). Our results also demonstrated that genetic diversity differs among the three wheat genomes; both the mean gene diversity and PIC value were higher in genome B than in genomes A and D, and similar trends were observed in previous studies (Chen et al. 2012).
Population structure and LD
Structural analysis conducted with two complementary methods suggested that the elite wheat varieties used in this work could be assigned to four subpopulations. This is consistent with the general views on the existence of genetic differences among wheat varieties or landraces (Sorrells and Yu 2009). Individuals were unequally distributed among the four subgroups, which indicated that there was a high extent of population structure in our mapping population. This may reflect the high number of bred varieties and breeder’s lines, which shared one or more founder parents in wheat breeding programs. Some elite lines are commonly used in many crosses and thus have a greater contribution to the population than other lines in breeding populations (Würschum et al. 2011). Population structure is particularly important in association mapping because it can cause spurious associations between markers and traits (Flint-Garcia et al. 2003; Pritchard et al. 2000). Numerous studies have proven that spurious MTAs could be reduced by treating the Q value (population structure) as a covariate during the marker-trait association analysis (Pritchard et al. 2000; Zhang et al. 2013). Consequently, to eliminate the spurious associations resulting from population structure, the association model MLM-Q-K in TASSEL3.0 was adopted in this study. Moreover, in the present study, the population structure analysis was used to obtain general knowledge of the biological background explaining the association mapping panel structure. In particularly, we were interested in the precise relatedness between lines included in the association mapping population, which is important not only for parental selection, but also for breeding system design.
The LD decay distance determines the marker density needed to effectively associate genotypes with traits, and influences the precision of association mapping (Cormier et al. 2014). Previous studies have reported LD decay distances ranging from 5 to 10 cM in various wheat collections using SSR markers (Breseghello and Sorrells 2006; Chen et al. 2012; Hao et al. 2011; Zhang et al. 2010). Recently, a few studies have revealed LD decay distances based on SNP markers. Based on a study of 287 elite spring wheat lines, the LD decay distance was approximately 5 cM in the D genome and 2 cM in the A and B genomes using 18,704 SNP markers (Sukumaran et al. 2014). In another study of 225 European elite varieties using 23,603 SNP markers, the mean LD decay distances for genomes A, B, and D were 0.52 cM, 0.70 cM, and 2.14 cM, respectively (Cormier et al. 2014). In our study, the LD decay distances were about 2 cM for the whole genome and approximately 1.5, 1.8, and 11 cM for the A, B, and D genomes, respectively. The above comparison shows that the LD decay distance was short in our mapping panel, which indicates the need for a higher marker density to locate genes. For the A and B genomes, the SNP density was sufficient with respect to LD decay. However, for the D genome, the lower SNP density may be compensated by the higher LD, but QTL detection is expected to be less precise. Furthermore, the average r 2 (0.21) values observed between linked locus pairs were higher than those of previous studies, e.g., a study of 94 elite common wheat varieties (r 2 = 0.05) (Hao et al. 2011) and a study of 90 Chinese winter wheat varieties (r 2 = 0.13) (Zhang et al. 2013). The higher r 2 observed in our study may be due to the fact that our mapping panel included many bred varieties and breeder’s lines. In plant breeding populations, LD is expected to be higher than in natural populations due to the selection of favorable genotypes and the shorter history of the germplasm (Reif et al. 2011). High r 2 values indicate that medium and small effect QTLs can be detected, whereas low r 2 values indicate that only QTLs with large effects can be detected (Würschum et al. 2011). MATs with medium and small effects were detected for TKW and related traits in our study, which suggests strong-QTL detection power and a high-mapping resolution.
Marker–trait associations
A fairly high number of significant loci were identified in this study, suggesting that MTAs with medium and small effects were detected for kernel weight-related traits. Therefore, we should focus on highly significant or stable MTAs and multi-trait MTAs for traits. A broad-scale comparison of the MTA results of our study and those of previous studies was performed using chromosome arms owing to differences in marker type and marker positions on different genetic maps.
We detected several MTAs for KL on chromosomes 1B, 2B, 2D, and 3A. Previous studies have reported QTLs for KL on chromosomes 1B, 2B, and 2D in different populations with SSR markers (Cui et al. 2015; Ramya et al. 2010). The marker Tdurum_contig4974_355, which was associated not only with KW, but also with KDR in the current study, was mapped to the short arm of chromosome 4B, similar to the QTL region for KW reported by Li et al. (2015). We detected the KT marker Tdurum_contig67399_676 on the short arm of chromosome 5A, which explained approximately 7.88 % of the trait variation. Li et al. (2015) also detected a QTL with significant effects on KT on chromosome 5A.
For TKW, we detected important MTAs on chromosomes 1B, 2A, 3A, 4B, 6A, and 6D. The markers on chromosomes 1B, 2A, and 3A are in pleiotropic regions affecting kernel weight-related traits. Zhang et al. (2015) also detected QTLs for TKW on chromosomes 1B, 2A, and 3A using both unconditional and conditional mapping analyses. The marker wsnp_BQ161779D_Ta_2_1 on chromosome 6D, a highly significant MTA in present study, was detected at similar location on the same chromosome by Wang et al. (2012). The marker BS00023893_51 on chromosome 6A, detected in two environments, was a stable MTA. Previous studies have reported major QTL for TKW on chromosome 6A (Huang et al. 2006; Sun et al. 2009; Zhang et al. 2014). Notably, this MTA was located near TaGW2, an important gene associated with TKW and KW located on chromosome 6A (Su et al. 2011). These results indicated that the region near TaGW2 on chromosome 6A may harbor a robust QTL cluster for kernel-related traits, and should be a focus of breeding programs. Interestingly, we also detected a number of MTAs for kernel weight-related traits that have not been described to date, some of which are major loci that explain more than 9 % of the observed genotypic variance (Table 1). These results show that the association mapping approach used in this study is a powerful tool to detect previously unknown QTL.
We also detected multi-trait MTAs. Two markers (Tdurum_contig4974_355 and CAP12_c4704_232) that were mapped to the short arm of chromosome 4B and were separated by approximately 14 cM were pleiotropic for KW and KDR and for TKW and KDR, respectively. Previous studies have demonstrated pleiotropic effects of QTLs on chromosome 4B for TKW and kernel size (Huang et al. 2006; Gegas et al. 2010; Sun et al. 2009). The marker wsnp_BE445431A_Td_2_1 on chromosome 2A was associated with TKW and TK in the present study. Patil et al. (2013) and Peng et al. (2003) reported multi-trait QTL on chromosome 2A for grain yield, TKW, KW, spikelet per spike, and kernels per spike in tetraploid wheat. Furthermore, multi-trait MTA for TKW and KT on chromosome 1B and multi-trait MTA for KL and KDR on chromosome 2B were observed in the same genomic region reported in previous studies (Patil et al. 2013; Snape et al. 2007; Zheng et al. 2010). These results confirmed that the QTLs identified in this region on chromosomes 1B, 2A, 2B, and 4B have pleiotropic effects and are highly likely also play key roles in determining wheat grain yield, and also partially explained the genotypic correlations of TKW and related traits, consistent with the results of a correlation analysis in this study.
SNPs can be intergenic or intragenic, accordingly, SNPs within a gene can directly affect protein structure or gene expression. Gene-based markers can be directly associated with genetic differences in a panel of diverse lines used in LD-based association mapping (Lu et al. 2009). Among SNP markers detected in the current study, the highly significant or stable MTAs and multi-trait MTAs, such as the four highly significant markers for KL and the stable MTA for TKW, can be used to develop cleaved amplified polymorphic sequence markers for marker-assisted selection and to identify kernel-related candidate genes via bioinformatics analyses, such as multiple sequence alignments and gene annotations.
In conclusion, we estimated the genetic diversity, population structure, and LD decay rate in a winter wheat association mapping panel. The SNP diversity was higher and LD decay distance was shorter than those of other wheat collections. Based on a GWAS, we detected 271 MTAs for TKW and related traits in four environments. The highly significant or stable MTAs and multi-trait MTAs can be used to develop cleaved amplified polymorphic sequence markers for molecular marker-assisted breeding. These results indicate that the association mapping panel with 24,355 SNPs is robust for association mapping for TKW and related traits.
References
Akhunov E, Nicolet C, Dvorak J (2009) Single nucleotide polymorphism genotyping in polyploid wheat with the Illumina GoldenGate assay. Theor Appl Genet 119:507–517
Al-Maskri AH, Sajjad M, Khan SH (2012) Association mapping: a step forward to discovering new alleles for crop improvement. Int J Agric Biol 14:1537–1546
Botstein D, Wlllte RL, Skolinck M (1980) Construction of a genetic linkage map in man using restriction fragment length polymorphisms. Am J Hum Genet 32:314–319
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2637–2645
Breseghello F, Sorrells ME (2006) Association mapping of kernel size and milling quality in wheat (Triticum aestivum L.) cultivars. Genetics 172:1165–1177
Buckler ES IV, Thornsberry JM (2002) Plant molecular diversity and applications to genomics. Curr Opin Plant Biol 5:107–111
Chao S, Zhang W, Akhunov E, Sherman J, Ma Y (2009) Analysis of gene-derived SNP marker polymorphism in US wheat (Triticum aestivum L.) cultivars. Mol Breed 23:23–33
Chen XJ, Min DH, Tauqeer AY, Hu YG (2012) Genetic diversity, population structure and linkage disequilibrium in elite Chinese winter wheat investigated with SSR Markers. PLoS ONE 7:e44510
Colasuonno P, Gadaleta A, Giancaspro A, Nigro D, Giove S, Incerti O, Mangini G, Signorile A, Simeone R, Blanco A (2014) Development of a high-density SNP-based linkage map and detection of yellow pigment content QTLs in durum wheat. Mol Breed 34:1563–1578
Cormier F, Gouis JL, Dubreuil P, Lafarge S, Praud S (2014) A genome-wide identification of chromosomal regions determining nitrogen use efficiency components in wheat (Triticum aestivum L.). Theor Appl Genet 127:2679–2693
Cui F, Ding A, Li J, Zhao C, Li X, Feng D, Wang X, Wang L, Gao J, Wang H (2011) Wheat kernel dimensions: how do they contribute to kernel weight at an individual QTL? J Genet 90:409–425
Cui F, Fan X, Chen M, Zhang N, Zhao C, Zhang W, Han J, Ji J, Zhao X, Yang L, Wang T, Li J (2015) QTL detection for wheat kernel size and quality and the responses of these traits to low nitrogen stress. Theor Appl Genet. doi:10.1007/s00122-015-2641-7
Eric M, James JM, Frank AM, Benjamin N, Stephen VF (2011) Genome-wide association study of blood pressure response to methylphenidate treatment of attention-deficit/hyperactivity disorder. Prog Neuro-Psychopharmacol Biol Psychiatr 35:4667–4672
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Flint-Garcia SA, Thornsberry JM, Buckler ES (2003) Structure of linkage disequilibrium in plants. Annu Rev Plant Biol 54:357–374
Gegas VC, Nazari A, Griffiths S, Simmonds J, Fish L, Orford S, Sayers L, Doonan JH, Snape JW (2010) A genetic framework for grain size and shape variation in wheat. Plant Cell 22:1046–1056
Gupta PK, Mir RR, Mohan A, Kumar J (2008) Wheat genomics: present status and future prospects. Int J Plant Genom 896451
Hao CY, Wang LF, Ge HM, Dong YC, Zhang XY (2011) Genetic diversity and linkage disequilibrium in chinese bread wheat (Triticum aestivum L.) revealed by SSR markers. PLoS ONE 6:1–13
Huang XQ, Cloutier S, Lycar L, Radovanovic N, Humphreys DG, Noll JS, Somers DJ, Brown PD (2006) Molecular detection of QTLs for agronomic and quality traits in adoubled haploid population derived from two Canadian wheats (Triticum aestivum L.). Theor Appl Genet 113:753–766
Huang XH, Wei XH, Sang T, Zhao Q, Feng Q, Zhao Y (2010) Genome-wide association studies of 14 agronomic traits in rice landraces. Nat Gene 42:961–967
Li QF, Zhang Y, Liu TT, Wang FF, Liu K, Chen JS, Tian JC (2015) Genetic analysis of kernel weight and kernel size in wheat (Triticum aestivum L.) using unconditional and conditional QTL mapping. Mol Breed 35:194
Liu K, Muse SV (2005) Power Marker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21:2128–2129
Lopes MS, Dreisigacker S, Peña RJ, Sukumaran S, Reynolds MP (2014) Genetic characterization of the wheat association mapping initiative (WAMI) panel for dissection of complex traits in spring wheat. Theor Appl Genet. doi:10.1007/s00122-014-2444-2
Lu YL, Yan JB, Claudia T, Guimaraes Suketoshi T, Hao ZF (2009) Molecular characterization of global maize breeding germplasm based on genome-wide single nucleotide polymorphisms. Theor Appl Genet 120:93–115
Nyquist WE, Nyquist RJ, Baker RJ (1991) Estimation of heritability and prediction of selection response in plant populations. Crit Rev Plant Sci 10:235–254
Patil RM, Tamhankar SA, Oak MD, Raut AL, Honrao BK, Rao VS, Misra SC (2013) Mapping of QTL for agronomic traits and kernel characters in durum wheat (Triticum durum Desf.). Euphytica 190:117–129
Peng J, Ronin Y, Fahima T, Röder MS, Li Y, Nevo E, Korol A (2003) Domestication quantitative trait loci in Triticum dicoccoides, the progenitor of wheat. Proc Natl Acad Sci USA 100:2489–2494
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure from multilocus genotype data. Genetics 155:945–959
Ramya P, Chaubal A, Kulkarni K, Gupta L, Kadoo N, Dhaliwal HS, Chhuneja P, Lagu M, Gupt P (2010) QTL mapping of 1000-kernel weight, kernel length, and kernel width in bread wheat (Triticum aestivum L.). J Appl Genet 51:421–429
Reif JC, Maurer HP, Korzun V, Ebmeyer E, Miedaner T, Würschum T (2011) Mapping QTLs with main and epistatic effects underlying grain yield and heading time in soft winter wheat. Theor Appl Genet. doi:10.1007/s00122-011-1583-y
Risch N, Merikangas K (1996) The future of genetic studies of complex human diseases. Science 80:1516–1517
Sajjad M, Khan SH, Kazi AM (2012) The low down on association mapping in hexaploid wheat (Triticum aestivum L.). J. Crop Sci. Biotech. (September) 15:147–158
Snape JW, Foulkes MJ, Simmonds J, Leverington M, Fish LJ, Wang Y, Ciavarrella M (2007) Dissecting gene 9 environmental effects on wheat yields via QTL and physiological analysis. Euphytica 154:401–408
Sorrells ME, Yu J (2009) Linkage disequilibrium and association mapping in the Triticeae. In: Muehlbaucer GJ (ed) Genetics and Genomics of the Triticeae Feuillet C. Springer, Gatersleben, pp 655–683
Sukumaran S, Yu J (2014) Association mapping of genetic resources: achievements and future perspectives. Genomics of plant genetic resources. Springer, Netherlands, pp 207–235
Sukumaran S, Dreisigacker S, Lopes M, Chavez P, Reynolds MP (2014) Genome wide association study for grain yield and related traits in an elite spring wheat population grown in temperate irrigated environments. Theor Appl Genet. doi:10.1007/s00122-014-2435-3
Sun XY, Wu K, Zhao Y, Kong FM, Han GZ, Jiang HM, Huang XJ, Li RJ, Wang HG, Li SS (2009) QTL analysis of kernel shape and weight using recombinant inbred lines in wheat. Euphytica 165:615–624
Tamura K, Dudley J, Nei M, Kumar S (2007) MEGA4: molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596–1599
Tommasini L, Schnurbusch T, Fossati D, Mascher F, Keller B (2007) Association mapping of Stagonospora nodorum blotch resistance in modern European winter wheat varieties. Theor Appl Genet 115:6977–6995
Tsilo TJ, Hareland GA, Simsek S, Chao S, Anderson JA (2010) Genome mapping of kernel characteristics in hard red spring wheat breeding lines. Theor Appl Genet 121:717–730
Wang L, Ge H, Hao C, Dong Y, Zhang X (2012) Identifying loci influencing 1,000-kernel weight in wheat by microsatellite screening for evidence of selection during breeding. PLoS ONE 7(2)
Wang SC, Wong D, Forrest K, Allen A, Chao S, Huang BE, Maccaferri M, Salvi S, Milner SG, Cattivelli L, Mastrangelo AM, Whan A, Stephen S, Barker G, Wieseke R, Plieske J, Lillemo M, Mather D, Appels R, Dolferus R, Guedira GB, Korol A, Akhunova AR, Feuillet C, Salse J, Morgante M, Pozniak C, Luo MC, Dvorak J, Morell M, Dubcovsky J, Ganal M, Tuberosa R, Lawley C, Mikoulitch I, Cavanagh C, Edwards KJ, Hayden M, Akhunov E (2014) Characterization of polyploid wheat genomic diversity using a high-density 90000 single nucleotide polymorphism array. Plant Biotechnol J 12:787–796
Wilson LM, Whitt SR, Ibanez AM, Rocheford TR, Goodman MM, Buckler ES (2004) Dissection of maize kernel composition and starch production by candidate gene associations. Plant Cell 16:2719–2733
Würschum T, Maurer HP, Kraft T, Janssen G, Nilsson C, Reif JC (2011) Genome-wide association mapping of agronomic traits in sugar beet. Theor Appl Genet 123:1121–1131
Yao J, Wang L, Liu L, Zhao C, Zheng Y (2009) Association mapping of agronomic traits on chromosome 2A of wheat. Genetica 137:677–687
Zanke CD, Ling J, Plieske J, Kollers S, Ebmeyer E, Korzun V, Argillier O, Stiewe G, Hinze M, Neumann F, Eichhorn A, Polley A, Jaenecke C, Ganal MW, Röder MS (2015) Analysis of main effect QTL for thousand grain weight in European winter wheat (Triticum aestivum L.) by genome-wide association mapping. Frontiers. Plant Sci 6:644
Zhang D, Bai G, Zhu C, Yu J, Carver BF (2010) Genetic diversity, population structure, linkage disequilibrium in U.S. elite winter wheat. Plant Genome 3:1177–1186
Zhang KP, Wang JJ, Zhang LY, Rong CW, Zhao FW, Peng T (2013) Association Analysis of Genomic Loci Important for Grain Weight Control in Elite Common Wheat Varieties Cultivated with Variable Water and Fertiliser Supply. PLoS ONE 8:e57853
Zhang XY, Deng ZY, Wang YR, Li JF, Tian JC (2014) Unconditional and conditional QTL analysis of kernel weight related traits in wheat (Triticum aestivum L.) in multiple genetic backgrounds. Genetica 142:371–379
Zheng BS, Le GJ, Leflon M, Rong WY, Laperche A, Brancourt HM (2010) Using probe genotypes to dissect QTL × environment interactions for grain yield components in winter wheat. Theor Appl Genet 121:1501–1517
Acknowledgments
This research was supported by the Shandong Provincial Agriculture Liangzhong Project Foundation of China (2014 No. 96) and National Natural Science Foundation of China (No. 31171554 and 31301315).
Author information
Authors and Affiliations
Corresponding author
Additional information
Guangfeng Chen and Han Zhang contributed equally to this study.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Chen, G., Zhang, H., Deng, Z. et al. Genome-wide association study for kernel weight-related traits using SNPs in a Chinese winter wheat population. Euphytica 212, 173–185 (2016). https://doi.org/10.1007/s10681-016-1750-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10681-016-1750-y