
Abstract
Neuroinflammation is one of the host defensive mechanisms through which the nervous system protects itself from pathogenic and or infectious insults. Moreover, neuroinflammation occurs as one of the most common pathological outcomes in various neurological disorders, makes it the promising target. The present review focuses on elaborating the recent advancement in understanding molecular mechanisms of neuroinflammation and its role in the etiopathogenesis of various neurological disorders, especially Alzheimer’s disease (AD), Parkinson’s disease (PD), and Epilepsy. Furthermore, the current status of anti-inflammatory agents in neurological diseases has been summarized in light of different preclinical and clinical studies. Finally, possible limitations and future directions for the effective use of anti-inflammatory agents in neurological disorders have been discussed.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Developing countries (like India) are going through a phase of epidemiological transition with a higher socio-economic burden of non-communicable disease. Among various non-communicable diseases, neurological disorders have been considered significant causes of mortality and morbidity, not only in India but also worldwide (Feigin et al. 2020). Owing to population overgrowth and aging, the absolute number of disabilities and deaths due to neurological diseases is piling up, suggesting prevention and management of primary neurological conditions are ineffective. The major reason could be a lack of clear understanding of the etiopathogenesis of these neurological conditions.
The neuroinflammatory cascade has been identified as a common etiopathogenic factor in different neurological disorders, including stroke, Alzheimer’s disease (AD), Parkinson’s disease (PD), Amyotrophic Lateral Sclerosis (ALS), Huntington’s disease (HD), migraine, epilepsy, Multiple Sclerosis (MS), ischemic/traumatic brain injury (TBI), spinal cord injury (SCI), and depression. Virtually, neuroinflammation appears to be involved in most neurological and neurodegenerative diseases and poses a common thread that connects their pathologies (Gilhus and Deuschl 2019; Brambilla 2019). The understanding of neuroinflammation is continuously under evolution; therefore, intermittent analysis of the current information is required.
The process of neuroinflammation originated by synchronized neural/non-neural cells, aiming to restore neuronal homeostasis and protect neuronal integrity (Degan et al. 2018). In the acute phase, such inflammatory response poses a protective effect by maintaining neuronal homeostasis, contributing to neurogenesis, repair, and clearance of protein plaques and damaged cells (Russo and McGavern 2016; Shabab et al. 2017). However, during chronic phase, the maladaptive outcome associated with neuroinflammation leads to worsening neuronal damage.
This review discusses the interrelationships between neuroinflammation and pathologies of several neurological disorders, mainly AD, PD, and epilepsy. We specifically focus on cellular and molecular events as significant players in neuroinflammation, including resident cells, immune cells, pro-inflammatory cytokines, inflammasomes, high mobility group box protein-1 (HMGB1), oxidative stress, mitochondrial dysfunction, neurotransmitters, and ion channel. Furthermore, modulation of neuroinflammation as a plausible therapeutic approach in neurological disorders has been summarized in light of various preclinical and clinical studies.
Molecular mechanism of neuroinflammation
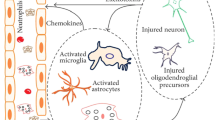
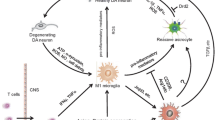
Neuroinflammation may be triggered by different biological mechanisms, including glial reactions and oxidative stress or brain injuries (stroke, trauma, chronic infection, etc.). Basic research to understand the mechanism of neuroinflammation is over-pouring; however, exact pathogenesis remains unclear. The current information on neuroinflammation and its role in different neurological disorders has been illustrated in Figs. 1 and 2.

Role of microglia, cytokines, inflammasome, mitochondrial dysfunction, in neuroinflammation and development of neurological diseases

Role of proinflammatory cytokines, HMGB1, DAMPs/PAMPs in neuroinflammation and associated neuropathological outcome
Glial cells in neuroinflammation
The CNS microenvironment is closely monitored by ramified microglia by sensing death/damage signals and proliferating microglia at the injury site. Microglial cells are very dynamic and play an essential role in maintaining neuronal homeostasis, neuronal growth, pruning of excess synapses to maintain optimal neuronal plasticity, and clearance of cellular debris, protein aggregates, several pathogens/antigens, and neural plasticity (Katsumoto et al. 2014). Under the influence of exogenous and endogenous factors (pathogen-associated molecular patterns (PAMP), and damage-associated molecular patterns (DAMP)) pattern recognition receptors (PPRs) on microglia may recognize pathogens, protein aggregates, or cellular debris, and lead to microglial activation (Leng and Edison 2021). During acute neuroinflammation, microglial cells get rapidly activated. Microglia activation leads to phagocytosis of pathogenic species and releases cytokines, chemokines, reactive oxygen/nitrogen species (ROS/RNS), and prostaglandins. Over time, progression to chronic inflammation causes degeneration of neurons and disruption of blood–brain barrier (BBB) possibly via over production of ROS/RNS and recruitment of peripheral immune cells to initiate cellular damage (Garden 2013). Generally, this process usually remains active until the immune response is eliminated.
In normal condition, microglia remains in a quiescent/resting state with ramified morphology and perform surveillance function in the CNS (Nimmerjahn et al. 2005; Davalos et al. 2005). However, under pathological conditions, dramatic morphological change (amoebic shape) activates the microglia. The activated microglia expresses various surface molecules (like Fc receptor, cluster of differentiation (CD-11b, CD-11c, CD-14), major histocompatibility complex (MHC), toll-like receptor (TLR), scavenger receptor, cytokine, and chemokine receptors), functions as antigen‐presenting cells (APC) and attracts various immunological cells (Rock et al. 2004). Thus, activated innate immunity exerts the neuroprotective effect of microglia via maintaining homeostasis, repair, neuroregeneration and, clearance of toxic substances. At first instance, activation of TLR and scavenger receptors dampens neurotoxicity by sequestering abnormal protein aggregates. At the same time, their downstream signaling (including cytokines/chemokines, excitatory amino acids, nucleic acids, ROS, and several proteases) activates the microglial neurotoxic effects. Overexpression of TLR/scavenger receptors has been reported in several neurological disorders (Cho et al. 2005; Carpentier et al. 2008; Tiwari et al. 2019). Thus, whether activated microglia would elicit a neuroprotective/neurotoxic effect depends on the microenvironment, type, and magnitude of stimuli (Nakanishi and Wu 2009; Sawada 2009).
The assessment of functional phenotype, M1/M2 dichotomy of microglia, was devised for a long, indicating M1 as pro-inflammatory phenotype while M2 as anti-inflammatory phenotype (Mantovani et al. 2002; Henkel et al. 2009). However, the experimental evidence contradicts such a microglia dichotomy. Still, these phenotypic dichotomy is widely used to refer M1 as neurotoxic and M2 as neuroprotective under certain pathological conditions (Ransohoff 2016). The activation of microglia depends on the type of stimuli received. Several experimental evidence has suggested, lipopolysaccharide (LPS) or interferon (IFN) γ may induce activation of detrimental M1 phenotype while IL-4/IL-13 might lead to induction of protective M2 phenotype (Loane and Kumar 2016).
During aging, sustained activation of microglia results in functional impairment and contributes to the emergence of neurodegenerative diseases. Various experimental evidence supports the chronic activation of microglia as a standard pathological marker of neurodegenerative disorders. In the early stage of neurodegeneration, disease-related proteins (Aβ or α-syn) serves as DAMPs and activates pattern recognition receptors. In contrast, soluble oligomers activate several other microglia receptors (CD-47, CD-14, CD-36, α6β1 integrin, TLR) (Fassbender et al. 2004; Tiwari et al. 2019). Activation of such receptors switches microglia from quiescent to the active state which proliferate near extracellular plaque and restrict its further growth (Edwards 2019). Therefore, in the early stage, microglial activation serves a neuro-supportive function by releasing several proteases for clearance of Aβ plaque (Jimenez et al. 2008).
Further, the M2 phenotype helps in phagocytosis of Aβ plaques and creates a physical barrier to prevent further spreading of plaque (Condello et al. 2015). Indeed, acute microglia activation appears neuroprotective, but the sustained chronic microglial activation exerts detrimental changes. Such microglial dysfunction results in the overproduction of pro-inflammatory cytokines leading to polarization of microglia towards M1. Furthermore, owing to M1 polarization, the accumulation of Aβ plaques leads to a neurotoxic effect and causes synaptic loss (Piccioni et al. 2021).
The inflammatory cytokines released from activated microglia may have a bit absolute neurotoxic effect, including most deleterious inflammatory cytokines (TNF-α and INF-γ). In contrast, their downstream signaling involves activation of MAPK and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) and exerts neuroprotection (Kamata et al. 2005). Indirectly, these mediators stimulate the glutamate release from activated microglia, resulting in neuronal damage via excitotoxicity (Barger and Basile 2001; Takeuchi et al. 2006, 2008). Such elevation in glutamate level might result from astrocytic glutamate transporters inhibition (critical for extracellular glutamate removal) and its potent neurotoxic effects (Liang et al. 2008; Takaki et al. 2012).
Thus, inhibition of microglial activation and blockade of glutamate signaling have been evaluated for their neuroprotective effect in several neurological disorders. However, such a hypothesis may have some potential side effects which need to be addressed simultaneously (Parsons et al. 2007). Overall, under pathological conditions, harmful impacts of microglia contribute to the development of various neurological disorders. Thus, a better understanding of the precise mechanism of neurotoxic behavior of microglia would support a desirable therapeutic approach inhibiting deleterious effects of microglia without affecting its neuroprotective effect for neurological disorders.
T cells and monocytes in neuroinflammation
The activated microglia and infiltrating T cells lead to Neuroinflammation (Denney et al. 2012). Neuroinflammation does get affected by stimulated testosterone T cells (Massa et al. 2017). The endogenous immune-suppressors are CD4+, Foxp3, and CD25 regulatory T cells (Tregs), suppresses T cell proliferation, and pro-inflammatory cytokine (Reynolds et al. 2010). IL-21 and IL-9 are produced by activation of stimulated CD4+ T cells, and it was significantly increasing in AD. The pro-inflammatory cytokines (IL-6, IL-21, IL-23) are involved in the differentiation of Th-17 cells and results in Neuroinflammation (Reynolds et al. 2010). Local administration of IL-17 was reported to enhance dendritic cells (activated) in the experimental model (Niranjan et al. 2016). Further research is required to establish therapeutic potential based on T cells in neuroinflammatory diseases.
According to recent research, monocyte plays crucial role in intitation of neuroinflammation (Jones et al. 2018). In experimental model of PD reported infiltration of monocytes from periphery to CNS results in neurodegeneration (Harms et al. 2018). In contrast, depletion of monocyte results in exacerbation of neurodegeneration in MPTP model of PD suggesting neuroprotective role of monocytes (Cote et al. 2015; Wattananit et al. 2016). Further, CXCL12, a chemokine, causes monocyte mediated stimulation of endothelial cells facilitating lymphocyte transmigration across BBB and deployment of immune cells towards brain (Schmitt et al. 2012). Monocytes contribute to phagocytosis of faulty proteins and debris in brain. The mediator of inflammation, like saturated fatty acids, causes toxicity towards neuronal cells of human monocytes. Infiltrating monocytes do not contribute to the resident microglia pool, but they trigger experimental autoimmune encephalitis progression (Ajami et al. 2011). The constitutive function of macrophages and monocytes in autoimmune neuroinflammation may be defined by activation of NF-κB (Karlmark et al. 2012).
Cytokines in neuroinflammation
Cytokines are a considerably more prominent, heterogeneous group of glycoproteins released from microglia, astrocytes, macrophages, monocytes, etc., and work in an autocrine and paracrine manner. Any infection, trauma, or ischaemic brain damage commences cellular responses that hold various hallmarks of neuroinflammation and are majorly responsible for neuronal repair and restoration of homeostasis (Alyu and Dikmen 2016). In such conditions, activation of glial cells, typically mediated via astrocytes and microglia, embrace enhanced production of an array of cytokines and chemokines.
Generally, cytokines mediate signaling is of two types: pro-inflammatory (IL-1β, IL-6, IFN-γ, and TNF-α) and anti-inflammatory cytokines (IL-4, IL-10, IL-13), which either facilitate or inhibit inflammatory responses. The equilibrium between these cytokines eliminates the underlying cause in acute phase, while the chronic release of pro-inflammatory cytokines appears as a critical pathogenic mechanism of various neurological diseases. In the acute stage, activation of astrocytes has been shown to integrate the signals from several cytokines (IL-4, IL-10, IL-13) and chemokines, including interferon-γ (IFN-γ), interleukin‑17 (IL‑17), IL‑1β, and CC-chemokine ligand 2 (CCL2). Furthermore, administration of pro-inflammatory cytokine has been reported to induce depression-like behavior, while antidepressant treatment has been found to reduce cytokine production (Himmerich et al. 2019).
Neuroinflammation is mainly triggered by blood-borne leukocytes invading CNS. Thus infiltrated leukocytes enhance the production of various cytokines, which further recruits more leukocytes and aggravates neuroinflammation. Cytokines produced by microglia and astrocyte activation, like- IL-1β, IL-6, TNF, IL-18, IL-12, IL-23, and IL-33, induce neuroinflammation and neurodegeneration (Becher et al. 2017).
APCs in secondary lymphoid tissues contain IL-12 and IL-23, which polarize differentiating T cells against different effecter function phenotypes. Initially, IL-12 was considered critical in the development of MS (Segal and Shevach 1996). Later role of IL-23 is recorded in migrating secondary lymphoid tissues in Neuroinflammation (Becher et al. 2002). After the pathogenic invasion of TH cells, the resident microglia release IL-23, which initiates Neuroinflammation (Li et al. 2007). Thus, IL-23 has emerged as a critical player in generating pathogenic TH cell phenotypes. There are both the genesis and maintenance of pathogenic TH cells, which function in both the peripheral immune compartment and the CNS. Both IL-1 and IL-6 appear to play critical peripheral roles in promoting pathogenic activity in TH cells capable of causing CNS inflammation. IL-6 is another crucial cytokine essential for the progression of neuroinflammation by inhibiting FoxP3 expression in differentiating TH cells (Eugster et al. 1998; Sonderegger et al. 2008). On the other hand, IL-6 induces the expression of IL-1 receptors on TH cells which helps them recognizing local IL-1 production in CNS (Chung et al. 2009).
Inflammasome in neuroinflammation
Inflammasomes are intracellular multi-protein complexes that recognize harmful substances (microbial pathogens or host-derived harmful substances) and activate inflammatory cascade and immune response against them. Upon detecting these dangerous signals, they induce host immune mechanisms by secreting inflammatory mediators like IL-1β and IL-18. They also induce pyroptosis (alytic cell death process), resulting in release of inflammatory mediators, thus potentiating the inflammasomes induced inflammatory response by many folds. Neuroinflammation and neurodegeneration are mostly a result of the deposition of aggregated host proteins like amyloid-β (Aβ), α-synuclein, and prions (Qi et al. 2018). These aggregations activate inflammasomes which in turn triggers neuroinflammation and neurodegeneration (Holbrook et al. 2021; Saresella et al. 2016).
Recognition of harmful pathogens and host-derived aggregation is mainly mediated by PRRs that sense PAMPs and host or environment-derived DAMPs (Lampron et al. 2013). PRRs can be of two types: membrane-bound PRRs (detects extracellular signals) and intracellular PRRs like- NLR and ALR families of receptors (Walsh et al. 2014). Detection of priming signals by these DAMPs and PAMPs and the detection of toxins and harmful chemicals by NLR and ALR activate the formation of various proteins that finally aggregate and form inflammasomes. Inflammasomes activate caspase-1 and induce secretion of pro-IL-1β, pro-IL-18, and gasdermin D. Caspase-1 cleaves them and produce IL-1β, IL-18, and gasdermin D N-terminal domain, which further activates the pyroptosis process (Fig. 1) (Lamkanfi and Dixit 2014; Shi et al. 2017). IL-1β and IL-18 have a vital role in CNS infections, cell death, brain injury, and neurodegenerative disease. Higher concentrations of IL-1β, IL-18 are observed in various neurodegenerative disorders (Broz and Dixit 2016).
Neuroinflammation-associated neuronal degeneration is a significant event in many neuronal disorders like- AD, PD, HD, TBI, SCI, etc. There is an increased expression of inflammasome markers like apoptosis-related speck-like protein containing a CARD (ASC), caspase 1, NLRP1, and NLRP3 levels in the CSF of patients (Wallisch et al. 2017). There is a steep increase in IL-1β levels initially during the disease development, whereas IL-18 levels gradually increased over several days. Thus, suggesting possible involvement of IL- 1β at early stages, while IL-18 might be involved in later stages of disease progression (Yatsiv et al. 2002). In spinal cord injury, higher levels of NLRP1, ASC, caspase1, IL-1β, and IL-18 are responsible for inflammasome activation and disease progression (de Rivero Vaccari et al. 2008).
The deposition of misfolded Aβ plaque is the main reason for AD development. Along with this, high expression of IL-1β in the microglia cells surrounding Aβ plaques was observed in AD patients (Simard et al. 2006). Also, high levels of inflammasome markers such as NLRP3, ASC-1, NLRC4, caspase 1, caspase 5, and cytokines IL-1β, IL-18 were reported in AD patients (Saresella et al. 2016). ALS, another fatal neurodegenerative disease, is associated with inflammasomes activation. NLRP3, ASC-1, NLRC4, caspase 1, and IL-18 levels increased in ALS patients (Gugliandolo et al. 2018). The pathology of this disease was proven to be driven by IL-18 or by DAMPs via pyroptosis (Maier et al. 2015). Higher expression of IL- 1β and NLRP3 is also associated with Parkinson’s disease. The pathology of PD is driven by progressive death of dopaminergic neurons in substantia nigra induced by IL- 1β and NLRP3 inflammasome activation (Ferrari et al. 2006; Mouton-Liger et al. 2018). HD has also been associated with higher expression of inflammasome, while treatment with caspase 1/3 inhibitor (tetracycline-derived minocycline) showed a delay in HD patients' disease progression (Voet et al. 2019).
HMGB1 proteins in neuroinflammation
HMGB1, a DAMP family protein, is a highly abundant protein released upon inflammasome activation by glial cells and neurons (Ravizza et al. 2018). They activate receptors for advanced glycine and end products (RAGE) and TLR4 on target cells. Thus, the HMGB1/TLR4/RAGE signaling axis plays an essential role in initiating neuroinflammation (Paudel et al. 2018).
During brain injury, HMGB1 acts as an inflammatory cytokine in case of brain injuries (Richard et al. 2017; Aucott et al. 2018). DAMPs influence synaptic organization and function within the hippocampus and might initiate epileptogenesis (Ravizza et al. 2018). It has been reported that immediately after neuronal injury, the amount of HMGB1 in the neuronal extracellular space gets increased several folds (Scaffidi et al. 2002).
HMGB1 may interact with numerous receptors (RAGE, TLR2/4/9, integrin, a-synuclein filaments, proteoglycans, T-cell immunoglobulin, TIM-3, TREM1, CD-24, etc.), present on extracellular domain (Kang et al. 2014). Among these receptors, RAGE and TLR4 play the leading role in the pathology of neuronal diseases via initiation of NF-κB mediated inflammatory cascade (Bianchi and Manfredi 2007). The role of HMGB1 within the growth and ailment of CNS has been well explained (Fang et al. 2012). At the site of neuronal injury, the released HMGB1 may get partially oxidized to give disulfide form of HMGB1 which further activate TLR/NF-κB mediated inflammatory cascade and generates free radicals leading to BBB damage and functional impairments (Gu et al. 2014; Paudel et al. 2018). Thus, HMGB1/TLR4/RAGE/NF-κB cell signaling activation causes neuroinflammation, hyperexcitability in neurons, development of seizures and neurodegeneration.
Mitochondrial dysfunction and oxidative stress in neuroinflammation
Mitochondria is a vital cell organelle that produces the energy required for almost all types of biological functions. Mitochondrial dysfunction can cause energy imbalances inside the cell, which leads to inflammation and cell death. There are several factors which induce mitochondrial damage.
The redox imbalance is one of the main reasons for mitochondrial damage. Oxidative stress refers to an imbalance favoring the development of ROS over antioxidant defenses factors. The generation of ROS (superoxide anions and hydrogen peroxide) is a result of electron transport system via mitochondrial oxidative phosphorylation (OXPHOS) complexes (H2O2). ROS are essential mediator of redox-sensitive signaling pathways, and may lead to oxidative damage to protein, lipids, and nucleic acids (West and Shadel 2017). Complex I/III of the OXPHOS chain are the most favourable sites of mitochondrial ROS processing (Chrysostomou et al. 2013). Oxidative stress is caused by interruption in one or more fuctions of mitochondria leading to increased mitochondrial membrane permeability resuting in lipid peroxidation, apoptosis and neuronal death (Kanamori et al. 2010; Karumuri et al. 2019; Quamar et al. 2019; Maan et al. 2020; Mandal et al. 2020a; Mandal et al. 2020b; Jaiswal et al. 2020).
Another consequence of oxidative stress is the accumulation of neurotoxic glutamate levels which may initiate autophagy (Lin and Kuang 2014). Deprivation of oxygen and nutrient makes the retinal ganglionic cells energy-deficient, which further dramatically increases ROS level. This rise is due to slower electron transport in the OXPHOS, which increases the reduction state of electron carriers and favors superoxide production at low oxygen levels (Chidlow et al. 2017). Abnormal cellular calcium influx also has been related to oxidative stress (Guo et al. 2013). Extensive genome-wide association studies (GWAS) have established susceptibility loci linked to mitochondrial dysfunction with mixed results (Van Bergen et al. 2015). Polymorphism in thioredoxin reductase 2, a mitochondrial protein necessary for redox homeostasis, is one example (Bailey et al. 2016). Mitophagy dysfunction is also a cause of mitochondrial disfunction, which leads to neuroinflammation. Mitophagy is the mechanism by which the autophagy system degrades damaged mitochondria. This process is essential for clearing debris and aggregating proteins from cells, shielding them from potentially harmful proteins. Association of PINK1 and Parkin activity helps recognize damaged mitochondria (Wu et al. 2015). Thus, defects in PINK1 and Parkin can cause impairment in mitophagy and cease inflammation.
As a result of mitochondrial dysfunction, mitochondrial membrane damage leads to release of DAMPs which initiates multiple inflammatory cascades resulting in chronic inflammation. These mitochondrial DAMPs are recognized by microglial PRRs which further activate TLR/ NF-κB inflammatory pathway promoting release of pro-inflammatory cytokines and chemokines. The inflammation caused by mitochondrial DAMPs can further lead to mitochondrial dysfunction and exacerbating the inflammatory cycle (Tezel and Wax 2000; Duarte 2021).
When PRRs recognize stress products like mitochondrial DAMPs like TLRs, TNF receptors, and inflammasomes, unique pathways involving NF-κB cascades are activated. One of the most important regulators of inflammatory immune responses is NF-κB. TLRs stimulate downstream transcription factors MyD88/TRIF. Various ligands may interaction with PRRs including TNF-α, IL-1, IL-6, and variety of chemokines (CCL2, CXCL1, and CXCL10) (Glass et al. 2010). The NOD-like receptors, which form protein complexes known as inflammasomes, are another group of PRRs that can modulate the microglial inflammatory response. The maturation and release of cytokines like IL-1 and IL-18 can be triggered by cooperative downstream cross-talks between TLRs and NOD-like receptors (Gurung et al. 2015).
Mitochondria can cause activation of inflammasome signaling directly. ROS generated by mitochondria or DAMPs activates the NLRP3 inflammasome pathway (Liu et al. 2018). Activated NLRP3 is further redistributed to membranes of nucleus and mitochondrial, where it conjugates with ASC and procaspase-1 and developes into NLRP3 inflammasome (Gurung et al. 2015). NLRP3 is usually associated with the endoplasmic reticulum membrane. Activated NLRP3 inflammasome results in caspase-1-dependent conversion of pro-IL1β/ pro-IL-18 into active IL-1β/ IL-18 and initiates inflammation meaditated cell death known as pyroptosis (Liu et al. 2018). The NLRP3 inflammasome senses mitochondrial dysfunction and blockade of mitophagy.
The activation of TLR/NLRP3/MyD88.NF-κB pathway leads to generation of cytokine storm. When NF-κB is activated, the IL-1 cytokine family is produced, which amplifies inflammatory signal manyfolds by inducing a progressive release of pro-inflammatory cytokines in astrocytes and microglia (Yang et al. 2011). TNF-α production is markedly increased in mitochondrial disfunction, indicating a pro-inflammatory imbalance. TNF-α release can impair the function of mitochondrial components, reduce ATP content, increase ROS, and depolarize the mitochondrial membrane potential, all of which can affect mitochondrial function. Increased ROS may then have even more negative consequences by keeping NF-κB activated and producing pro-inflammatory signaling. Thus, the activation of neuroinflammation may negatively impact mitochondrial function, resulting in a vicious cycle (Wilkins et al. 2017). The complement system is also activated in mitochondrial disfunction, which leads to induction in the production of pro-inflammatory factors like IL-1β and TNF-α. It augments the release of mtDAMPs from mitochondria, thus, causing Neuroinflammation (Duarte 2021).
Autophagy in neuroinflammation
Autophagy is a biological process of degradation of cellular proteins and organelles inside the lysozyme in multicellular organisms. The damaged organelles and proteins are incorporated inside a membrane structure, known as an autophagosome. This autophagosome then binds to the lysozymes and results in the degradation of cellular organelles. There are mainly three types of autophagy, macroautophagy, microautophagy, and chaperone-mediated autophagy (Trempe and Fon 2013). Macroautophagy (also known as autophagy) is a conserved eukaryotic cell pathway that allows for the bulk degradation of cytoplasmic components. The goal components are kept apart from the rest of the system by an autophagosome (Glick et al. 2010).
Autophagy machinery consists of various steps, including initiation, elongation, and maturation of autophagic vesicles and ultimately fusion with lysosomes to form an autolysosome (Alavian et al. 2011). The procedure entails ULK complexes with ATG1, ATG13, ATG17, and ATG9, regulatory class III PI3 kinase complexes with beclin-1 (ATG6), and ATG5-ATG12-ATG16 multimerization complexes are formed de novo. ATG9, a transmembrane protein required for lipidation of phagophore membrane, is recruited by the ATG1-ATG13 complex (Ravikumar et al. 2010). Depending on the interaction ligands, the PI3 kinase-beclin1 complex may activate or suppress the autophagy and further engage other ATG proteins necessary for phagosome growth. When ultraviolet radiation absorption group (UVRAG) is combined with activating molecule in beclin-1-regulated autophagy (AMBRA) and ATG14, it promotes autophagy by interacting with the beclin-1 complex (Ghavami et al. 2014). On the other hand, UVRAG complexed with RUN domain and cysteine-rich domain-containing (RUBICON) interaction, on the other side, inhibits autophagy (Ghavami et al. 2014).
At the endoplasmic reticulum, Bcl2 releases beclin-1 which forms a complex with UVRAG/AMBRA, and initiates the development of the multimeric complex (ATG5-ATG12-ATG16) (Glick et al. 2010). The LC3bII, a lipidated product of LC3bI (ATG8), is cleaved by ATG4 and further complexed with phosphatidylethanolamine unevenly on both sides of the membrane ATG9 of the ULK complex, and then incorporated into the initiating membrane (Ghavami et al. 2012). During elongation and development of autophagosomes, around the selected complex for degradation, the membrane of mitochondria, and endoplasmic reticulum, are recruited (Ghavami et al. 2014). The release of LC3bII from the exterior membrane surface signifies the completion of the autophagosome. As a result, LC3bII appears as a standard marker for autophagic flux monitoring (Glick et al. 2010). The newly developed autophagosome merges with the lysosome and turn into autophagolysosome. The momentary formation of amphisomes offers the requisite pH for proper lysosomal protease activity. The merger of lysosome and autophagosome is mediated through cytoskeletal microtubules which helps transferring autophagosomes to various lysosomal membrane proteins (Atlashkin et al. 2003; Lee et al. 2007).
Neuroinflammation has been a common feature in almost all lysosomal storage diseases with a neurological component (Farfel-Becker et al. 2011). Autophagy is essential in the regulation of inflammation and immunity. Autophagy selectively degraded catalase, resulting in ROS generation and macrophage non-apoptotic death (Yu et al. 2006). As a result, therapeutic strategies targeting autophagy mediated inhibition of neuroinflammation may be helpful in the quest to develop a novel target for the management of neurodegenerative diseases. Neuroinflammation and oxidative stress have been implicated in the development of AD. They are interested in the advancement of cognitive destruction in AD because they can promote Aβ and neurofibrillary tangles (NFT) development and thus contribute to progressive cognitive decline in AD (Cai et al. 2013; Guan et al. 2018). Rapamycin, autophagy inducer, has shown improvement in learning and memory via reduction in Aβ,tau aggregation, oxidative stress, and neuroinflammation signaling cascades (Liu et al. 2013; Ji et al. 2018).
Neurotransmitters in neuroinflammation
Glutamate, most abundant excitatory neurotransmitter in CNS, is known to play a vital role in astrocyte activation (Perea et al. 2009). Excitatory amino acid transporters (EAATs) present on neuron and astrocyte cells maintain the glutamate release (Bak et al. 2006). Astrocytes maintain the glutamate level in the synaptic cleft by Glutamate Aspartate Transporter (GLAST), and the excess glutamate is primarily regulated by GLU transporters on astrocytes (Chaudhry et al. 2002). Animal studies suggested that knockdown of GLAST results in increased expression of glutamate in the synaptic cleft, which activates the astrocyte cell to maintain the glutamate levels by GLU transporters (Danbolt 2001). This astrocyte activation plays a central role in the neuroinflammation process. Further, glutamate toxicity leads to increased expression of inflammatory markers, microglial activation in the brain leading to neuroinflammation (Boka et al. 1994; Tsai et al. 2012).
Recent studies suggested that peripheral and central neuroinflammation is associated with an altered GABAergic system (Montoliu et al. 2015; Dadsetan et al. 2016). This study indicated an increased expression of GABA transporter GAT-3 in the cerebellum of rats having neuroinflammation. Also, there was an increase in the extracellular GABA concentration in the cerebellum. This increased GAT-3 and GABA expression lead to neuroinflammation and motor disfunction in the rats. However, the neuroinflammation gets reversed when the expression of GAT-3 and GABA gets normalized (Dadsetan et al. 2016).
Dopamine is associated with cytokine production. Decrease dopamine release leads to increase cytokine production, thus, results in neuroinflammation (Abd Wahab et al. 2019). Serotonin (5-HT) plays an essential role in innate and adaptive immunity and modulates neuroinflammation process. 5-HT can trigger the lymphocyte and monocytes and impact cytokines' secretion (Dürk et al. 2005). They modulate the release of IL-1β, IL-6, IL-8/CXCL8, IL-12p40, and TNF- α. Activation of the 5-HT3 receptor upregulates IL-1β, IL-6, IL-8/CXCL8 cytokines, which leads to Neuroinflammation (Dürk et al. 2005). In contrast, activation of 5-HT2A has been reported to inhibit TNF-α release and cause reduction of Neuroinflammation (Vanover et al. 2008). The expression of 5-HT2B receptor has been widely recognized in microglia cells which play an essential role in their activation and proliferation, resulting in Neuroinflammation (Kolodziejczak et al. 2015).
Role of neuroinflammation in various neurological diseases
Given chronic neuroinflammation, anti-inflammatory medications in neurological diseases have shown efficacy with varying observations in clinical investigations. Non-steroidal anti-inflammatory drugs (NSAIDs) have been found to inhibit prostaglandin synthesis by inhibiting the COX-2 enzyme. On the other hand, they have also been found to regulate mitochondrial calcium homeostasis. PPARs, inflammasome, HMGB1, and thus might limit the disease progression. Neuroinflammation in the pathogenesis of AD, PD, epilepsy, and miscellaneous neurological conditions has been elaborated.
Neuroinflammation in Alzheimer’s disease
AD is one of the highly prevalent chronic neurodegenerative disorders, contributing around 60–70% of dementia worldwide. According to prevailing amyloid and tau cascade hypotheses, the accumulation of Aβ deposits and neurofibrillary tangle containing hyperphosphorylated Tau in the brain initiates the cellular events in AD (Leng and Edison 2021; Cummings 2021).
AD is characterized by an intracellular deposition of NFTs, extracellular deposition of Aβ plague, and microglial activation. NFTs are paired helical filaments made up of hyperphosphorylated tau proteins, and Aβ is derived from Amyloid precursor proteins (APP) by the action of β and γ secretase. Beta-site APP cleaving enzyme 1 (BACE1) is one major β-secretase enzyme found in the human brain (Webers et al. 2020). There are mainly two types of Aβ proteins Aβ1-40 and Aβ1-42, containing 40 and 42 amino acid residues. Among these two types, Aβ1-42 is more amyloidogenic and tends to aggregate to form Aβ plague. The Aβ plagues are cleared by microglia cells. Microglia cells remove the Aβ aggregates by uptake, local degradation, or degradation by releasing a neprilysin enzyme. So, there is a delicate balance between the removal and synthetic pathways of Aβ proteins (Webers et al. 2020). Disruption of this balance by neuronal insult or overproduction leads to aggregation of Aβ proteins and formation of plaques in the extracellular compartment in the brain. A wide variety of receptors, like the different toll-like receptors (TLR4 and TLR6) and the NACHT, LRR, CD36, and pyrin-protein-containing (NLRP3) domains, are resent in microglia, which can bind to Aβ. They are stimulated by DAMPs, including adenosine triphosphate (Calsolaro and Edison 2016; Rivers-Auty et al. 2020). When Aβ binds to CD36, TLR4, or TLR6 receptors, microglia get activated, and proinflammatory cytokines and chemokines are produced. The cytokines released by the activation of microglia cells are mainly TNF-α and IL-1β (Kaur et al. 2019). TNF-α binds to the TNF receptor on the cell membrane, induces the extrinsic pathway of apoptosis. Extracellular Aβ can stimulate pro-inflammatory gene expression by activation of the MAPK/NF-kB pathway (Solito and Sastre 2012).
Aβ can also induce NADPH oxidase-mediated priming in microglial cells, leading to generation of ROS. Production of ROS leads to increased oxidative stress and mitochondrial dysfunction (Simpson and Oliver 2020). This mitochondrial dysfunction increased the cell membrane permeability of mitochondria and leads to the secretion of mitochondrial DAMPs, and cytochrome-c. Mitochondrial DAMPs leads to activation of NF-kB, which promotes the development of pro-inflammatory cytokines and chemokines (pro-IL-1 and TNF-α). The release of cytochrome-c further initiates the apoptosis cascade (Duarte 2021). Microglia can also interact with astrocytes to activate them. Although TNF-α and IL-1β can also directly stimulate and activate astrocytes. Activation of astrocytes leads to the formation of iNOS, thus can cause prolonged neuroinflammation. The C-terminal 100 amino acid of bAPP in Aβ plaques can cause astrogliosis and neurodegeneration in the existence of amyloid pathology (Tiwari et al. 2019).
Aβ itself can initiate a cascade of relations which leads to cytokine release and neuroinflammation. Aβ induces the release of various pro-inflammatory cytokines like IL-1α, IL-1β, IL-6, and TNF-α. Aβ sensitizes astrocytes and microglia cells to secrete IL-1, which influences the production of Aβ plagues. Thus, by this mechanism, Aβ induces its own production several-folds resulting in intensified cytokine mediated neuroinflammation. IL-1β induces astrogliosis in Aβ treated astrocytes by activating IL-1R. The IL-1β was primarily triggered by astrocytes APP and neurotoxic Aβ manufacturing by neurons.IL-1β also activates astrocytes and microglia cells and induces them to secrete various pro-inflammatory cytokines like- IL-1β, IL-6, and IL-18. IL-1β shows cell-specific activity; like in glia cells, IL-1β activates the NF-kβ mediated cytokine production. It stimulates the cascading mitogen-activated protein kinases (MAPK)-p38, enhances the secreted APP fragment clamped with BACE1 (Sawikr et al. 2017). IL-6, synthesized by astrocytes, microglia, neuron, and endothelial, on the other hand, increased IL-1β mediated inflammatory response, and elevated IL-6 mRNA concentration was found in the brain of AD patients (Fernandes et al. 2017).
Tau protein is also one of the significant drivers of AD pathogenesis. Tau corresponds to the microtube-associated protein (MAP) family and is mainly expressed in axon-preferred neurons. Hyperpolarization of tau protein is associated with the prognosis of AD. IL-1β can cause tau protein phosphorylation and NFT creation via the MAPK-p38 pathway. The neuronal hyperphosphorylation of Tau protein, is engaged in the IL-1β-induced up-regulation of MAPK-p38. in AD, hyperphosphorylated Tau exists in coupled helical filaments, dystrophic neuritis, and NFTs. This finding demonstrated a loss of axonal integrity as well as a decrease in synapse connection, which is the reason for dementia in AD. AD observes a loss of synaptophysin in triangular neurons, and the activated microglia correlates with the neuronal disease (Laurent et al. 2018). The anti-inflammatory approaches targeting neuroinflammation have been bolstered by several preclinical findings listed in Table 1.
Neuroinflammation in Parkinson’s disease
PD is the second most common neurodegenerative disease majorly affecting the geriatric population and is characterized by various motor and non-motor symptoms. Major motor manifestations include bradykinesia, rigidity, and tremor, while these symptoms are preceded by premotor symptoms, including constipation, bladder disorders, hyposmia, autonomic dysfunction, depression, and sleep disorders (Angelopoulou et al. 2021; Hirsch and Standaert 2021; Mann et al. 2020).
The main characteristic of PD pathology is the accumulation of α-synuclein and neuroinflammation. The primary role of α-synuclein is to maintain synaptic vesicle dynamics, mitochondrial function, intracellular trafficking, and act as a chaperon. When soluble α-synuclein monomers start aggregating, it gives rise to large insoluble α-synuclein fibers and Lewy body formation (Rocha et al. 2018). Generally, the insoluble α-synuclein is removed from cells by majorly two pathways, ubiquitin–proteasome pathway and lysosomal autophagy system (LAS). LAS seems even more significant to concise oligomeric assemblies than the ubiquitin–proteasome system (Panicker et al. 2021). It has been proposed that LAS mediates α-synuclein degradation with chaperone-mediated autophagy as well as macroautophagy. Alteration of this process leads to the accumulation of α-synuclein inside the cell (Brás and Outeiro 2021).
Aggregation of α-synuclein fibers leads to the formation of toxic β-plated sheets, which ultimately leads to the formation of Lewy bodies. These β-plated sheets further block the LAS system, thus, induce the aggregation of α-synuclein. The appearance of β-plated sheets further increases the production of α-synuclein inside the cell (Poewe et al. 2017). This trans-synaptic transmission of β-plated sheets spreads the pathology to other unaffected cells.
Aggregation of toxic oligomers leads to mitochondrial dysfunction by inhibiting mitochondrial complex-I/III. This further leads to the downregulation of mitochondrial master transcriptional regulator, peroxisome proliferator-activated receptor-gamma coactivator 1-α (PGC-1α) (Das et al. 2021). The downregulation of PGC-1α leads to the genetiaon of ROS, further adding to more misfolding of α-synuclein and reduction in its proteolysis. In addition, oxidative stress increases the calcium ion influx into the cell, which leads to impaired calcium homeostasis. This imbalance leads to caspase activation and cell death via apoptosis (Bose and Beal 2016). Toxic oligomers can also activate microglia. Activated microglia then releases a pro-inflammatory cytokine (TNF-α, IL-1β), which can induce the inflammatory process in the brain. TNF-α then acts on the TNF receptor and activates the extrinsic pathway of apoptosis (Duarte 2021), resulting in neuronal cell death. Inflammation can also induce the misfolding of α-synuclein and can disturb proteolysis of α-synuclein, thus influencing the whole cycle many-fold a lead to dopaminergic cell death (Poewe et al. 2017).
The activation of M1 type of microglia cells, various pro-inflammatory cytokines are released like- iNOS, IL-1β, IL-6, TNF-α, IFN-γ, C1q, IL-1α, and IL-1β (Janda et al. 2018). The release of these cytokines leads to inflammatory reactions inside the brain. Activated microglia then interacts with astrocytes and triggers them to produce pro-inflammatory factors. Furthermore, α-synuclein can also directly induce astrocytes and increase the production of pro-inflammatory cytokines and chemokines such as CCL2, CCL20, CXCL1, CX3CL1, IL-1β, IL-6, TNF-α (Miyazaki and Asanuma 2020). Overall, the release of these pro-inflammatory mediators results in the degeneration of dopaminergic neurons. Preclinical evidence supporting the role of various anti-inflammatory agents against the experimental model of PD has listed in Table 2.
Neuroinflammation in epilepsy
Epilepsy is one of the common neurological disorders characterized affecting around 70 million people worldwide with identified causes like acute brain injury (stroke, neurotrauma, tumor, and status epilepticus), chronic CNS infection, and metabolic altercations or idiopathic (Akyuz et al. 2021; Vezzani et al. 2019; Mishra and Goel 2016, 2019). Neuroinflammation has been conversed as one of the plausible pathogenmic feature in partial, generalized and refractory seizures (Vezzani et al. 2019; Vezzani 2020; Löscher et al. 2020; Terrone et al. 2020).
Neuroinflammation has been recorded in CNS before the onset of epilepsy in the human brain (Prabowo et al. 2013; Pauletti et al. 2019) and experimental models (Ravizza et al. 2008). In refractory epilepsy brain, activation of TORC1 pathway was associated with increased expression of inflammatory markers such as MHC-I/II, TLR-2/4 for advanced glycation end products (RAGE), IL-1β, HMGB1, and inducible nitric oxide synthase (iNOS) (Pauletti et al. 2019; Shi et al. 2017; Prabowo et al. 2013; Ravizza et al. 2008; Gorter et al. 2006; Turrin and Rivest 2004). These pro-inflammatory mediators cause BBB damage and recruit peripheral immune cells (Vezzani and Friedman 2011).
Systemic inflammatory cascade causing peripheral inflammation can contribute further to developing inflammatory mediators and the accumulation of leukocytes, neutrophils around the BBB (Rana and Musto 2018). Then leukocytes secrete chemokine CCL2 and inflammatory mediator IL-1β, which leads to upregulation of MMP9 (Bronisz and Kurkowska-Jastrzębska 2016). MMP-9 has different roles in the brain, from structural changes to inflammatory processes. Chronic MMP-9 levels lead to dilution and dendritic spine elongation, leading to morphological synapse modifications and related to affected person plasticity of synaptic (Acar et al. 2015). In addition, MMP-9 weakens the BBB by destroying the tight junction interconnections directly. Thus, BBB becomes weak, which allows the migration of leucocytes by traffic cups. The elevated CD44, VCAM-1, and ICAM-1 levels support trans-endothelial migration of leucocytes through the BBB (Acar et al. 2015).
Albumins like serum proteins also infiltrate the loosened BBB and enter into the CNS. Albumin enhances the production of TGF-β1 via activation of astrocytes. This binding also activates the astrocytes and induces secretion of TGF-β1 and MMP9. Produced MMP9 further loosens the BBB, and TGF-β1 impairs the potassium glutamate buffer system inside the cells (Webster et al. 2017). TGF-β1 additionally induces excitatory synaptogenesis, thus helps in the development of hyperactivity of neurons. A pro-inflammatory, platelet-activating factor (PAF) is released from the postsynaptic neurons. PAF induces the per synaptic sites to release glutamine, and it also upregulates COX2 release from microglia and astrocytes by acting upon the PAF receptor present on them (Hammond et al. 2016; Guan and Wang 2019).
Microglia are activated by various cytokines present in the brain like TNF-α, IL-1β, and partly by PAF. Induction of COX2 mediated production of PGE2 level stimulates astrocytic glutamate causing hyperexcitability mediated neuronal death (Shimada et al. 2014). COX2 also upregulates various chemokines like CCL2 and CXCL12 in the brain, further weakening the BBB and stimulating TNF-α release from microglial cells (Sharma et al. 2019). The released TNF-α from microglia then induces neuroinflammation, cell death and upregulates AMPA receptors (Takeuchi et al. 2006; Galic et al. 2012). IL-1β upregulates NMDA receptor expression (Viviani et al. 2003) and reduces GABAA current (Roseti et al. 2015) results in ictogenesis. In addition, activation of TLR, present in astrocytes and microglia, induces the release of pro-inflammatory cytokines like TNF-α, IL-1β from microglial cells (Iori et al. 2017; Rana and Musto 2018; Li et al. 2021). IL-1β also induces the production of MMP-9 by upregulating its transcription (Bronisz and Kurkowska-Jastrzębska 2016). Thus, IL-1β plays a role in epileptogenesis and BBB damage. Activation of TLR3 potentiates TNF-α level and induces endocytosis of GABA receptors, leading to epileptogenesis (Rana and Musto 2018). Results of various anti-inflammatory agents supporting the protective effect against experimental models of epilepsy are listed in Table 3.
Limitations of anti-inflammatory approaches
Various anti-inflammatory approaches have illustrated neuroprotection against degenerating neurons in experimental models of AD, PD, and epilepsy by mitigating neuroinflammation, however clinical relevance of NSAIDs in neurological diseases appears conflicting. Summary of few clinical investigations exploring the role of anti-inflammatory agents in neurological disorders in clinical samples has been mentioned in Table 4.
Chronic treatment with selective/nonselective COX2 inhibitors in placebo-controlled clinical trials has suggested no protective effect on mild-moderate AD symptoms (Aisen et al. 2003; Reines et al. 2004). In addition, rofecoxib treatment has shown discouraging findings in patients with mild cognitive impairment reported in a secondary preventive study (Thal et al. 2005). Meanwhile, some preclinical investigations suggested the protective effect of NSAIDs and raised hope for futuristic clinical investigations (Varvel et al. 2009). However, randomized clinical trials have failed to corroborate these findings in AD patients (Ali et al. 2019). Anti-inflammatory agents have also exhibited protective effect in PD patients. A meta-analysis suggested pronounced protective effect of nonaspirin NSAIDs (ibuprofen) as compared to aspirin in PD patients (Gagne and Power 2010). In contrast, a recent meta-analysis suggested no association of NSAIDs and risk of PD (Poly et al. 2019). Overall, the clinical impact of anti-inflammatory agents (NSAIDs) remain ambiguous. In epilepsy, pre-clinical and clinical studies supported targeting the anti-inflammatory approach as a complementary approach for symptomatic treatment of recurrent seizures from conventional and refractory seizures (Radu et al. 2017). Till date only low dose aspirin had suggested protection against seizures. Moreover, limited information is available regarding the use of anti-inflammatory agents in clinical epilepsy.
NSAIDs have been negated to have protective role in various neurological disorders and thus, should not be used as a therapeutic option in the clinical setup (Miguel-Álvarez et al. 2015). In a clinical trial, minocycline, an antibiotic with anti-inflammatory properties, failed to exert disease-modifying or delay traverse disease progression in patients with mild AD (Howard et al. 2020). Howard and colleagues explained the first reason as “neuroinflammation might be a reaction to pathological characteristics of the disease rather than important factor in neurodegeneration” and second reason as microglial activation and neurodegeneration is a complex phenomenon, whose inactivation might interfere with its supportive function (Howard et al. 2020). Gyengesi and Münch suggested the future trials to be more specific for cytokine-suppressive anti-inflammatory drugs, which can directly inhibit the production of cytotoxic cytokines (TNF-α, IL-1β, iNOS), and free radicals (Gyengesi and Münch 2020).
The possible explanations behind these varied outcomes may incorporate anti-inflammatory agents organization, selectivity of COX variant, improper utilization of anti-inflammatory agents for a given neurological condition and their severity, inappropriateness in the target site, or limited entry to the CNS through BBB. This way, the plan of new anti-inflammatory approach for targeting neurological disorders, in view of improved BBB permeability, may deduce a viable treatment option. However, the anti-inflammatory agents (especially with COX inhibitors) have been considered as major sources of drug induced complications (renal and gastro-intestinal side effects). Use of specific COX2 inhibitor (rofecoxib and valdecoxib) have been associated with vascular complications along with higher risk of stroke (Auriel et al. 2014). Moreover, use of such agents in elderly, asthama, pregnant, breastfeeding. patients with liver/kidney/heart diseases are contraindicated. Furthermore, more research is expected to comprehend the mechanism devoid of such adverse outcomes.
Conclusion and future perspective
Neuroinflammation plays a significant role in the development of neurological disease. The concept of neuroinflammation theory covers a wide range. The glial cells, cytokines, inflamosome, autophagy, ion channels, monocytes function as a sensor for homeostasis of disrupted brain tissue and accumulate locally in response to neuronal injury or foreign pathogen entry into the brain. Still, it is very arduous to give a universal conclusion. Preclinical evidences appear commendable suggesting utilization of anti-inflammatory approach to alleviate neurological disease progression, either alone or as combination therapy. However, the finding of clinical investigations is obscure in their result. Thus, until authoritative clinical evidence is established, used of anti-inflammatory agents should be restricted to the approved drugs. Simultaneously exploration of novel anti-inflammatory approaches (other than COX inhibitors) may be given due considereation in the management of disease progression. Further, use of nanoformulation approach also attact attention of researchers for improved brain delivery of anti-inflammatory agents.
Data Availability
No datasets were generated or analyzed during the current study.
References
Abd Wahab DYA, Gau CH, Zakaria R, Muthu Karuppan MK, A-Rahbi BS, Abdullah Z, Alrafiah A, Abdullah JM, Muthuraju S (2019) Review on Cross talk between neurotransmitters and neuroinflammation in striatum and cerebellum in the mediation of motor behaviour. Biomed Res Int 2019:1767203. https://doi.org/10.1155/2019/1767203
Abuelezz SA, Hendawy N (2021) HMGB1/RAGE/TLR4 axis and glutamate as novel targets for PCSK9 inhibitor in high fat cholesterol diet induced cognitive impairment and amyloidosis. Life Sci 273:119310. https://doi.org/10.1016/j.lfs.2021.119310
Acar G, Tanriover G, Acar F, Demir R (2015) Increased expression of matrix metalloproteinase-9 in patients with temporal lobe epilepsy. Turk Neurosurg 25(5):749–756. https://doi.org/10.5137/1019-5149.JTN.10738-14.0
Aguirre-Vidal Y, Morales-Montor J, de León CTG, Ostoa-Saloma P, Díaz-Zaragoza M, Montes S, Arteaga-Silva M, Monroy-Noyola A (2020) Protection induced by estradiol benzoate in the MPP+ rat model of Parkinson’s disease is associated with the regulation of the inflammatory cytokine profile in the nigro striatum. J Neuroimmunol 349:577426. https://doi.org/10.1016/j.jneuroim.2020.577426
Ahuja M, Ammal Kaidery N, Yang L, Calingasan N, Smirnova N, Gaisin A, Gaisina IN, Gazaryan I, Hushpulian DM, Kaddour-Djebbar I, Bollag WB, Morgan JC, Ratan RR, Starkov AA, Beal MF, Thomas B (2016) Distinct Nrf2 signaling mechanisms of fumaric acid esters and their role in neuroprotection against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced experimental Parkinson’s-like disease. J Neurosci 36(23):6332–6351. https://doi.org/10.1523/JNEUROSCI.0426-16.2016
Aisen PS, Schafer KA, Grundman M, Pfeiffer E, Sano M, Davis KL, Farlow MR, Jin S, Thomas RG, Thal LJ, Alzheimer’s Disease Cooperative Study (2003) Effects of rofecoxib or naproxen vs placebo on Alzheimer disease progression: a randomized controlled trial. JAMA 289(21):2819–2826. https://doi.org/10.1001/jama.289.21.2819
Ajami B, Bennett JL, Krieger C, McNagny KM, Rossi FM (2011) Infiltrating monocytes trigger EAE progression, but do not contribute to the resident microglia pool. Nat Neurosci 14:1142–1149
Akyuz E, Polat AK, Eroglu E, Kullu I, Angelopoulou E, Paudel YN (2021) Revisiting the role of neurotransmitters in epilepsy: an updated review. Life Sci 265:118826. https://doi.org/10.1016/j.lfs.2020.118826
Alavian SM, Ande SR, Coombs KM, Yeganeh B, Davoodpour P, Hashemi M, Los M, Ghavami S (2011) Virus-triggered autophagy in viral hepatitis-possible novel strategies for drug development. J Viral Hepat 18(12):821–830. https://doi.org/10.1111/j.1365-2893.2011.01530.x
Alcoreza O, Tewari BP, Bouslog A, Savoia A, Sontheimer H, Campbell SL (2019) Sulfasalazine decreases mouse cortical hyperexcitability. Epilepsia 60(7):1365–1377. https://doi.org/10.1111/epi.16073
Ali MM, Ghouri RG, Ans AH, Akbar A, Toheed A (2019) Recommendations for anti-inflammatory treatments in Alzheimer’s disease: a comprehensive review of the literature. Cureus 11(5):e4620. https://doi.org/10.7759/cureus.4620
Alyu F, Dikmen M (2016) Inflammatory aspects of epileptogenesis: contribution of molecular inflammatory mechanisms. Acta Neuropsychiatrica 29(01):1–16
Angelopoulou E, Paudel YN, Piperi C, Mishra A (2021) Neuroprotective potential of cinnamon and its metabolites in Parkinson’s disease: mechanistic insights, limitations, and novel therapeutic opportunities. J Biochem Mol Toxicol 35(4):e22720. https://doi.org/10.1002/jbt.22720
Ardestani PM, Evans AK, Yi B, Nguyen T, Coutellier L, Shamloo M (2017) Modulation of neuroinflammation and pathology in the 5XFAD mouse model of Alzheimer’s disease using a biased and selective beta-1 adrenergic receptor partial agonist. Neuropharmacology 116:371–386. https://doi.org/10.1016/j.neuropharm.2017.01.010
Atlashkin V, Kreykenbohm V, Eskelinen EL, Wenzel D, Fayyazi A, von Mollard GF (2003) Deletion of the SNARE vti1b in mice results in the loss of a single SNARE partner, syntaxin 8. Mol Cell Biol 23(15):5198–5207. https://doi.org/10.1128/mcb.23.15.5198-5207.2003
Attia GM, Elmansy RA, Elsaed WM (2019) Neuroprotective effect of nilotinib on pentylenetetrazol-induced epilepsy in adult rat hippocampus: involvement of oxidative stress, autophagy, inflammation, and apoptosis. Folia Neuropathol 57(2):146–160. https://doi.org/10.5114/fn.2019.84423
Aucott H, Sowinska A, Harris HE, Lundback P (2018) Ligation of free HMGB1 to TLR2 in the absence of ligand is negatively regulated by the C-terminal tail domain. Mol Med 24(1):19. https://doi.org/10.1186/s10020-018-0021-x
Auriel E, Regev K, Korczyn AD (2014) Nonsteroidal anti-inflammatory drugs exposure and the central nervous system. Handb Clin Neurol 119:577–584. https://doi.org/10.1016/B978-0-7020-4086-3.00038-2
Azmand MJ, Rajaei Z (2021) Effects of crocin on spatial or aversive learning and memory impairments induced by lipopolysaccharide in rats. Avicenna J Phytomed 11(1):79–90
Babaei P, Eyvani K, Kouhestani S (2021) Sex-independent cognition improvement in response to kaempferol in the model of sporadic Alzheimer’s disease. Neurochem Res. https://doi.org/10.1007/s11064-021-03289-y
Bailey JN, Loomis SJ, Kang JH, Allingham RR, Gharahkhani P, Khor CC, Burdon KP, Aschard H, Chasman DI, Igo RP Jr, Hysi PG, Glastonbury CA, Ashley-Koch A, Brilliant M, Brown AA, Budenz DL, Buil A, Cheng CY, Choi H, Christen WG, Curhan G, De Vivo I, Fingert JH, Foster PJ, Fuchs C, Gaasterland D, Gaasterland T, Hewitt AW, Hu F, Hunter DJ, Khawaja AP, Lee RK, Li Z, Lichter PR, Mackey DA, McGuffin P, Mitchell P, Moroi SE, Perera SA, Pepper KW, Qi Q, Realini T, Richards JE, Ridker PM, Rimm E, Ritch R, Ritchie M, Schuman JS, Scott WK, Singh K, Sit AJ, Song YE, Tamimi RM, Topouzis F, Viswanathan AC, Verma SS, Vollrath D, Wang JJ, Weisschuh N, Wissinger B, Wollstein G, Wong TY, Yaspan BL, Zack DJ, Zhang K, EPIC-Norfolk Eye Study, ANZRAG Consortium, Weinreb RN, Pericak-Vance MA, Small K, Hammond CJ, Aung T, Liu Y, Vithana EN, MacGregor S, Craig JE, Kraft P, Howell G, Hauser MA, Pasquale LR, Haines JL, Wiggs JL (2016) Genome-wide association analysis identifies TXNRD2, ATXN2 and FOXC1 as susceptibility loci for primary open-angle glaucoma. Nat Genet 48(2):189–194. https://doi.org/10.1038/ng.3482
Bak LK, Schousboe A, Waagepetersen HS (2006) The glutamate/GABA-glutamine cycle: aspects of transport, neurotransmitter homeostasis and ammonia transfer. J Neurochem 98(3):641–653. https://doi.org/10.1111/j.1471-4159.2006.03913.x
Barbiero JK, Santiago R, Tonin FS, Boschen S, da Silva LM, Werner MF, da Cunha C, Lima MM, Vital MA (2014) PPAR-α agonist fenofibrate protects against the damaging effects of MPTP in a rat model of Parkinson’s disease. Prog Neuropsychopharmacol Biol Psychiatry 53:35–44. https://doi.org/10.1016/j.pnpbp.2014.02.009
Barger SW, Basile AS (2001) Activation of microglia by secreted amyloid precursor protein evokes release of glutamate by cystine exchange and attenuates synaptic function. J Neurochem 76(3):846–854
Bartels AL, Willemsen AT, Doorduin J, de Vries EF, Dierckx RA, Leenders KL (2010) [11C]-PK11195 PET: quantification of neuroinflammation and a monitor of anti-inflammatory treatment in Parkinson’s disease? Parkinsonism RelatDisord 16(1):57–59. https://doi.org/10.1016/j.parkreldis.2009.05.005
Bassani TB, Turnes JM, Moura ELR, Bonato JM, Cóppola-Segovia V, Zanata SM, Oliveira RMMW, Vital MABF (2017) Effects of curcumin on short-term spatial and recognition memory, adult neurogenesis and neuroinflammation in a streptozotocin-induced rat model of dementia of Alzheimer’s type. Behav Brain Res 335:41–54. https://doi.org/10.1016/j.bbr.2017.08.014
Becher B, Durell BG, Noelle RJ (2002) Experimental autoimmune encephalitis and inflammation in the absence of interleukin-12. J Clin Invest 110(4):493–497. https://doi.org/10.1172/JCI15751
Becher B, Spath S, Goverman J (2017) Cytokine networks in neuroinflammation. Nat Rev Immunol 17(1):49–59. https://doi.org/10.1038/nri.2016.123
Begum AN, Jones MR, Lim GP, Morihara T, Kim P, Heath DD, Rock CL, Pruitt MA, Yang F, Hudspeth B, Hu S, Faull KF, Teter B, Cole GM, Frautschy SA (2008) Curcumin structure-function, bioavailability, and efficacy in models of neuroinflammation and Alzheimer’s disease. J Pharmacol Exp Ther 326(1):196–208. https://doi.org/10.1124/jpet.108.137455
Bianchi ME, Manfredi AA (2007) High-mobility group box 1 (HMGB1) protein at the crossroads between innate and adaptive immunity. Immunol Rev 220:35–46. https://doi.org/10.1111/j.1600-065X.2007.00574.x
Boiangiu RS, Mihasan M, Gorgan DL, Stache BA, Petre BA, Hritcu L (2020) Cotinine and 6-hydroxy-L-nicotine reverses memory deficits and reduces oxidative stress in Aβ25-35-induced rat model of Alzheimer’s disease. Antioxidants (basel) 9(8):768. https://doi.org/10.3390/antiox9080768
Boka G, Anglade P, Wallach D, Javoy-Agid F, Agid Y, Hirsch EC (1994) Immunocytochemical analysis of tumor necrosis factor and its receptors in Parkinson’s disease. Neurosci Lett 172(1–2):151–154. https://doi.org/10.1016/0304-3940(94)90684-x
Bose A, Beal MF (2016) Mitochondrial dysfunction in Parkinson’s disease. J Neurochem 139(Suppl 1):216–231. https://doi.org/10.1111/jnc.13731
Brambilla R (2019) Neuroinflammation, the thread connecting neurological disease: cluster: “Neuroinflammatory mechanisms in neurodegenerative disorders.” Acta Neuropathol 137(5):689–691. https://doi.org/10.1007/s00401-019-02009-9
Brás IC, Outeiro TF (2021) Alpha-synuclein: mechanisms of release and pathology progression in synucleinopathies. Cells 10(2):375. https://doi.org/10.3390/cells10020375
Breitner J, Meyer PF (2020) Author response: INTREPAD: a randomized trial of naproxen to slow progress of presymptomatic Alzheimer disease. Neurology 94(13):594. https://doi.org/10.1212/WNL.0000000000009185
Bronisz E, Kurkowska-Jastrzębska I (2016) Matrix metalloproteinase 9 in epilepsy: the role of neuroinflammation in seizure development. Mediators Inflamm 2016:7369020. https://doi.org/10.1155/2016/7369020
Broz P, Dixit VM (2016) Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol 16(7):407–420. https://doi.org/10.1038/nri.2016.58
Cai Z, Yan LJ, Ratka A (2013) Telomere shortening and Alzheimer’s disease. Neuromolecular Med 15(1):25–48. https://doi.org/10.1007/s12017-012-8207-9
Calsolaro V, Edison P (2016) Neuroinflammation in Alzheimer’s disease: current evidence and future directions. Alzheimers Dement 12(6):719–732. https://doi.org/10.1016/j.jalz.2016.02.010
Cao W, Dong Y, Zhao W, Lu X, Sun L (2019) Mulberrin attenuates 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced Parkinson’s disease by promoting Wnt/β-catenin signaling pathway. J Chem Neuroanat 98:63–70. https://doi.org/10.1016/j.jchemneu.2019.04.003
Cao J, Tang C, Gao M, Rui Y, Zhang J, Wang L, Wang Y, Xu B, Yan BC (2020) Hyperoside alleviates epilepsy-induced neuronal damage by enhancing antioxidant levels and reducing autophagy. J Ethnopharmacol 257:112884. https://doi.org/10.1016/j.jep.2020.112884
Carpentier PA, Duncan DS, Miller SD (2008) Glial toll-like receptor signaling in central nervous system infection and autoimmunity. Brain Behav Immun 22(2):140–147
Castro AA, Wiemes BP, Matheus FC, Lapa FR, Viola GG, Santos AR, Tasca CI, Prediger RD (2013) Atorvastatin improves cognitive, emotional and motor impairments induced by intranasal 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) administration in rats, an experimental model of Parkinson’s disease. Brain Res 1513:103–116. https://doi.org/10.1016/j.brainres.2013.03.029
Chaudhry FA, Reimer RJ, Edwards RH (2002) The glutamine commute: take the N line and transfer to the A. J Cell Biol 157(3):349–355. https://doi.org/10.1083/jcb.200201070
Chen Y, Qi Z, Qiao B, Lv Z, Hao Y, Li H (2019) Oxymatrine can attenuate pathological deficits of Alzheimer’s disease mice through regulation of neuroinflammation. J Neuroimmunol 334:576978. https://doi.org/10.1016/j.jneuroim.2019.576978
Cheng C, Zhu X (2019) Cordycepin mitigates MPTP-induced Parkinson’s disease through inhibiting TLR/NF-κB signaling pathway. Life Sci 223:120–127. https://doi.org/10.1016/j.lfs.2019.02.037
Chidlow G, Wood JPM, Casson RJ (2017) Investigations into hypoxia and oxidative stress at the optic nerve head in a rat model of glaucoma. Front Neurosci 11:478. https://doi.org/10.3389/fnins.2017.00478
Cho S, Park EM, Febbraio M, Anrather J, Park L, Racchumi G et al (2005) The class B scavenger receptor CD36 mediates free radical production and tissue injury in cerebral ischemia. J Neurosci 25(10):2504–2512
Chrysostomou V, Rezania F, Trounce IA, Crowston JG (2013) Oxidative stress and mitochondrial dysfunction in glaucoma. Curr Opin Pharmacol 13(1):12–15. https://doi.org/10.1016/j.coph.2012.09.008
Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, Ma L, Watowich SS, Jetten AM, Tian Q, Dong C (2009) Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity 30(4):576–587. https://doi.org/10.1016/j.immuni.2009.02.007
Citraro R, Leo A, Marra R, De Sarro G, Russo E (2015) Antiepileptogenic effects of the selective COX-2 inhibitor etoricoxib, on the development of spontaneous absence seizures in WAG/Rij rats. Brain Res Bull 113:1–7. https://doi.org/10.1016/j.brainresbull.2015.02.004
Condello C, Yuan P, Schain A, Grutzendler J (2015) Microglia constitute a barrier that prevents neurotoxic protofibrillar Aβ42 hotspots around plaques. Nat Commun 6:6176. https://doi.org/10.1038/ncomms7176
Cote M, Poirier AA, Aube B, Jobin C, Lacroix S, Soulet D (2015) Partial depletion of the proinflammatory monocyte population is neuroprotective in the myenteric plexus but not in the basal ganglia in a MPTP mouse model of Parkinson’s disease. Brain Behav Immun 46:154–167
Cummings J (2021) New approaches to symptomatic treatments for Alzheimer's disease. Mol Neurodegener 16(1):2. https://doi.org/10.1186/s13024-021-00424-9. Erratum in: Mol Neurodegener. 2021 Apr 1;16(1):21
Dadsetan S, Balzano T, Forteza J, Agusti A, Cabrera-Pastor A, Taoro-Gonzalez L, Hernandez-Rabaza V, Gomez-Gimenez B, ElMlili N, Llansola M, Felipo V (2016) Infliximab reduces peripheral inflammation, neuroinflammation, and extracellular GABA in the cerebellum and improves learning and motor coordination in rats with hepatic encephalopathy. J Neuroinflam 13(1):245. https://doi.org/10.1186/s12974-016-0710-8
Danbolt NC (2001) Glutamate uptake. Prog Neurobiol 65(1):1–105. https://doi.org/10.1016/s0301-0082(00)00067-8
Daniels MJ, Rivers-Auty J, Schilling T, Spencer NG, Watremez W, Fasolino V, Booth SJ, White CS, Baldwin AG, Freeman S, Wong R, Latta C, Yu S, Jackson J, Fischer N, Koziel V, Pillot T, Bagnall J, Allan SM, Paszek P, Galea J, Harte MK, Eder C, Lawrence CB, Brough D (2016) Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against Alzheimer’s disease in rodent models. Nat Commun 7:12504. https://doi.org/10.1038/ncomms12504
Das B, Dash SP, Mohanty S, Patel P (2021) Parkinson’s disease and impairment in mitochondrial metabolism: a pathognomic signature. Adv Exp Med Biol 1286:65–76. https://doi.org/10.1007/978-3-030-55035-6_4
Davalos D, Grutzendler J, Yang G, Kim JV, Zuo Y, Jung S et al (2005) ATP mediates rapid microglial response to local brain injury in vivo. Nat Neurosci 8(6):752–758
de Rivero Vaccari JP, Lotocki G, Marcillo AE, Dietrich WD, Keane RW (2008) A molecular platform in neurons regulates inflammation after spinal cord injury. J Neurosci 28(13):3404–3414. https://doi.org/10.1523/JNEUROSCI.0157-08.2008
Degan D, Ornello R, Tiseo C, Carolei A, Sacco S, Pistoia F (2018) The role of inflammation in neurological disorders. Curr Pharm Des 24(14):1485–1501. https://doi.org/10.2174/1381612824666180327170632
Denney L, Kok WL, Cole SL, Sanderson S, McMichael AJ, Ho LP (2012) Activation of invariant NKT cells in early phase of experimental autoimmune encephalomyelitis results in differentiation of Ly6Chi inflammatory monocyte to M2 macrophages and improved outcome. J Immunol 189:551–557
Dhir A, Naidu PS, Kulkarni SK (2006a) Effect of cyclooxygenase inhibitors on pentylenetetrazol (PTZ)-induced convulsions: possible mechanism of action. Prog Neuropsychopharmacol Biol Psychiatry 30(8):1478–1485. https://doi.org/10.1016/j.pnpbp.2006.06.003
Dhir A, Naidu PS, Kulkarni SK (2006b) Effect of cyclooxygenase-2 (COX-2) inhibitors in various animal models (bicuculline, picrotoxin, maximal electroshock-induced convulsions) of epilepsy with possible mechanism of action. Indian J Exp Biol 44(4):286–291
Duarte JN (2021) Neuroinflammatory mechanisms of mitochondrial dysfunction and neurodegeneration in glaucoma. J Ophthalmol 2021:4581909. https://doi.org/10.1155/2021/4581909
Dürk T, Panther E, Müller T, Sorichter S, Ferrari D, Pizzirani C, Di Virgilio F, Myrtek D, Norgauer J, Idzko M (2005) 5-Hydroxytryptamine modulates cytokine and chemokine production in LPS-primed human monocytes via stimulation of different 5-HTR subtypes. Int Immunol 17(5):599–606. https://doi.org/10.1093/intimm/dxh242
Edwards FA (2019) A unifying hypothesis for Alzheimer’s disease: from plaques to neurodegeneration. Trends Neurosci 42(5):310–322. https://doi.org/10.1016/j.tins.2019.03.003
Ehrenreich H, Fischer B, Norra C, Schellenberger F, Stender N, Stiefel M, Sirén AL, Paulus W, Nave KA, Gold R, Bartels C (2007) Exploring recombinant human erythropoietin in chronic progressive multiple sclerosis. Brain 130(Pt 10):2577–2588. https://doi.org/10.1093/brain/awm203
Elfakhri KH, Abdallah IM, Brannen AD, Kaddoumi A (2019) Multi-faceted therapeutic strategy for treatment of Alzheimer’s disease by concurrent administration of etodolac and α-tocopherol. Neurobiol Dis 125:123–134. https://doi.org/10.1016/j.nbd.2019.01.020
Erdogan MA, Yigitturk G, Erbas O, Taskıran D (2021) Neuroprotective effects of dexpanthenol on streptozotocin-induced neuronal damage in rats. Drug Chem Toxicol. https://doi.org/10.1080/01480545.2021.1914464
Eugster HP, Frei K, Kopf M, Lassmann H, Fontana A (1998) IL-6-deficient mice resist myelin oligodendrocyte glycoprotein-induced autoimmune encephalomyelitis. Eur J Immunol 28(7):2178–2187. https://doi.org/10.1002/(SICI)1521-4141(199807)28:07%3c2178::AID-IMMU2178%3e3.0.CO;2-D
Fang P, Schachner M, Shen YQ (2012) HMGB1 in development and diseases of the central nervous system. Mol Neurobiol 45(3):499–506. https://doi.org/10.1007/s12035-012-8264-y
Farfel-Becker T, Vitner EB, Pressey SN, Eilam R, Cooper JD, Futerman AH (2011) Spatial and temporal correlation between neuron loss and neuroinflammation in a mouse model of neuronopathic Gaucher disease. Hum Mol Genet 20(7):1375–1386. https://doi.org/10.1093/hmg/ddr019
Fassbender K, Walter S, Kühl S, Landmann R, Ishii K, Bertsch T, Stalder AK, Muehlhauser F, Liu Y, Ulmer AJ, Rivest S, Lentschat A, Gulbins E, Jucker M, Staufenbiel M, Brechtel K, Walter J, Multhaup G, Penke B, Adachi Y, Hartmann T, Beyreuther K (2004) The LPS receptor (CD14) links innate immunity with Alzheimer’s disease. FASEB J 18(1):203–205. https://doi.org/10.1096/fj.03-0364fje
Feigin VL, Vos T, Nichols E, Owolabi MO, Carroll WM, Dichgans M, Deuschl G, Parmar P, Brainin M, Murray C (2020) The global burden of neurological disorders: translating evidence into policy. Lancet Neurol 19(3):255–265. https://doi.org/10.1016/S1474-4422(19)30411-9
Fernandes GL, Araujo P, Tufik S, Andersen ML (2017) The role of IL-6 and STAT in sleep and neuroinflammation. Clin Immunol 180:58–59. https://doi.org/10.1016/j.clim.2017.04.004
Ferrari CC, Pott Godoy MC, Tarelli R, Chertoff M, Depino AM, Pitossi FJ (2006) Progressive neurodegeneration and motor disabilities induced by chronic expression of IL-1beta in the substantia nigra. Neurobiol Dis 24(1):183–193. https://doi.org/10.1016/j.nbd.2006.06.013
Gagne JJ, Power MC (2010) Anti-inflammatory drugs and risk of Parkinson disease: a meta-analysis. Neurology 74(12):995–1002. https://doi.org/10.1212/WNL.0b013e3181d5a4a3
Galic MA, Riazi K, Pittman QJ (2012) Cytokines and brain excitability. Front Neuroendocrinol 33(1):116–125. https://doi.org/10.1016/j.yfrne.2011.12.002
Garcez ML, Mina F, Bellettini-Santos T, Carneiro FG, Luz AP, Schiavo GL, Andrighetti MS, Scheid MG, Bolfe RP, Budni J (2017) Minocycline reduces inflammatory parameters in the brain structures and serum and reverses memory impairment caused by the administration of amyloid β (1–42) in mice. Prog Neuropsychopharmacol Biol Psychiatry 77:23–31. https://doi.org/10.1016/j.pnpbp.2017.03.010
García E, Santana-Martínez R, Silva-Islas CA, Colín-González AL, Galván-Arzate S, Heras Y, Maldonado PD, Sotelo J, Santamaría A (2014) S-allyl cysteine protects against MPTP-induced striatal and nigral oxidative neurotoxicity in mice: participation of Nrf2. Free Radic Res 48(2):159–167. https://doi.org/10.3109/10715762.2013.857019
Garcia-Curran MM, Hall AM, Patterson KP, Shao M, Eltom N, Chen K, Dubé CM, Baram TZ (2019) Dexamethasone attenuates hyperexcitability provoked by experimental febrile status epilepticus. eNeuro. https://doi.org/10.1523/ENEURO.0430-19.2019
Garden GA (2013) Epigenetics and the modulation of neuroinflammation. Neurotherapeutics 10(4):782–788. https://doi.org/10.1007/s13311-013-0207-4
Ghavami S, Cunnington RH, Yeganeh B, Davies JJ, Rattan SG, Bathe K, Kavosh M, Los MJ, Freed DH, Klonisch T, Pierce GN, Halayko AJ, Dixon IM (2012) Autophagy regulates trans fatty acid-mediated apoptosis in primary cardiac myofibroblasts. Biochim Biophys Acta 1823(12):2274–2286. https://doi.org/10.1016/j.bbamcr.2012.09.008
Ghavami S, Shojaei S, Yeganeh B, Ande SR, Jangamreddy JR, Mehrpour M, Christoffersson J, Chaabane W, Moghadam AR, Kashani HH, Hashemi M, Owji AA, Łos MJ (2014) Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog Neurobiol 112:24–49. https://doi.org/10.1016/j.pneurobio.2013.10.004
Gilhus NE, Deuschl G (2019) Neuroinflammation—a common thread in neurological disorders. Nat Rev Neurol 15(8):429–430. https://doi.org/10.1038/s41582-019-0227-8
Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH (2010) Mechanisms underlying inflammation in neurodegeneration. Cell 140(6):918–934. https://doi.org/10.1016/j.cell.2010.02.016
Glick D, Barth S, Macleod KF (2010) Autophagy: cellular and molecular mechanisms. J Pathol 221(1):3–12. https://doi.org/10.1002/path.2697
Gobbo OL, O’Mara SM (2004) Post-treatment, but not pre-treatment, with the selective cyclooxygenase-2 inhibitor celecoxib markedly enhances functional recovery from kainic acid-induced neurodegeneration. Neuroscience 125(2):317–327. https://doi.org/10.1016/j.neuroscience.2004.01.045
Godfred RM, Parikh MS, Haltiner AM, Caylor LM, Sepkuty JP, Doherty MJ (2013) Does aspirin use make it harder to collect seizures during elective video-EEG telemetry? Epilepsy Behav 27(1):115–117. https://doi.org/10.1016/j.yebeh.2012.12.031
Gorter JA, van Vliet EA, Aronica E, Breit T, Rauwerda H, da Silva FHL, Wadman WJ (2006) Potential new antiepileptogenic targets indicated by microarray analysis in a rat model for temporal lobe epilepsy. J Neurosci 26(43):11083–11110
Goudarzi R, Zamanian G, Partoazar A, Dehpour A (2019) Novel effect of Arthrocen (avocado/soy unsaponifiables) on pentylenetetrazole-induced seizure threshold in mice: role of GABAergic pathway. Epilepsy Behav 104(Pt A):106500. https://doi.org/10.1016/j.yebeh.2019.106500
Gu XJ, Xu J, Ma BY, Chen G, Gu PY, Wei D, Hu WX (2014) Effect of glycyrrhizin on traumatic brain injury in rats and its mechanism. Chin J Traumatol 17(1):1–7
Guan PP, Wang P (2019) Integrated communications between cyclooxygenase-2 and Alzheimer’s disease. FASEB J 33(1):13–33. https://doi.org/10.1096/fj.201800355RRRR
Guan Y, Li Y, Zhao G (2018) HMGB1 promotes the starvation-induced autophagic degradation of alpha-synuclein in SH-SY5Y cells Atg 5-dependently. Life Sci 202:1–10
Gugliandolo A, Giacoppo S, Bramanti P, Mazzon E (2018) NLRP3 inflammasome activation in a transgenic amyotrophic lateral sclerosis model. Inflammation 41(1):93–103. https://doi.org/10.1007/s10753-017-0667-5
Guo D, Bi H, Wang D, Wu Q (2013) Zinc oxide nanoparticles decrease the expression and activity of plasma membrane calcium ATPase, disrupt the intracellular calcium homeostasis in rat retinal ganglion cells. Int J Biochem Cell Biol 45(8):1849–1859. https://doi.org/10.1016/j.biocel.2013.06.002
Gupta A, Dhir A, Kumar A, Kulkarni SK (2009) Protective effect of cyclooxygenase (COX)-inhibitors against drug-induced catatonia and MPTP-induced striatal lesions in rats. Pharmacol Biochem Behav 94(2):219–226. https://doi.org/10.1016/j.pbb.2009.07.018
Gupta A, Dhir A, Kumar A, Kulkarni SK (2010) Effect of preferential cyclooxygenase-2 (COX-2) inhibitor against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced striatal lesions in rats: behavioral, biochemical and histological evidences. Indian J Exp Biol 48(6):577–585
Gurung P, Lukens JR, Kanneganti TD (2015) Mitochondria: diversity in the regulation of the NLRP3 inflammasome. Trends Mol Med 21(3):193–201. https://doi.org/10.1016/j.molmed.2014.11.008
Gyengesi E, Münch G (2020) In search of an anti-inflammatory drug for Alzheimer disease. Nat Rev Neurol 16(3):131–132. https://doi.org/10.1038/s41582-019-0307-9
Hammond JW, Lu SM, Gelbard HA (2016) Platelet activating factor enhances synaptic vesicle exocytosis via PKC, elevated intracellular calcium, and modulation of synapsin 1 dynamics and phosphorylation. Front Cell Neurosci 8(9):505. https://doi.org/10.3389/fncel.2015.00505.Erratum.In:FrontCellNeurosci.2016;10:113
Harms AS, Thome AD, Yan Z, Schonhoff AM, Williams GP, Li X, Liu Y, Qin H, Benveniste EN, Standaert DG (2018) Peripheral monocyte entry is required for alpha-Synuclein induced inflammation and Neurodegeneration in a model of Parkinson disease. Exp Neurol 300:179–218
Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, Bar-Or A, Panzara M, Sarkar N, Agarwal S, Langer-Gould A, Smith CH, HERMES Trial Group (2008) B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med 358(7):676–688. https://doi.org/10.1056/NEJMoa0706383
Hawker K, O’Connor P, Freedman MS, Calabresi PA, Antel J, Simon J, Hauser S, Waubant E, Vollmer T, Panitch H, Zhang J, Chin P, Smith CH, OLYMPUS trial group (2009) Rituximab in patients with primary progressive multiple sclerosis: results of a randomized double-blind placebo-controlled multicenter trial. Ann Neurol 66(4):460–471. https://doi.org/10.1002/ana.21867
He P, Cheng X, Staufenbiel M, Li R, Shen Y (2013) Long-term treatment of thalidomide ameliorates amyloid-like pathology through inhibition of β-secretase in a mouse model of Alzheimer’s disease. PLoS ONE 8(2):e55091. https://doi.org/10.1371/journal.pone.0055091
He D, Fu S, Zhou A, Su Y, Gao X, Zhang Y, Huang B, Du J, Liu D (2021) Camptothecin regulates microglia polarization and exerts neuroprotective effects via activating AKT/Nrf2/HO-1 and inhibiting NF-κB pathways in vivo and in vitro. Front Immunol 12:619761. https://doi.org/10.3389/fimmu.2021.619761
Henkel JS, Beers DR, Zhao W, Appel SH (2009) Microglia in ALS: the good, the bad, and the resting. J Neuroimmune Pharmacol 4(4):389–398
Himmerich H, Patsalos O, Lichtblau N, Ibrahim MAA, Dalton B (2019) Cytokine research in depression: principles, challenges, and open questions. Front Psychiatry 10:30. https://doi.org/10.3389/fpsyt.2019.00030
Hirsch EC, Standaert DG (2021) Ten unsolved questions about neuroinflammation in Parkinson’s disease. Mov Disord 36(1):16–24. https://doi.org/10.1002/mds.28075
Holbrook JA, Jarosz-Griffiths HH, Caseley E, Lara-Reyna S, Poulter JA, Williams-Gray CH, Peckham D, McDermott MF (2021) Neurodegenerative disease and the NLRP3 inflammasome. Front Pharmacol 12:643254. https://doi.org/10.3389/fphar.2021.643254
Howard R, Zubko O, Bradley R, Harper E, Pank L, O’Brien J, Fox C, Tabet N, Livingston G, Bentham P, McShane R, Burns A, Ritchie C, Reeves S, Lovestone S, Ballard C, Noble W, Nilforooshan R, Wilcock G, Gray R, Minocycline in Alzheimer Disease Efficacy (MADE) Trialist Group (2020) Minocycline at 2 different dosages vs placebo for patients with mild Alzheimer disease: a randomized clinical trial. JAMA Neurol 77(2):164–174. https://doi.org/10.1001/jamaneurol.2019.3762
Hwang CJ, Lee HP, Choi DY, Jeong HS, Kim TH, Lee TH, Kim YM, Moon DB, Park SS, Kim SY, Oh KW, Hwang DY, Han SB, Lee HJ, Hong JT (2016) Inhibitory effect of thiacremonone on MPTP-induced dopaminergic neurodegeneration through inhibition of p38 activation. Oncotarget 7(30):46943–46958. https://doi.org/10.18632/oncotarget.10504
Iori V, Iyer AM, Ravizza T, Beltrame L, Paracchini L, Marchini S, Cerovic M, Hill C, Ferrari M, Zucchetti M, Molteni M, Rossetti C, Brambilla R, Steve White H, D’Incalci M, Aronica E, Vezzani A (2017) Blockade of the IL-1R1/TLR4 pathway mediates disease-modification therapeutic effects in a model of acquired epilepsy. Neurobiol Dis 99:12–23
Jaiswal P, Mandal M, Mishra A (2020) Effect of hesperidin on fluoride-induced neurobehavioral and biochemical changes in rats. J Biochem Mol Toxicol 34(11):e22575. https://doi.org/10.1002/jbt.22575
Janda E, Boi L, Carta AR (2018) Microglial phagocytosis and its regulation: a therapeutic target in parkinson’s disease? Front Mol Neurosci 11:144. https://doi.org/10.3389/fnmol.2018.00144
Ji J, Xue TF, Guo XD, Yang J, Guo RB, Wang J, Huang JY, Zhao XJ, Sun XL (2018) Antagonizing peroxisome proliferator-activated receptor gamma facilitates M1-to-M2 shift of microglia by enhancing autophagy via the LKB1-AMPK signaling pathway. Aging Cell 17:e12774
Ji Y, Han J, Lee N, Yoon JH, Youn K, Ha HJ, Yoon E, Kim DH, Jun M (2020) Neuroprotective effects of Baicalein, Wogonin, and Oroxylin A on amyloid beta-induced toxicity via NF-κB/MAPK pathway modulation. Molecules 25(21):5087. https://doi.org/10.3390/molecules25215087
Jiang L, Wu X, Wang S, Chen SH, Zhou H, Wilson B, Jin CY, Lu RB, Xie K, Wang Q, Hong JS (2016) Clozapine metabolites protect dopaminergic neurons through inhibition of microglial NADPH oxidase. J Neuroinflammation 13(1):110. https://doi.org/10.1186/s12974-016-0573-z
Jimenez S, Baglietto-Vargas D, Caballero C, Moreno-Gonzalez I, Torres M, Sanchez-Varo R, Ruano D, Vizuete M, Gutierrez A, Vitorica J (2008) Inflammatory response in the hippocampus of PS1M146L/APP751SL mouse model of Alzheimer’s disease: age-dependent switch in the microglial phenotype from alternative to classic. J Neurosci 28(45):11650–11661. https://doi.org/10.1523/JNEUROSCI.3024-08.2008
Jones KA, Maltby S, Plank MW, Kluge M, Nilsson M, Foster PS, Walker FR (2018) Peripheral immune cells infiltrate into sites of secondary neurodegeneration after ischemic stroke. Brain Behav Immun 67:299–307
Kalra J, Kumar P, Majeed AB, Prakash A (2016) Modulation of LOX and COX pathways via inhibition of amyloidogenesis contributes to mitoprotection against β-amyloid oligomer-induced toxicity in an animal model of Alzheimer’s disease in rats. Pharmacol Biochem Behav 146–147:1–12. https://doi.org/10.1016/j.pbb.2016.04.002
Kamata H, Honda S, Maeda S, Chang L, Hirata H, Karin M (2005) Reactive oxygen species promote TNFalpha-induced death and sustained JNK activation by inhibiting MAP kinase phosphatases. Cell 120(5):649–661
Kanamori A, Catrinescu MM, Kanamori N, Mears KA, Beaubien R, Levin LA (2010) Superoxide is an associated signal for apoptosis in axonal injury. Brain 133(9):2612–2625. https://doi.org/10.1093/brain/awq105
Kang R, Chen R, Zhang Q, Hou W, Wu S, Cao L, Huang J, Yu Y, Fan XG, Yan Z, Sun X, Wang H, Wang Q, Tsung A, Billiar TR, Zeh HJ 3rd, Lotze MT, Tang D (2014) HMGB1 in health and disease. Mol Aspects Med 40:1–116. https://doi.org/10.1016/j.mam.2014.05.001
Kappos L, Hartung HP, Freedman MS, Boyko A, Radü EW, Mikol DD, Lamarine M, Hyvert Y, Freudensprung U, Plitz T, van Beek J (2014) Atacicept in multiple sclerosis (ATAMS): a randomised, placebo-controlled, double-blind, phase 2 trial. Lancet Neurol 13(4):353–363. https://doi.org/10.1016/S1474-4422(14)70028-6
Karlmark KR, Tacke F, Dunay IR (2012) Monocytes in health and disease—minireview. Eur J Microbiol Immunol 2:97–102
Karson A, Utkan T, Şahin TD, Balcı F, Arkan S, Ateş N (2021) Etanercept rescues cognitive deficits, depression-like symptoms, and spike-wave discharge incidence in WAG/Rij rat model of absence epilepsy. Epilepsy Behav 115:107532. https://doi.org/10.1016/j.yebeh.2020.107532
Katsumoto A, Lu H, Miranda AS, Ransohoff RM (2014) Ontogeny and functions of central nervous system macrophages. J Immunol 193:2615–2621. https://doi.org/10.4049/jimmunol.1400716
Katyal J, Kumar H, Gupta YK (2015) Anticonvulsant activity of the cyclooxygenase-2 (COX-2) inhibitor etoricoxib in pentylenetetrazole-kindled rats is associated with memory impairment. Epilepsy Behav 44:98–103. https://doi.org/10.1016/j.yebeh.2014.12.032
Kaur D, Sharma V, Deshmukh R (2019) Activation of microglia and astrocytes: a roadway to neuroinflammation and Alzheimer’s disease. Inflammopharmacology 27(4):663–677. https://doi.org/10.1007/s10787-019-00580-x
Karumuri SB, Singh H, Naqvi S, Mishra A, Flora SJS (2019) Impact of chronic low dose exposure of monocrotophos in rat brain: Oxidative/ nitrosative stress, neuronal changes and cholinesterase activity. Toxicol Rep 6:1295–1303. https://doi.org/10.1016/j.toxrep.2019.11.005
Kim HG, Ju MS, Kim DH, Hong J, Cho SH, Cho KH, Park W, Lee EH, Kim SY, Oh MS (2010) Protective effects of Chunghyuldan against ROS-mediated neuronal cell death in models of Parkinson’s disease. Basic Clin Pharmacol Toxicol 107(6):958–964. https://doi.org/10.1111/j.1742-7843.2010.00612.x
Kim BW, Koppula S, Kumar H, Park JY, Kim IW, More SV, Kim IS, Han SD, Kim SK, Yoon SH, Choi DK (2015) α-Asarone attenuates microglia-mediated neuroinflammation by inhibiting NF kappa B activation and mitigates MPTP-induced behavioral deficits in a mouse model of Parkinson's disease. Neuropharmacology 97:46–57. https://doi.org/10.1016/j.neuropharm.2015.04.037. Epub 2015 May 15. Erratum in: Neuropharmacology. 2017;116:444–445
Kim YE, Hwang CJ, Lee HP, Kim CS, Son DJ, Ham YW, Hellström M, Han SB, Kim HS, Park EK, Hong JT (2017) Inhibitory effect of punicalagin on lipopolysaccharide-induced neuroinflammation, oxidative stress and memory impairment via inhibition of nuclear factor-kappaB. Neuropharmacology 117:21–32. https://doi.org/10.1016/j.neuropharm.2017.01.025
Kimizu T, Takahashi Y, Oboshi T, Horino A, Omatsu H, Koike T, Yoshitomi S, Yamaguchi T, Otani H, Ikeda H, Imai K, Shigematsu H (2020) Methylprednisolone pulse therapy in 31 patients with refractory epilepsy: a single-center retrospective analysis. Epilepsy Behav 109:107116. https://doi.org/10.1016/j.yebeh.2020.107116
Kolodziejczak M, Béchade C, Gervasi N, Irinopoulou T, Banas SM, Cordier C, Rebsam A, Roumier A, Maroteaux L (2015) Serotonin modulates developmental microglia via 5-HT2B receptors: potential implication during synaptic refinement of retinogeniculate projections. ACS Chem Neurosci 6(7):1219–1230. https://doi.org/10.1021/cn5003489
Kong ZH, Chen X, Hua HP, Liang L, Liu LJ (2017) The oral pretreatment of glycyrrhizin prevents surgery-induced cognitive impairment in aged mice by reducing neuroinflammation and Alzheimer’s-related pathology via HMGB1 inhibition. J Mol Neurosci 63(3–4):385–395. https://doi.org/10.1007/s12031-017-0989-7
Kostoula C, Shaker T, Cerovic M, Craparotta I, Marchini S, Butti E, Pascente R, Iori V, Garlanda C, Aronica E, Martino G, Ravizza T, Carmant L, Vezzani A (2019) TLR3 preconditioning induces anti-inflammatory and anti-ictogenic effects in mice mediated by the IRF3/IFN-β axis. Brain Behav Immun 81:598–607. https://doi.org/10.1016/j.bbi.2019.07.021
Kübra Elçioğlu H, Kabasakal L, Tufan F, Elçioğlu ÖH, Solakoglu S, Kotil T, Karan MA (2015) Effects of systemic Thalidomide and intracerebroventricular Etanercept and Infliximab administration in a Streptozotocin induced dementia model in rats. Acta Histochem 117(2):176–181. https://doi.org/10.1016/j.acthis.2014.12.002
Kunnanayaka V, Jain P, Sharma S, Seth A, Aneja S (2018) Addition of pyridoxine to prednisolone in the treatment of infantile spasms: a pilot, randomized controlled trial. Neurol India 66(2):385–390. https://doi.org/10.4103/0028-3886.227281
Kurt S, Aygun H (2021) Anticonvulsive effects of edaravone on penicillin-induced focal onset seizure model in the conscious rats. Fundam Clin Pharmacol. https://doi.org/10.1111/fcp.12651
Lamkanfi M, Dixit VM (2014) Mechanisms and functions of inflammasomes. Cell 157(5):1013–1022. https://doi.org/10.1016/j.cell.2014.04.007
Lampron A, Elali A, Rivest S (2013) Innate immunity in the CNS: redefining the relationship between the CNS and Its environment. Neuron 78(2):214–232. https://doi.org/10.1016/j.neuron.2013.04.005
Laurent C, Buée L, Blum D (2018) Tau and neuroinflammation: what impact for Alzheimer’s disease and tauopathies? Biomed J 41(1):21–33. https://doi.org/10.1016/j.bj.2018.01.003
Lecca D, Nevin DK, Mulas G, Casu MA, Diana A, Rossi D, Sacchetti G, Fayne D, Carta AR (2015) Neuroprotective and anti-inflammatory properties of a novel non-thiazolidinedione PPARγ agonist in vitro and in MPTP-treated mice. Neuroscience 302:23–35. https://doi.org/10.1016/j.neuroscience.2015.04.026. Epub 2015 Apr 20. Erratum in: Neuroscience. 2016 Dec 17;339:678. Fayne D [added]
Lee HK, Lund JM, Ramanathan B, Mizushima N, Iwasaki A (2007) Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science 315(5817):1398–1401. https://doi.org/10.1126/science.1136880
Lee EY, Lee JE, Park JH, Shin IC, Koh HC (2012) Rosiglitazone, a PPAR-γ agonist, protects against striatal dopaminergic neurodegeneration induced by 6-OHDA lesions in the substantia nigra of rats. Toxicol Lett 213(3):332–344. https://doi.org/10.1016/j.toxlet.2012.07.016
Lee KM, Lee Y, Chun HJ, Kim AH, Kim JY, Lee JY, Ishigami A, Lee J (2016) Neuroprotective and anti-inflammatory effects of morin in a murine model of Parkinson’s disease. J Neurosci Res 94(10):865–878. https://doi.org/10.1002/jnr.23764
Leng F, Edison P (2021) Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat Rev Neurol 17(3):157–172. https://doi.org/10.1038/s41582-020-00435-y
Leo A, Nesci V, Tallarico M, Amodio N, Gallo Cantafio EM, De Sarro G, Constanti A, Russo E, Citraro R (2020) IL-6 receptor blockade by tocilizumab has anti-absence and anti-epileptogenic effects in the WAG/Rij rat model of absence epilepsy. Neurotherapeutics 17(4):2004–2014. https://doi.org/10.1007/s13311-020-00893-8
Li Y, Chu N, Hu A, Gran B, Rostami A, Zhang GX (2007) Increased IL-23p19 expression in multiple sclerosis lesions and its induction in microglia. Brain 130(Pt 2):490–501. https://doi.org/10.1093/brain/awl273
Li S, Luo Z, Lu B, Xia S, Li C, Guan X, Zhang J, Huang K, Xian F (2019) Protective effects of lycopene on kainic acid-induced seizures. Epilepsy Res 151:1–6. https://doi.org/10.1016/j.eplepsyres.2019.01.010
Li L, Acioglu C, Heary RF, Elkabes S (2021) Role of astroglial toll-like receptors (TLRs) in central nervous system infections, injury and neurodegenerative diseases. Brain Behav Immun 91:740–755. https://doi.org/10.1016/j.bbi.2020.10.007
Liang J, Takeuchi H, Doi Y, Kawanokuchi J, Sonobe Y, Jin S et al (2008) Excitatory amino acid transporter expression by astrocytes is neuroprotective against microglial excitotoxicity. Brain Res 1210:11–19
Lim HW, Park JI, More SV, Park JY, Kim BW, Jeon SB, Yun YS, Park EJ, Yoon SH, Choi DK (2015) Anti-neuroinflammatory effects of DPTP, a novel synthetic clovamide derivative in in vitro and in vivo model of neuroinflammation. Brain Res Bull 112:25–34. https://doi.org/10.1016/j.brainresbull.2015.01.004
Lin WJ, Kuang HY (2014) Oxidative stress induces autophagy in response to multiple noxious stimuli in retinal ganglion cells. Autophagy 10(10):1692–1701. https://doi.org/10.4161/auto.36076
Liu D, Pitta M, Jiang H, Lee JH, Zhang G, Chen X, Kawamoto EM, Mattson MP (2013) Nicotinamide forestalls pathology and cognitive decline in Alzheimer mice: evidence for improved neuronal bioenergetics and autophagy procession. Neurobiol Aging 34(6):1564–1580. https://doi.org/10.1016/j.neurobiolaging.2012.11.020. Epub 2012 Dec 25. Erratum in: Neurobiol Aging. 2013;34(9):e3
Liu Q, Zhang D, Hu D, Zhou X, Zhou Y (2018) The role of mitochondria in NLRP3 inflammasome activation. Mol Immunol 103:115–124. https://doi.org/10.1016/j.molimm.2018.09.010
Liu DH, Agbo E, Zhang SH, Zhu JL (2019) Anticonvulsant and neuroprotective effects of paeonol in epileptic rats. Neurochem Res 44(11):2556–2565. https://doi.org/10.1007/s11064-019-02874-6
Liu R, Wu S, Guo C, Hu Z, Peng J, Guo K, Zhang X, Li J (2020) Ibuprofen exerts antiepileptic and neuroprotective effects in the rat model of pentylenetetrazol-induced epilepsy via the COX-2/NLRP3/IL-18 pathway. Neurochem Res 45(10):2516–2526. https://doi.org/10.1007/s11064-020-03109-9
Loane DJ, Kumar A (2016) Microglia in the TBI brain: the good, the bad, and the dysregulated. Exp Neurol 275 Pt 3(03):316–327. https://doi.org/10.1016/j.expneurol.2015.08.018
Lonnemann N, Hosseini S, Marchetti C, Skouras DB, Stefanoni D, D’Alessandro A, Dinarello CA, Korte M (2020) The NLRP3 inflammasome inhibitor OLT1177 rescues cognitive impairment in a mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA 117(50):32145–32154. https://doi.org/10.1073/pnas.2009680117
Löscher W, Potschka H, Sisodiya SM, Vezzani A (2020) Drug resistance in epilepsy: clinical impact, potential mechanisms, and new innovative treatment options. Pharmacol Rev 72(3):606–638. https://doi.org/10.1124/pr.120.019539
Luft JG, Steffens L, Morás AM, da Rosa MS, Leipnitz G, Regner GG, Pflüger PF, Gonçalves D, Moura DJ, Pereira P (2019) Rosmarinic acid improves oxidative stress parameters and mitochondrial respiratory chain activity following 4-aminopyridine and picrotoxin-induced seizure in mice. Naunyn Schmiedebergs Arch Pharmacol 392(11):1347–1358. https://doi.org/10.1007/s00210-019-01675-6
Maan G, Sikdar B, Kumar A, Shukla R, Mishra A (2020) Role of flavonoids in neurodegenerative diseases: limitations and future perspectives. Curr Top Med Chem 20(13):1169–1194. https://doi.org/10.2174/1568026620666200416085330
Maier A, Deigendesch N, Müller K, Weishaupt JH, Krannich A, Röhle R, Meissner F, Molawi K, Münch C, Holm T, Meyer R, Meyer T, Zychlinsky A (2015) Interleukin-1 antagonist anakinra in amyotrophic lateral sclerosis—a pilot study. PLoS ONE 10(10):e0139684. https://doi.org/10.1371/journal.pone.0139684
Mandal M, Jaiswal P, Mishra A (2020a) Role of curcumin and its nanoformulations in neurotherapeutics: a comprehensive review. J Biochem Mol Toxicol 34(6):e22478
Mandal M, Jaiswal P, Mishra A (2020b) Curcumin loaded nanoparticles reversed monocrotophos induced motor impairment and memory deficit: role of oxidative stress and intracellular calcium level. J Drug Deliv Sci Technol 56:101559. https://doi.org/10.1016/j.jddst.2020.101559
Mantovani A, Sozzani S, Locati M, Allavena P, Sica A (2002) Macrophage polarization: tumor-associated macrophages as a paradigm for polarized M2 mononuclear phagocytes. Trends Immunol 23(11):549–555
Marques NF, Castro AA, Mancini G, Rocha FL, Santos ARS, Prediger RD, De Bem AF, Tasca CI (2018) Atorvastatin prevents early oxidative events and modulates inflammatory mediators in the striatum following intranasal 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) administration in rats. Neurotox Res 33(3):549–559. https://doi.org/10.1007/s12640-017-9840-8
Massa MG, David C, Jorg S, Berg J, Gisevius B, Hirschberg S, Linker RA, Gold R, Haghikia A (2017) Testosterone differentially affects T cells and neurons in murine and human models of neuroinflammation and neurodegeneration. Am J Pathol 187:1613–1622
Mei M, Zhou Y, Liu M, Zhao F, Wang C, Ding J, Lu M, Hu G (2019) Antioxidant and anti-inflammatory effects of dexrazoxane on dopaminergic neuron degeneration in rodent models of Parkinson’s disease. Neuropharmacology 160:107758. https://doi.org/10.1016/j.neuropharm.2019.107758
Mhillaj E, Morgese MG, Tucci P, Furiano A, Luongo L, Bove M, Maione S, Cuomo V, Schiavone S, Trabace L (2018) Celecoxib prevents cognitive impairment and neuroinflammation in soluble amyloid β-treated rats. Neuroscience 372:58–73. https://doi.org/10.1016/j.neuroscience.2017.12.046
Mhillaj E, Papi M, Paciello F, Silvestrini A, Rolesi R, Palmieri V, Perini G, Fetoni AR, Trabace L, Mancuso C (2020) Celecoxib exerts neuroprotective effects in β-amyloid-treated SH-SY5Y Cells through the regulation of heme oxygenase-1: novel insights for an old drug. Front Cell Dev Biol 8:561179. https://doi.org/10.3389/fcell.2020.561179
Miguel-Álvarez M, Santos-Lozano A, Sanchis-Gomar F, Fiuza-Luces C, Pareja-Galeano H, Garatachea N, Lucia A (2015) Non-steroidal anti-inflammatory drugs as a treatment for Alzheimer’s disease: a systematic review and meta-analysis of treatment effect. Drugs Aging 32(2):139–147. https://doi.org/10.1007/s40266-015-0239-z
Mishra A, Goel RK (2016) Chronic 5-HT3 receptor antagonism ameliorates seizures and associated memory deficit in pentylenetetrazole-kindled mice. Neuroscience 339:319–328. https://doi.org/10.1016/j.neuroscience.2016.10.010
Mishra A, Goel RK (2019) Modulatory effect of serotonergic system in pentylenetetrazole-induced seizures and associated memory deficit: role of 5-HT1A and 5-HT2A/2C. J Epilepsy Res 9(2):119–125. https://doi.org/10.14581/jer.19012
Miyazaki I, Asanuma M (2020) Neuron-astrocyte interactions in Parkinson’s disease. Cells 9(12):2623. https://doi.org/10.3390/cells9122623
Montoliu C, Llansola M, Felipo V (2015) Neuroinflammation and neurological alterations in chronic liver diseases. Neuroimmunol Neuroinflam 2:138–144
Moraes RCM, Singulani MP, Gonçalves AC, Portari GV, Torrão ADS (2020) Oral benfotiamine reverts cognitive deficit and increase thiamine diphosphate levels in the brain of a rat model of neurodegeneration. Exp Gerontol 141:111097. https://doi.org/10.1016/j.exger.2020.111097
Morales-Sosa M, Orozco-Suárez S, Vega-García A, Caballero-Chacón S, Feria-Romero IA (2018) Immunomodulatory effect of Celecoxib on HMGB1/TLR4 pathway in a recurrent seizures model in immature rats. Pharmacol Biochem Behav 170:79–86. https://doi.org/10.1016/j.pbb.2018.05.007
Morihara T, Teter B, Yang F, Lim GP, Boudinot S, Boudinot FD, Frautschy SA, Cole GM (2005) Ibuprofen suppresses interleukin-1beta induction of pro-amyloidogenic alpha1-antichymotrypsin to ameliorate beta-amyloid (Abeta) pathology in Alzheimer’s models. Neuropsychopharmacology 30(6):1111–1120. https://doi.org/10.1038/sj.npp.1300668
Moussa C, Hebron M, Huang X, Ahn J, Rissman RA, Aisen PS, Turner RS (2017) Resveratrol regulates neuro-inflammation and induces adaptive immunity in Alzheimer’s disease. J Neuroinflam 14(1):1. https://doi.org/10.1186/s12974-016-0779-0
Mouton-Liger F, Rosazza T, Sepulveda-Diaz J, Ieang A, Hassoun SM, Claire E, Mangone G, Brice A, Michel PP, Corvol JC, Corti O (2018) Parkin deficiency modulates NLRP3 inflammasome activation by attenuating an A20-dependent negative feedback loop. Glia 66(8):1736–1751. https://doi.org/10.1002/glia.23337
Nakanishi H, Wu Z (2009) Microglia-aging: roles of microglial lysosome- and mitochondria-derived reactive oxygen species in brain aging. Behav Brain Res 201(1):1–7
Nimmerjahn A, Kirchhoff F, Helmchen F (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308(5726):1314–1318
Niranjan R, Thakur AK, Mishra A (2016) Food allergy and eosinophilic esophagitis in India: lack of diagnosis. Indian J Gastroenterol 35(1):72–73
Ota M, Matsuo J, Ishida I, Takano H, Yokoi Y, Hori H, Yoshida S, Ashida K, Nakamura K, Takahashi T, Kunugi H (2019) Effects of a medium-chain triglyceride-based ketogenic formula on cognitive function in patients with mild-to-moderate Alzheimer’s disease. Neurosci Lett 690:232–236. https://doi.org/10.1016/j.neulet.2018.10.048
Panicker N, Ge P, Dawson VL, Dawson TM (2021) The cell biology of Parkinson’s disease. J Cell Biol 220(4):e202012095. https://doi.org/10.1083/jcb.202012095
Park G, Kim HG, Ju MS, Ha SK, Park Y, Kim SY, Oh MS (2013) 6-Shogaol, an active compound of ginger, protects dopaminergic neurons in Parkinson’s disease models via anti-neuroinflammation. Acta Pharmacol Sin 34(9):1131–1139. https://doi.org/10.1038/aps.2013.57
Park J, Lee SY, Shon J, Kim K, Lee HJ, Kim KA, Lee BY, Oh SH, Kim NK, Kim OJ (2019) Adalimumab improves cognitive impairment, exerts neuroprotective effects and attenuates neuroinflammation in an Aβ1-40-injected mouse model of Alzheimer’s disease. Cytotherapy 21(6):671–682. https://doi.org/10.1016/j.jcyt.2019.04.054
Parsons CG, Stoffler A, Danysz W (2007) Memantine: a NMDA receptor antagonist that improves memory by restoration of homeostasis in the glutamatergic system–too little activation is bad, too much is even worse. Neuropharmacology 53(6):699–723
Pasqualetti P, Bonomini C, Dal Forno G, Paulon L, Sinforiani E, Marra C, Zanetti O, Rossini PM (2009) A randomized controlled study on effects of ibuprofen on cognitive progression of Alzheimer’s disease. Aging Clin Exp Res 21(2):102–110. https://doi.org/10.1007/BF03325217
Paudel YN, Shaikh MF, Chakraborti A, Kumari Y, Aledo-Serrano Á, Aleksovska K, Alvim MKM, Othman I (2018) HMGB1: a common biomarker and potential target for TBI, neuroinflammation, epilepsy, and cognitive dysfunction. Front Neurosci 12:628. https://doi.org/10.3389/fnins.2018.00628
Pauletti A, Terrone G, Shekh-Ahmad T, Salamone A, Ravizza T, Rizzi M, Pastore A, Pascente R, Liang LP, Villa BR, Balosso S, Abramov AY, van Vliet EA, Del Giudice E, Aronica E, Patel M, Walker MC, Vezzani A (2019) Targeting oxidative stress improves disease outcomes in a rat model of acquired epilepsy. Brain 142(7):e39. https://doi.org/10.1093/brain/awz130
Peng J, Wu S, Guo C, Guo K, Zhang W, Liu R, Li J, Hu Z (2019) Effect of Ibuprofen on autophagy of astrocytes during pentylenetetrazol-induced epilepsy and its significance: an experimental study. Neurochem Res 44(11):2566–2576. https://doi.org/10.1007/s11064-019-02875-5
Perea G, Navarrete M, Araque A (2009) Tripartite synapses: astrocytes process and control synaptic information. Trends Neurosci 32(8):421–431. https://doi.org/10.1016/j.tins.2009.05.001
Piccioni G, Mango D, Saidi A, Corbo M, Nisticò R (2021) Targeting microglia-synapse interactions in Alzheimer’s disease. Int J Mol Sci 22(5):2342. https://doi.org/10.3390/ijms22052342
Poewe W, Seppi K, Tanner CM, Halliday GM, Brundin P, Volkmann J, Schrag AE, Lang AE (2017) Parkinson disease. Nat Rev Dis Primers 3:17013. https://doi.org/10.1038/nrdp.2017.13
Polascheck N, Bankstahl M, Löscher W (2010) The COX-2 inhibitor parecoxib is neuroprotective but not antiepileptogenic in the pilocarpine model of temporal lobe epilepsy. Exp Neurol 224(1):219–233. https://doi.org/10.1016/j.expneurol.2010.03.014
Poly TN, Islam MMR, Yang HC, Li YJ (2019) Non-steroidal anti-inflammatory drugs and risk of Parkinson’s disease in the elderly population: a meta-analysis. Eur J Clin Pharmacol 75(1):99–108. https://doi.org/10.1007/s00228-018-2561-y
Prabowo AS, Anink JJ, Lammens M, Nellist M, van den Ouweland AM, Adle-Biassette H, Sarnat HB, Flores-Sarnat L, Crino PB, Aronica E (2013) Fetal brain lesions in tuberous sclerosis complex: TORC1 activation and inflammation. Brain Pathol 23(1):45–59. https://doi.org/10.1111/j.1750-3639.2012.00616.x
Qi Y, Klyubin I, Cuello AC, Rowan MJ (2018) NLRP3-dependent synaptic plasticity deficit in an Alzheimer’s disease amyloidosis model in vivo. Neurobiol Dis 114:24–30
Qian X, Wang ZR, Zheng JJ, Ding JQ, Zhong JG, Zhang TY, Li W, Zhang M (2019) Baicalein improves cognitive deficits and hippocampus impairments in temporal lobe epilepsy rats. Brain Res 1714:111–118. https://doi.org/10.1016/j.brainres.2019.02.028
Quamar S, Kumar J, Mishra A, Flora SJS (2019) Oxidative stress and neurobehavioural changes in rats following copper exposure and their response to MiADMSA and D-penicillamine. Toxicol Res Appl 3:2397847319844782. https://doi.org/10.1177/2397847319844782
Radu BM, Epureanu FB, Radu M, Fabene PF, Bertini G (2017) Nonsteroidal anti-inflammatory drugs in clinical and experimental epilepsy. Epilepsy Res 131:15–27. https://doi.org/10.1016/j.eplepsyres.2017.02.003
Rana A, Musto AE (2018) The role of inflammation in the development of epilepsy. J Neuroinflam 15(1):144. https://doi.org/10.1186/s12974-018-1192-7
Ransohoff RM (2016) A polarizing question: do M1 and M2 microglia exist? Nat Neurosci 19(8):987–991. https://doi.org/10.1038/nn.4338
Ravikumar B, Sarkar S, Davies JE, Futter M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M, Korolchuk VI, Lichtenberg M, Luo S, Massey DC, Menzies FM, Moreau K, Narayanan U, Renna M, Siddiqi FH, Underwood BR, Winslow AR, Rubinsztein DC (2010) Regulation of mammalian autophagy in physiology and pathophysiology. Physiol Rev 90(4):1383–1435. https://doi.org/10.1152/physrev.00030.2009
Ravizza T, Gagliardi B, Noé F, Boer K, Aronica E, Vezzani A (2008) Innate and adaptive immunity during epileptogenesis and spontaneous seizures: evidence from experimental models and human temporal lobe epilepsy. Neurobiol Dis 29(1):142–160. https://doi.org/10.1016/j.nbd.2007.08.012
Ravizza T, Terrone G, Salamone A, Frigerio F, Balosso S, Antoine DJ, Vezzani A (2018) High Mobility Group Box 1 is a novel pathogenic factor and a mechanistic biomarker for epilepsy. Brain Behav Immun 72:14–21. https://doi.org/10.1016/j.bbi.2017.10.008
Reines SA, Block GA, Morris JC, Liu G, Nessly ML, Lines CR, Norman BA, Baranak CC, Rofecoxib Protocol 091 Study Group (2004) Rofecoxib: no effect on Alzheimer’s disease in a 1-year, randomized, blinded, controlled study. Neurology 62(1):66–71. https://doi.org/10.1212/wnl.62.1.66
Reksidler AB, Lima MM, Zanata SM, Machado HB, da Cunha C, Andreatini R, Tufik S, Vital MA (2007) The COX-2 inhibitor parecoxib produces neuroprotective effects in MPTP-lesioned rats. Eur J Pharmacol 560(2–3):163–175. https://doi.org/10.1016/j.ejphar.2006.12.032
Ren B, Zhang YX, Zhou HX, Sun FW, Zhang ZF, Wei Z, Zhang CY, Si DW (2015) Tanshinone IIA prevents the loss of nigrostriatal dopaminergic neurons by inhibiting NADPH oxidase and iNOS in the MPTP model of Parkinson’s disease. J Neurol Sci 348(1–2):142–152. https://doi.org/10.1016/j.jns.2014.11.026
Reynolds AD, Stone DK, Hutter JA, Benner EJ, Mosley RL, Gendelman HE (2010) Regulatory T cells attenuate Th17 cell-mediated nigrostriatal dopaminergic neurodegeneration in a model of Parkinson’s disease. J Immunol 184:2261–2271
Richard SA, Min W, Su Z, Xu H (2017) High mobility group box 1 and traumatic brain injury. J Behav Brain Sci 7(02):50
Rivers-Auty J, Mather AE, Peters R, Lawrence CB, Brough D (2020) Anti-inflammatories in Alzheimer’s disease-potential therapy or spurious correlate? Brain Commun 2(2):fcaa109. https://doi.org/10.1093/braincomms/fcaa109
Rocha EM, De Miranda B, Sanders LH (2018) Alpha-synuclein: pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol Dis 109(Pt B):249–257. https://doi.org/10.1016/j.nbd.2017.04.004
Rock RB, Gekker G, Hu S, Sheng WS, Cheeran M, Lokensgard JR et al (2004) Role of microglia in central nervous system infections. Clin Microbiol Rev 17(4):942–964
Roseti C, van Vliet E, Cifelli P et al (2015) GABAA currents are decreased by IL-1β in epileptogenic tissue of patients with temporal lobe epilepsy: implications for ictogenesis. Neurobiol Dis 82:311–320
Russo MV, McGavern DB (2016) Inflammatory neuroprotection following traumatic brain injury. Science 353:783–785
Sachdeva AK, Chopra K (2015) Lycopene abrogates Aβ(1–42)-mediated neuroinflammatory cascade in an experimental model of Alzheimer’s disease. J Nutr Biochem 26(7):736–744. https://doi.org/10.1016/j.jnutbio.2015.01.012
Saini N, Akhtar A, Chauhan M, Dhingra N, Pilkhwal SS (2020) Protective effect of indole-3-carbinol, an NF-κB inhibitor in experimental paradigm of Parkinson’s disease: in silico and in vivo studies. Brain Behav Immun 90:108–137. https://doi.org/10.1016/j.bbi.2020.08.001
Sánchez-Sarasúa S, Fernández-Pérez I, Espinosa-Fernández V, Sánchez-Pérez AM, Ledesma JC (2020) Can we treat neuroinflammation in Alzheimer’s disease? Int J Mol Sci 21(22):8751. https://doi.org/10.3390/ijms21228751
Saresella M, La Rosa F, Piancone F, Zoppis M, Marventano I, Calabrese E, Rainone V, Nemni R, Mancuso R, Clerici M (2016) The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol Neurodegener 11:23. https://doi.org/10.1186/s13024-016-0088-1
Sawada M (2009) Neuroprotective and toxic changes in microglia in neurodegenerative disease. Parkinsonism Relat Disord 15(Suppl. 1):S39-41
Sawikr Y, Yarla NS, Peluso I, Kamal MA, Aliev G, Bishayee A (2017) Neuroinflammation in Alzheimer’s disease: the preventive and therapeutic potential of polyphenolic nutraceuticals. Adv Protein Chem Struct Biol 108:33–57. https://doi.org/10.1016/bs.apcsb.2017.02.001
Scaffidi P, Misteli T, Bianchi ME (2002) Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature 418(6894):191–195. https://doi.org/10.1038/nature00858. Erratum in: Nature. 2010 Sep 30;467(7315):622
Schlichtiger J, Pekcec A, Bartmann H, Winter P, Fuest C, Soerensen J, Potschka H (2010) Celecoxib treatment restores pharmacosensitivity in a rat model of pharmacoresistant epilepsy. Br J Pharmacol 160(5):1062–1071. https://doi.org/10.1111/j.1476-5381.2010.00765.x
Schmitt C, Strazielle N, Ghersi-Egea JF (2012) Brain leukocyte infiltration initiated by peripheral inflammation or experimental autoimmune encephalomyelitis occurs through pathways connected to the CSF-filled compartments of the forebrain and midbrain. J Neuroinflam 9:187
Segal BM, Shevach EM (1996) IL-12 unmasks latent autoimmune disease in resistant mice. J Exp Med 184(2):771–775. https://doi.org/10.1084/jem.184.2.771
Shabab T, Khanabdali R, Moghadamtousi SZ, Kadir HA, Mohan G (2017) Neuroinflammation pathways: a general review. Int J Neurosci 127:624–633
Sharma R, Leung WL, Zamani A, O’Brien TJ, Casillas Espinosa PM, Semple BD (2019) Neuroinflammation in post-traumatic epilepsy: pathophysiology and tractable therapeutic targets. Brain Sci 9(11):318. https://doi.org/10.3390/brainsci9110318
Shi X, Chen YH, Liu H, Qu HD (2016) Therapeutic effects of paeonol on methyl-4-phenyl-1,2,3,6-tetrahydropyridine/probenecid-induced Parkinson’s disease in mice. Mol Med Rep 14(3):2397–2404. https://doi.org/10.3892/mmr.2016.5573
Shi J, Gao W, Shao F (2017) Pyroptosis: gasdermin-mediated programmed necrotic cell death. Trends Biochem Sci 42(4):245–254. https://doi.org/10.1016/j.tibs.2016.10.004
Shimada T, Takemiya T, Sugiura H, Yamagata K (2014) Role of inflammatory mediators in the pathogenesis of epilepsy. Mediators Inflamm 2014:901902. https://doi.org/10.1155/2014/901902
Shunan D, Yu M, Guan H, Zhou Y (2021) Neuroprotective effect of Betalain against AlCl3-induced Alzheimer’s disease in Sprague Dawley Rats via putative modulation of oxidative stress and nuclear factor kappa B (NF-κB) signaling pathway. Biomed Pharmacother 137:111369. https://doi.org/10.1016/j.biopha.2021.111369
Sil S, Goswami AR, Dutta G, Ghosh T (2014) Effects of naproxen on immune responses in a colchicine-induced rat model of Alzheimer’s disease. NeuroImmunoModulation 21(6):304–321. https://doi.org/10.1159/000357735
Silk M, Nantz E (2018) Efficacy and safety of tabalumab in patients with relapsing-remitting multiple sclerosis: a randomized, double-blind, placebo-controlled study (P3.397). Neurology 90(15 Suppl):1567–1570
Simard AR, Soulet D, Gowing G, Julien JP, Rivest S (2006) Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer’s disease. Neuron 49(4):489–502. https://doi.org/10.1016/j.neuron.2006.01.022
Simpson DSA, Oliver PL (2020) ROS Generation in microglia: understanding oxidative stress and inflammation in neurodegenerative disease. Antioxidants (basel) 9(8):743. https://doi.org/10.3390/antiox9080743
Solito E, Sastre M (2012) Microglia function in Alzheimer’s disease. Front Pharmacol 3:14. https://doi.org/10.3389/fphar.2012.00014
Sonderegger I, Iezzi G, Maier R, Schmitz N, Kurrer M, Kopf M (2008) GM-CSF mediates autoimmunity by enhancing IL-6-dependent Th17 cell development and survival. J Exp Med 205(10):2281–2294. https://doi.org/10.1084/jem.20071119
Stuve O, Weideman RA, McMahan DM, Jacob DA, Little BB (2020) Diclofenac reduces the risk of Alzheimer’s disease: a pilot analysis of NSAIDs in two US veteran populations. Ther Adv Neurol Disord 13:1756286420935676. https://doi.org/10.1177/1756286420935676
Sung S, Yang H, Uryu K, Lee EB, Zhao L, Shineman D, Trojanowski JQ, Lee VM, Praticò D (2004) Modulation of nuclear factor-kappa B activity by indomethacin influences A beta levels but not A beta precursor protein metabolism in a model of Alzheimer’s disease. Am J Pathol 165(6):2197–2206. https://doi.org/10.1016/s0002-9440(10)63269-5
Takaki J, Fujimori K, Miura M, Suzuki T, Sekino Y, Sato K (2012) L-glutamate released from activated microglia downregulates astrocytic L-glutamate transporter expression in neuroinflammation: the ‘collusion’ hypothesis for increased extracellular L-glutamate concentration in neuroinflammation. J Neuroinflam 9:275
Takeuchi H, Jin S, Wang J, Zhang G, Kawanokuchi J, Kuno R, Sonobe Y, Mizuno T, Suzumura A (2006) Tumor necrosis factor-alpha induces neurotoxicity via glutamate release from hemichannels of activated microglia in an autocrine manner. J Biol Chem 281(30):21362–21368. https://doi.org/10.1074/jbc.M600504200
Takeuchi H, Jin S, Suzuki H, Doi Y, Liang J, Kawanokuchi J et al (2008) Blockade of microglial glutamate release protects against ischemic brain injury. Exp Neurol 214:144–146
Temp FR, Marafiga JR, Milanesi LH, Duarte T, Rambo LM, Pillat MM, Mello CF (2017) Cyclooxygenase-2 inhibitors differentially attenuate pentylenetetrazol-induced seizures and increase of pro- and anti-inflammatory cytokine levels in the cerebral cortex and hippocampus of mice. Eur J Pharmacol 810:15–25. https://doi.org/10.1016/j.ejphar.2017.05.013
Terrone G, Balosso S, Pauletti A, Ravizza T, Vezzani A (2020) Inflammation and reactive oxygen species as disease modifiers in epilepsy. Neuropharmacology 167:107742. https://doi.org/10.1016/j.neuropharm.2019.107742
Tezel G, Wax MB (2000) Increased production of tumor necrosis factor-alpha by glial cells exposed to simulated ischemia or elevated hydrostatic pressure induces apoptosis in cocultured retinal ganglion cells. J Neurosci 20(23):8693–8700. https://doi.org/10.1523/JNEUROSCI.20-23-08693.2000
Thal LJ, Ferris SH, Kirby L, Block GA, Lines CR, Yuen E, Assaid C, Nessly ML, Norman BA, Baranak CC, Reines SA (2005) Rofecoxib Protocol 078 study group. A randomized, double-blind, study of rofecoxib in patients with mild cognitive impairment. Neuropsychopharmacology 30(6):1204–1215. https://doi.org/10.1038/sj.npp.1300690
Tiwari S, Atluri V, Kaushik A, Yndart A, Nair M (2019) Alzheimer’s disease: pathogenesis, diagnostics, and therapeutics. Int J Nanomedicine 14:5541–5554. https://doi.org/10.2147/IJN.S200490
Trandafir CC, Pouliot WA, Dudek FE, Ekstrand JJ (2015) Co-administration of subtherapeutic diazepam enhances neuroprotective effect of COX-2 inhibitor, NS-398, after lithium pilocarpine-induced status epilepticus. Neuroscience 284:601–610. https://doi.org/10.1016/j.neuroscience.2014.10.021
Trempe JF, Fon EA (2013) Structure and function of Parkin, PINK1, and DJ-1, the three musketeers of neuroprotection. Front Neurol 4:38. https://doi.org/10.3389/fneur.2013.00038
Tsai MC, Tanaka K, Overstreet-Wadiche L, Wadiche JI (2012) Neuronal glutamate transporters regulate glial excitatory transmission. J Neurosci 32(5):1528–1535. https://doi.org/10.1523/JNEUROSCI.5232-11.2012
Turrin NP, Rivest S (2004) Innate immune reaction in response to seizures: implications for the neuropathology associated with epilepsy. Neurobiol Dis 16:321–334
Van Bergen NJ, Crowston JG, Craig JE, Burdon KP, Kearns LS, Sharma S, Hewitt AW, Mackey DA, Trounce IA (2015) Measurement of systemic mitochondrial function in advanced primary open-angle glaucoma and leber hereditary optic neuropathy. PLoS ONE 10(10):e0140919. https://doi.org/10.1371/journal.pone.0140919
Van Dam D, Coen K, De Deyn PP (2010) Ibuprofen modifies cognitive disease progression in an Alzheimer’s mouse model. J Psychopharmacol 24(3):383–388. https://doi.org/10.1177/0269881108097630
van Stuijvenberg M, Derksen-Lubsen G, Steyerberg EW, Habbema JD, Moll HA (1998) Randomized, controlled trial of ibuprofen syrup administered during febrile illnesses to prevent febrile seizure recurrences. Pediatrics 102(5):E51. https://doi.org/10.1542/peds.102.5.e51
Vanover KE, Betz AJ, Weber SM, Bibbiani F, Kielaite A, Weiner DM, Davis RE, Chase TN, Salamone JD (2008) A 5-HT2A receptor inverse agonist, ACP-103, reduces tremor in a rat model and levodopa-induced dyskinesias in a monkey model. Pharmacol Biochem Behav 90(4):540–544. https://doi.org/10.1016/j.pbb.2008.04.010
Varvel NH, Bhaskar K, Kounnas MZ, Wagner SL, Yang Y, Lamb BT, Herrup K (2009) NSAIDs prevent, but do not reverse, neuronal cell cycle reentry in a mouse model of Alzheimer disease. J Clin Investig 119(12):3692–3702. https://doi.org/10.1172/JCI39716
Verma V, Singh D, Kh R (2020) Sinapic acid alleviates oxidative stress and neuro-inflammatory changes in sporadic model of Alzheimer’s disease in rats. Brain Sci 10(12):923. https://doi.org/10.3390/brainsci10120923
Vezzani A (2020) Brain inflammation and seizures: evolving concepts and new findings in the last 2 decades. Epilepsy Curr. 20(6_suppl):40S-43S. https://doi.org/10.1177/1535759720948900
Vezzani A, Friedman A (2011) Brain inflammation as a biomarker in epilepsy. Biomark Med 5(5):607–614. https://doi.org/10.2217/bmm.11.61
Vezzani A, Balosso S, Ravizza T (2019) Neuroinflammatory pathways as treatment targets and biomarkers in epilepsy. Nat Rev Neurol 15(8):459–472. https://doi.org/10.1038/s41582-019-0217-x
Viviani B, Bartesaghi S, Gardoni F, Vezzani A, Behrens MM, Bartfai T, Binaglia M, Corsini E, Di Luca M, Galli CL, Marinovich M (2003) Interleukin-1beta enhances NMDA receptor-mediated intracellular calcium increase through activation of the Src family of kinases. J Neurosci 23(25):8692–8700. https://doi.org/10.1523/JNEUROSCI.23-25-08692.2003
Voet S, Srinivasan S, Lamkanfi M, van Loo G (2019) Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol Med 11(6):e10248. https://doi.org/10.15252/emmm.201810248
Wallisch JS, Simon DW, Bayır H, Bell MJ, Kochanek PM, Clark RSB (2017) Cerebrospinal fluid NLRP3 is increased after severe traumatic brain injury in infants and children. Neurocrit Care 27(1):44–50. https://doi.org/10.1007/s12028-017-0378-7
Walsh JG, Muruve DA, Power C (2014) Inflammasomes in the CNS. Nat Rev Neurosci 15(2):84–97. https://doi.org/10.1038/nrn3638
Wattananit S, Tornero D, Graubardt N, Memanishvili T, Monni E, Tatarishvili J, Miskinyte G, Ge R, Ahlenius H, Lindvall O, Schwartz M, Kokaia Z (2016) Monocyte-derived macrophages contribute to spontaneous long-term functional recovery after stroke in mice. J Neurosci 36:4182–4195
Webers A, Heneka MT, Gleeson PA (2020) The role of innate immune responses and neuroinflammation in amyloid accumulation and progression of Alzheimer’s disease. Immunol Cell Biol 98(1):28–41. https://doi.org/10.1111/imcb.12301
Webster KM, Sun M, Crack P, O’Brien TJ, Shultz SR, Semple BD (2017) Inflammation in epileptogenesis after traumatic brain injury. J Neuroinflammation 14(1):10. https://doi.org/10.1186/s12974-016-0786-1
Webster KM, Shultz SR, Ozturk E, Dill LK, Sun M, Casillas-Espinosa P, Jones NC, Crack PJ, O’Brien TJ, Semple BD (2019) Targeting high-mobility group box protein 1 (HMGB1) in pediatric traumatic brain injury: chronic neuroinflammatory, behavioral, and epileptogenic consequences. Exp Neurol 320:112979. https://doi.org/10.1016/j.expneurol.2019.112979
West AP, Shadel GS (2017) Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat Rev Immunol 17(6):363–375. https://doi.org/10.1038/nri.2017.21
Wilcock GK, Black SE, Hendrix SB, Zavitz KH, Swabb EA, Laughlin MA, Tarenflurbil Phase II Study investigators (2008) Efficacy and safety of tarenflurbil in mild to moderate Alzheimer's disease: a randomised phase II trial. Lancet Neurol 7(6):483–493. https://doi.org/10.1016/S1474-4422(08)70090-5. Epub 2008 Apr 29. Erratum in: Lancet Neurol. 2008;7(7):575. Erratum in: Lancet Neurol. 2011;10(4):297
Wilkins HM, Weidling IW, Ji Y, Swerdlow RH (2017) Mitochondria-derived damage-associated molecular patterns in neurodegeneration. Front Immunol 8:508. https://doi.org/10.3389/fimmu.2017.00508
Wu W, Xu H, Wang Z, Mao Y, Yuan L, Luo W, Cui Z, Cui T, Wang XL, Shen YH (2015) PINK1-Parkin-mediated mitophagy protects mitochondrial integrity and prevents metabolic stress-induced endothelial injury. PLoS ONE 10(7):e0132499. https://doi.org/10.1371/journal.pone.0132499
Yabuki Y, Ohizumi Y, Yokosuka A, Mimaki Y, Fukunaga K (2014) Nobiletin treatment improves motor and cognitive deficits seen in MPTP-induced Parkinson model mice. Neuroscience 259:126–141. https://doi.org/10.1016/j.neuroscience.2013.11.051
Yang X, Luo C, Cai J, Powell DW, Yu D, Kuehn MH, Tezel G (2011) Neurodegenerative and inflammatory pathway components linked to TNF-α/TNFR1 signaling in the glaucomatous human retina. Invest Ophthalmol vis Sci 52(11):8442–8454. https://doi.org/10.1167/iovs.11-8152
Yang JS, Wu XH, Yu HG, Teng LS (2017) Tangeretin inhibits neurodegeneration and attenuates inflammatory responses and behavioural deficits in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced Parkinson’s disease dementia in rats. Inflammopharmacology 25(4):471–484. https://doi.org/10.1007/s10787-017-0348-x
Yang C, Mo Y, Xu E, Wen H, Wei R, Li S, Zheng J, Li W, Le B, Chen Y, Pan H, Huang S, Wang S, Wang Q (2019) Astragaloside IV ameliorates motor deficits and dopaminergic neuron degeneration via inhibiting neuroinflammation and oxidative stress in a Parkinson’s disease mouse model. Int Immunopharmacol 75:105651. https://doi.org/10.1016/j.intimp.2019.05.036
Yatsiv I, Morganti-Kossmann MC, Perez D, Dinarello CA, Novick D, Rubinstein M, Otto VI, Rancan M, Kossmann T, Redaelli CA, Trentz O, Shohami E, Stahel PF (2002) Elevated intracranial IL-18 in humans and mice after traumatic brain injury and evidence of neuroprotective effects of IL-18-binding protein after experimental closed head injury. J Cereb Blood Flow Metab 22(8):971–978. https://doi.org/10.1097/00004647-200208000-00008
Yeo IJ, Yun J, Son DJ, Han SB, Hong JT (2020) Antifungal drug miconazole ameliorated memory deficits in a mouse model of LPS-induced memory loss through targeting iNOS. Cell Death Dis 11(8):623. https://doi.org/10.1038/s41419-020-2619-5
Yin J, Zhao F, Chojnacki JE, Fulp J, Klein WL, Zhang S, Zhu X (2018) NLRP3 inflammasome inhibitor ameliorates amyloid pathology in a mouse model of Alzheimer’s disease. Mol Neurobiol 55(3):1977–1987. https://doi.org/10.1007/s12035-017-0467-9
You SJ, Jung DE, Kim HD, Lee HS, Kang HC (2008) Efficacy and prognosis of a short course of prednisolone therapy for pediatric epilepsy. Eur J Paediatr Neurol 12(4):314–320. https://doi.org/10.1016/j.ejpn.2007.09.003
Yu L, Wan F, Dutta S, Welsh S, Liu Z, Freundt E, Baehrecke EH, Lenardo M (2006) Autophagic programmed cell death by selective catalase degradation. Proc Natl Acad Sci USA 103(13):4952–4957. https://doi.org/10.1073/pnas.0511288103
Yu Y, Li L, Nguyen DT, Mustafa SM, Moore BM, Jiang J (2020) Inverse agonism of cannabinoid receptor type 2 confers anti-inflammatory and neuroprotective effects following status epileptics. Mol Neurobiol 57(6):2830–2845. https://doi.org/10.1007/s12035-020-01923-4
Yuan C, Shin M, Park Y, Choi B, Jang S, Lim C, Yun HS, Lee IS, Won SY, Cho KS (2021) Linalool alleviates Aβ42-induced neurodegeneration via suppressing ROS production and inflammation in fly and rat models of Alzheimer’s disease. Oxid Med Cell Longev 2021:8887716. https://doi.org/10.1155/2021/8887716
Yue J, He J, Wei Y, Shen K, Wu K, Yang X, Liu S, Zhang C, Yang H (2020) Decreased expression of Rev-Erbα in the epileptic foci of temporal lobe epilepsy and activation of Rev-Erbα have anti-inflammatory and neuroprotective effects in the pilocarpine model. J Neuroinflamm 17(1):43. https://doi.org/10.1186/s12974-020-1718-7
Zhang W, Shin EJ, Wang T, Lee PH, Pang H, Wie MB, Kim WK, Kim SJ, Huang WH, Wang Y, Zhang W, Hong JS, Kim HC (2006) 3-Hydroxymorphinan, a metabolite of dextromethorphan, protects nigrostriatal pathway against MPTP-elicited damage both in vivo and in vitro. FASEB J 20(14):2496–2511. https://doi.org/10.1096/fj.06-6006com
Zhang Z, Lai D, Wang L, Yu P, Zhu L, Guo B, Xu L, Zhou L, Sun Y, Lee SM, Wang Y (2014) Neuroprotective effects of the andrographolide analogue AL-1 in the MPP+/MPTP-induced Parkinson’s disease model in vitro and in mice. Pharmacol Biochem Behav 122:191–202. https://doi.org/10.1016/j.pbb.2014.03.028
Zhang C, Qin H, Zheng R, Wang Y, Yan T, Huan F, Han Y, Zhu W, Zhang L (2018) A new approach for Alzheimer’s disease treatment through P-gp regulation via ibuprofen. Pathol Res Pract 214(11):1765–1771
Zhang SH, Liu D, Hu Q, Zhu J, Wang S, Zhou S (2019) Ferulic acid ameliorates pentylenetetrazol-induced seizures by reducing neuron cell death. Epilepsy Res 156:106183. https://doi.org/10.1016/j.eplepsyres.2019.106183
Zhang LY, Jin QQ, Hölscher C, Li L (2021) Glucagon-like peptide-1/glucose-dependent insulinotropic polypeptide dual receptor agonist DA-CH5 is superior to exendin-4 in protecting neurons in the 6-hydroxydopamine rat Parkinson model. Neural Regen Res 16(8):1660–1670. https://doi.org/10.4103/1673-5374.303045
Zhou T, Zhu M, Liang Z (2018) (−)-Epigallocatechin-3-gallate modulates peripheral immunity in the MPTP-induced mouse model of Parkinson’s disease. Mol Med Rep 17(4):4883–4888. https://doi.org/10.3892/mmr.2018.8470
Zhu X, Liu J, Chen O, Xue J, Huang S, Zhu W, Wang Y (2019) Neuroprotective and anti-inflammatory effects of isoliquiritigenin in kainic acid-induced epileptic rats via the TLR4/MYD88 signaling pathway. Inflammopharmacology 27(6):1143–1153. https://doi.org/10.1007/s10787-019-00592-7
Acknowledgements
The authors would like to acknowledge the Head of School, School of Pharmaceutical Sciences, Lovely Professional University, Phagwara for the necessary infrastructure and facility. The figures in the manuscript are "Created by BioRender.com".
Funding
Declared none.
Author information
Authors and Affiliations
Contributions
AM: Concept designing, execution, data collection, interpretation, writing; RB: Data collection; PKS: Data Collection; PSM: Data collection and conceptualizing figures; NS: Prrof reading, language editing; NK: Proof reading, language editing; All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This article does not contain any studies with human participants or animals performed by any of the authors.
Conflict of interest
The authors declare that there is no conflict of interest related to this work.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Mishra, A., Bandopadhyay, R., Singh, P.K. et al. Neuroinflammation in neurological disorders: pharmacotherapeutic targets from bench to bedside. Metab Brain Dis 36, 1591–1626 (2021). https://doi.org/10.1007/s11011-021-00806-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11011-021-00806-4