
Abstract
Epilepsy is one of the most common diseases of the central nervous system. Recent studies have shown that a variety of inflammatory mediators play a key role in the pathogenesis of the disease. Ibuprofen (IBP) is a well-known anti-inflammatory agent that reduces the neuroinflammatory response and neuronal damage. In this study, we examined the effect of IBP in a rat model of pentylenetetrazol (PTZ)-induced chronic epilepsy. PTZ injection was given a total of 15 times on alternate days (over a period of 29 days) to induce epilepsy. The effects of IBP were evaluated by behavioral observation, EEG recording, Nissl staining, immunohistochemistry, Western blot analysis, and electrophysiological recording. The results showed that IBP alone affected the expression of cyclooxygenase-2 (COX-2) and neuronal excitability but did not cause epilepsy. IBP reduced seizure scores in the PTZ-treated rats, and it minimized the loss of hippocampal neurons. In addition, IBP decreased the secretion of COX-2, inhibited the activation of the NOD-like receptor 3 inflammasome, and reduced the secretion of the inflammatory cytokine interleukin-18. Furthermore, the results of whole-cell patch-clamp revealed that IBP affected action potential properties, including frequency, latency and duration in epileptic rats, suggesting that it may impact neuronal excitability. These effects of IBP may underlie its antiepileptic and neuroprotective actions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Epilepsy is among the most common and most complex of all neurological diseases, affecting over 70 million people worldwide. The vast majority of patients with epilepsy show long-term repetitive seizures that result in cognitive and behavioral disorders. The patients suffering from epilepsy usually requires lifelong medication therapy. However, approximately 30% of them do not respond to antiepileptic drugs. Therefore, a better understanding of the pathogenesis of epilepsy and novel more effective treatments are urgently needed. Accumulating evidence suggests that inflammatory processes in the brain play a key role in this disease [1, 2]. Thus, anti-inflammatory therapies could have potential for the prevention and treatment of epilepsy.
The inflammasome is a multi-protein complex that was discovered in 2002 [3]. The inflammasome activates inflammatory mediators that in turn induce the secretion of inflammatory cytokines such as interleukin-1β (IL-1β), IL-18, IL-6 and TNF-α [4]. The well-known NOD-like receptor 3 (NLRP3) inflammasome consists of NLRP3, apoptosis-associated speck-like protein (ASC) and pro-caspase-1 [5]. NLRP3 has been linked to many neurological diseases such as Parkinson disease [6], Alzheimer’s disease [7], amyotrophic lateral sclerosis (ALS) [8] and epilepsy. Rong et al. found that inhibiting NLRP3 in a mouse model of pentylenetetrazol (PTZ)-induced epilepsy reduced seizure susceptibility, ameliorated cognitive dysfunction, and decreased the levels of inflammatory cytokines [9]. Cyclooxygenase-2 (COX-2) is a membrane-bound rate-limiting enzyme in the production of prostaglandins and is also involved in the mechanism of inflammation [10]. It has been reported that the level of COX-2 increases rapidly during seizures, which may induce repetitive seizures and cause secondary damage to the brain [11]. Furthermore, both inhibition and knockdown of COX-2 have been shown to inhibit the NLRP3 inflammasome and reduce IL-1β secretion [12, 13]. These observations strongly suggest that inhibition of COX-2, as a strategy to reduce activation of the NLRP3 inflammasome, may have potential for epilepsy treatment.
Ibuprofen (IBP) is one of the safest and oldest non-steroidal anti-inflammatory drugs (NSAIDs). Our previous study showed that IBP decreases the occurrence of seizures [14]. It has been reported that IBP inhibits Ca2+ channels, and this action is related to neuroprotection [15]. IBP also inhibits the neuroinflammatory response and reduces damage to neurons [16]. The anti-inflammatory action of IBP is related to the inhibition of COX-2 [17]. However, the effects of IBP in epilepsy have not been fully researched. In the present study, we investigate the effects and mechanisms of action of IBP in a rat model of PTZ-induced chronic epilepsy.
Materials and Methods
Experimental Animals
Sixty-four Sprague–Dawley (SD) rats (200–250 g), 4–5 weeks-old, were provided by Shandong Jinan Pengyue Experimental Animal Breeding Co. Ltd. The rats were housed in the specific pathogen free (SPF) experimental animal room of Binzhou Medical University Hospital, at a temperature of 20–24 °C, humidity of 50–60%, and a 12 h light/dark cycle. All animals were acclimated for 1 week before experiments. All procedures were performed in accordance with the National Institutes of Health Guide for the care and use of laboratory animals and recommendations of the Ethics Committee of Binzhou Medical University Hospital.
Rat Chronic Epilepsy Model
The chronic epilepsy model was produced by injection of PTZ. This model is widely used to simulate clinical epilepsy [18]. Using a random number table, the rats were randomly divided into the following four groups (n = 16/group): (i) Control group—rats received normal saline (NS) administered intraperitoneally (i.p.); (ii) IBP group—rats received IBP (30 mg/kg, i.p.); (iii) PTZ group—rats received PTZ (35 mg/kg, i.p.; Sigma-Aldrich, St. Louis, MO, USA); (iv) IBP + PTZ group—rats received IBP (30 mg/kg, i.p.) 30 min before PTZ (35 mg/kg, i.p.) injection [19]. NS and all drugs were administered 15 times on alternate days (over a period of 29 days).
Rats were observed for 30 min after each injection. The seizure intensity was scored as follows [20]: stage 0, no response; stage 1, facial rhythmic spat, ear or facial muscle twitch; stage 2, myoclonic convulsions without rearing; stage 3, forelimb clonus, but not upright; stage 4, forelimb clonus with upright posture; stage 5, generalized clonic-tonic seizures with loss of postural control; stage 6, death. Rats with over three consecutive seizures of stage 4 were considered complete kindling seizures [21].
Behavioral Observation and EEG Recording
The behavior during a 30 min period after drug intervention was observed, and the seizure score, complete kindling rate and latency to seizure were analyzed. Abnormal brain discharges after drug intervention were recorded with an electroencephalogram (EEG) apparatus (Nicolet EEG V32 recorder, CareFusion, San Diego, USA) for 30 min. The rats were anesthetized with 4% chloral hydrate (0.4 ml/100 g, i.p.). The rats were fixed to a brain stereotaxic apparatus and implanted with a hypodermic needle electrode. The implantation point was centered on the top, 3.8 mm behind the anterior fontanelle, 2.0 mm before the anterior fontanelle and 2.0 mm either side of the midline, using a binaural electrode as the reference electrode. The recording parameters were as follows: filter at 40 Hz, sensitivity of 70 μV/mm, and a paper speed of 30 mm/s [22].
Tissue Preparation
Each group of 16 rats was randomly divided into two subgroups. The first subgroup of 10 rats was used for immunohistochemistry and Western blot analysis, while the remaining 6 rats were used for whole-cell patch-clamp. The rats were anesthetized with 10% chloral hydrate (0.4 ml/100 g, i.p.) within 4 h of the last drug intervention. After the blink reflex disappeared, the rats were decapitated, and the brain tissue was dissected and placed in an ice tray along the sagittal suture. The brain tissue was then cut into two halves. One half of the brain tissue was removed and quickly stored in the refrigerator at − 80 °C for further experiments. The other half was fixed with neutral formalin buffer, routinely processed for embedding, and sliced (RM2245 paraffin slicer, Leica, Germany).
Nissl Staining
The sections were routinely dewaxed, rehydrated, and stained with toluidine blue (Beijing Leagene Biotechnology Co., Ltd., China) following the manufacturer’s instructions. The sections were then rinsed in distilled water, dehydrated by gradient concentrations of ethanol (70%, 80%, 90%, and 100%), cleared in xylene, and finally coverslipped with neutral balsam. Cell counts in the CA1 area were calculated and analyzed by ImageJ software (NIH, Bethesda, USA). The number of positive cells from the above field (400× fields) was averaged.
Immunohistochemistry (IHC)
The sections were dewaxed, rehydrated, treated with 3% hydrogen peroxide, and subjected to heat for antigen retrieval. The sections were then incubated with rabbit anti-COX-2 polyclonal antibody (1:200, Affinity Biosciences, OH, USA), rabbit anti-NLRP3 polyclonal antibody (1:150, Boster Biological Technology Co., Ltd., Wuhan, China), rabbit anti-caspase-1 polyclonal antibody (1:200, Affinity Biosciences) or rabbit anti-IL-18 polyclonal antibody (1:200, Proteintech Europe, Manchester, UK) overnight at 4 °C. The sections were then incubated with goat anti-rabbit IgG antibody (1:400, OriGene Technologies, Inc., Rockville, USA) at 37 °C for 40 min. Subsequently, all sections were visualized with the DAB kit (Zhongshan Golden Bridge Biotechnology, Beijing, China) and counterstained with hematoxylin. Images were captured on an HPLAS-1000 pathological image analysis system, and 10 visual fields of the hippocampus were randomly selected in each section. (400×). The expression levels of COX-2, NLRP3, caspase-1 and IL-18 were observed, and the mean optical density (MOD) was measured with ImageJ software.
Western Blot
Proteins were extracted from hippocampal tissue on ice, and protein concentrations were measured with the bicinchoninic acid (BCA) assay. The proteins were resolved by SDS-PAGE (10 μg/well) and transferred to PVDF membranes (Millipore, Massachusetts, USA). The membranes were blocked with TBST containing 5% skimmed milk at room temperature for 2 h, and then incubated with rabbit anti-COX-2 polyclonal antibody (1:1500; Affinity Biosciences), rabbit anti-NLRP3 polyclonal antibody (1:1000; Boster Biological Technology Co., Ltd.), rabbit anti-caspase-1 polyclonal antibody (1:2000; Affinity Biosciences) or rabbit anti-IL-18 polyclonal antibody (1:2000; Proteintech Europe) overnight at 4 °C. The blots were thereafter washed with TBST and incubated with HRP-labeled goat anti-rabbit lgG (1:8000; Abcam, CA, USA) at room temperature for 1 h. Signals were detected with enhanced chemiluminescence (ECL; Thermo Fisher, USA) and analyzed.
Slice Recordings and Analysis
Electrophysiological recordings were performed as previously described [23]. Briefly, rats were anesthetized with isoflurane, and the brains were quickly transferred to an ice-cold solution (254 mM sucrose, 3 mM KCl, 2 mM MgCl2, 2 mM CaCl2, 1.25 mM NaH2PO4, 10 mM d-glucose, and 24 mM NaHCO3). Coronal brain slices (300 μm) containing the hippocampus were prepared with a Leica VT1000S vibratome (Leica Microsystems) and allowed to recover at 30 °C for at least 1 h in an oxygenated (95% O2/5% CO2) artificial cerebrospinal fluid solution (124 mM NaCl, 3 mM KCl, 2 mM CaCl2, 1.3 mM MgSO4, 25 mM NaHCO3, 1.2 mM NaH2PO4, 10 mM d-glucose). Patch electrodes with tip resistances between 4 and 7 MΩ were filled with a potassium gluconate-based internal solution (120 mM potassium gluconate, 20 mM KCl, 2 mM MgCl2, 10 mM HEPES, 2 mM ATP, 0.25 mM GTP, and 0.1 mM EGTA adjusted to pH 7.4 and an osmolarity of 295 mOsm). Neurons in the CA1 area of the hippocampus were visualized with a 40× water-immersion lens. Current signals were recorded with a Multiclamp 700B apparatus (molecular devices) and filtered at 2 kHz using a built-in Bessel filter and digitized at 10 kHz. To investigate the firing properties of these neurons, incremental stepwise positive current injections (10 pA) of 1 s duration were given. Data acquisition was terminated when series resistance was >15 MΩ. Data were exported and analyzed with Clampfit 10.6 software. The rheobase is the minimum current to evoke an AP. The input resistance was calculated from voltage-dependent steady-state currents. The latency of APs is the time between the onset of the stimulus and the first evoked AP. The frequency of APs is the number of APs generated over the 1 s current pulse. The amplitude of APs is the voltage difference between the threshold and the peak of the AP. The half-width of APs is the duration of half amplitude.
Statistical Analysis
The data were analyzed with SPSS 26.0 software and expressed as the mean ± SEM. Statistical analysis was performed with Student’s t-test, one-way ANOVA or two-way ANOVA. Values of P < 0.05 were considered significant.
Results
IBP Affects Behavior in Epileptic Rats
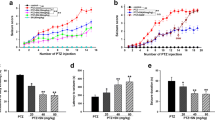
First, we investigated the antiepileptic effect of IBP. Seizure scores were recorded after every administration. We also recorded the complete kindling rate and the latency (the time from PTZ injection to complete kindling seizures occurance) of rats. As shown in Fig. 1a, the control group and the IBP group had no epilepsy. The seizure score of the PTZ group was high, and significant changes were observed in the IBP + PTZ group. IBP (the IBP + PTZ group) decreased the seizure score after every administration compared with the PTZ group. IBP also decreased the rate of complete kindling (Fig. 1b). Furthermore, IBP significantly prolonged the latency to complete kindling seizures (P < 0.01) (Fig. 1c). These results suggest that IBP exerts an antiepileptic effect in rats with PTZ-induced seizures.

Effects of IBP on PTZ-induced seizure. a Comparison of seizure score in each group of rats after every injection (*P < 0.05, **P < 0.01, compared with PTZ group; two-way ANOVA). b Comparison of the complete kindling rate in each group of rats. c Comparison of the latency to complete kindling seizures in each group of rats (**P < 0.01; Student’s t-test). d EEG of each group of rats
IBP Affects EEG Characteristics in Epileptic Rats
Epilepsy is caused by the excessive and synchronous discharge of neurons in the brain, which is observable as abnormal EEG during seizures. The waveform of the EEG in the control group and the IBP group was normal, and no epileptic spike-waves or sharp waves were detected, unlike the PTZ group. The EEG peak in the IBP + PTZ group was lower than that in the PTZ group and showed low amplitude small spike-waves and spike slow complex waves (Fig. 1d). These results reveal that IBP reduces the intensity of seizures in rats with PTZ-induced epilepsy.
IBP Reduces Damage to Hippocampal Neurons in Epileptic Rats
Because spatial learning and memory functions are strongly hippocampus-dependent, we investigated the effects of IBP on hippocampal neuronal damage caused by PTZ-induced kindling. Our results showed that the number of Nissl positive cells was similar between the control group and the IBP group (P > 0.05), but it was decreased in the PTZ group compared with the control group (P < 0.01) (Fig. 2). After IBP intervention (the IBP + PTZ group), the number of Nissl positive cells was increased compared with the PTZ group (P < 0.01), which meaning neuronal cell loss was inhibited (Fig. 2). These results indicating that IBP may have a neuroprotective effect in the rat model of PTZ-induced epilepsy.

Hippocampal cytoarchitecture visualized with Nissl staining. a Nissl-positive hippocampal neurons in the CA1 area in each group. Calibration bars: 50 μm. b Quantitative data of the number of Nissl-positive hippocampal neurons in the CA1 area (**P < 0.01; one-way ANOVA)
IBP Affects the Expression of COX-2 in Epileptic Rats
A previous study demonstrated that COX-2 is activated in rats with temporal lobe epilepsy (TLE). The results of immunohistochemical staining and Western blotting demonstrated that the expression of COX-2 in the IBP group was decreased compared with the control group (P < 0.05), but it tended to be significantly higher in the PTZ group than in the control group (P < 0.01). After the administration of IBP (the IBP + PTZ group), the expression of COX-2 significantly decreased compared with the PTZ group (P < 0.01) (Figs. 3, 4). These results suggest that IBP inhibits the expression of COX-2 in rats with PTZ-induced epilepsy.

Immunohistochemistry showing the expression of COX-2 in the brain tissue of rats in each group. a Immunohistochemistry of COX-2 in the CA1 area of the hippocampus. Calibration bars: 50 μm. b The comparison of the mean optical density (MOD) of COX-2 in different groups (*P < 0.05, **P < 0.01; one-way ANOVA)

Western blot for COX-2 in the brain tissue of rats in each group (*P < 0.05, **P < 0.01; one-way ANOVA)
IBP Impacts the Expression of NLRP3, Caspase-1 and IL-18 in Epileptic Rats
The inflammasome and its downstream inflammatory cytokines play a role in the pathophysiology of epilepsy. Immunohistochemistry and Western blot analysis revealed that a slight reduction of the expression of NLRP3, caspase-1 and IL-18 were observed in the IBP group that was not statistically significant compared with the control group (P > 0.05). The expression of NLRP3, caspase-1 and IL-18 were increased in the PTZ group (P < 0.01). However, they were significantly downregulated in the IBP + PTZ group compared with the PTZ group (P < 0.05) (Figs. 5, 6). These results show that IBP inhibits the expression of NLRP3, caspase-1 and IL-18 in PTZ-kindled epileptic rats.

Immunohistochemistry showing the expression of NLRP3, caspase-1, and IL-18 in the brain tissue of rats in each group. a Immunohistochemistry of NLRP3, caspase-1, and IL-18 in the CA1 area of the hippocampus. Calibration bars: 50 μm. b Comparison of the mean optical densities (MOD) of NLRP3, caspase-1 and IL-18 in the various groups (**P < 0.01; one-way ANOVA)

Western blot for NLRP3, caspase-1 and IL-18 in the brain tissue of rats in each group (*P < 0.05, **P < 0.01; one-way ANOVA)
IBP Affects Action Potentials in Hippocampal Neurons in Epileptic Rats
Neuronal hyperexcitability is an important pathological contributor to seizures. To evaluate intrinsic excitability, the firing responses of hippocampal CA1 neurons to depolarizing current injections were analyzed under current-clamp mode. Our experiments demonstrate that there was a slight difference in the number of action potentials between the control group and the IBP group (Fig. 7a, b). The latency (P < 0.01) and duration (P < 0.05) of action potentials and rheobase (P < 0.05) were significantly higher in the IBP group compared with the control group (Fig. 7c–e). Neurons from the IBP + PTZ group rats generated fewer action potentials than neurons from rats in the PTZ group in response to the same current injection (Fig. 7a, b). The rheobase of hippocampal CA1 neurons was raising in the IBP + PTZ group rats (P < 0.01) (Fig. 7c). Meanwhile, the latency and duration of action potentials were also significantly increased (P < 0.01) (Fig. 7d, e). However, the frequency of action potentials was significantly decreased in neurons from the IBP + PTZ group rats (P < 0.05) (Fig. 7f). Other membrane properties, including input resistance and resting membrane potential, showed no difference between the four groups (P > 0.05) (Fig. 7g, h). In addition, we analyzed the properties of the first action potential evoked by current injection, which revealed that the amplitude and half-width did not differ between the four groups (P > 0.05) (Fig. 7i, j). These results show that IBP affects the action potential of hippocampal neurons in PTZ-kindled epileptic rats, which may, at least in part, underlie its antiepileptic and neuroprotective properties.

Effect of IBP on neuronal hyperexcitability. a Representative traces of action potentials from neurons in the hippocampal CA1 in the various groups. b The number of action potentials (APs) in each group (*P < 0.05, **P < 0.01, compared with control group; #P < 0.05, ##P < 0.01, compared with PTZ group; two-way ANOVA). c–j The rheobase, latency of APs, duration of APs, frequency of APs, resting membrane potential, input resistance, amplitude of APs and half-width of APs, respectively, in each group (*P < 0.05, **P < 0.01; one-way ANOVA)
Discussion
Epilepsy is a highly prevalent disease, which is characterized by recurrent and spontaneous seizures. It can lead to significant impairments in neurobiology, cognition, psychology, and behavior [24]. However, the pathogenesis of epilepsy is complex and still unclear. Studies have shown that inflammatory processes in the brain may result in seizures and the establishment of chronic epileptic foci [25]. The expression of pro-inflammatory cytokines such as IL-1β, IL-2, and IL-6 increased after seizures, while the concentrations were usually low in the brain [26]. It is reported that PTZ-kindled chronic epileptic rats show neuronal damage in the hippocampus, upregulation of pro-inflammatory factors, and an increase in escape latency. Our study showed that, compared with the control group, the rats in the PTZ group had seizures, abnormal EEG characteristics, damaged hippocampal CA1 neurons and increased expression of inflammatory factors such as NLRP3.
NLRP3 inflammasome has been implicated in the pathogenesis of many central nervous system diseases. Various stimuli can activate this cytosolic innate immune signaling receptor. Upon activation, NLRP3 nucleates the assembly of the inflammasome, leading to caspase-1-mediated proteolytic activation of the interleukin family of cytokines, which in turn induces pyroptotic cell death [27]. Studies have shown that the expression of the NLRP3 inflammasome is significantly increased in patients with brain and spinal cord injuries [28]. It was reported that the NLRP3 inflammasome is activated in a rat model of subarachnoid hemorrhage, and that inhibition of the NLRP3 inflammasome improved brain edema and neurological impairment [29]. Notably, studies have shown that NLRP3 and caspase-1 are upregulated in animal models of epilepsy and that inhibition and silencing of NLRP3 and caspase-1 have antiepileptic and neuroprotective effects [30, 31]. In addition, the increase of NLRP3 will cause an increase in the expression of pro-inflammatory cytokines IL-18 [32]. It has been shown that IL-18 inhibition contributes to nerve recovery after craniocerebral injury [33]. These observations strongly suggest that the NLRP3 inflammasome and its downstream inflammatory factors play a key role in neuronal damage in epilepsy. And our study showed that the expression of NLRP3, caspase-1 and IL-18 in the PTZ group was higher than that in the control group. Therefore, effectively inhibiting the expression of NLRP3 can become an important method for the prevention and treatment of epilepsy.
The expression of NLRP3 is regulated by COX-2. COX-2 is an important mediator of neuroinflammation. Evidence shows that COX-2 is elevated in a variety of nervous system diseases. The activation of COX-2 is in connection with neuroinflammation in colchicine induced Alzheimer’s disease rats [34]. COX-2 is expressed at low levels in hippocampal neurons but is significantly upregulated within 1 h after seizures [35]. In rodent models of TLE, the activation of COX-2 in the brain has been shown to promote delayed neuronal damage [36]. In agreement with previous reports, we found here that PTZ upregulated the expression of COX-2 compared with the control group. Therefore, COX-2 is considered a potential neurotherapeutic target for the treatment of epilepsy.
IBP is a classical NSAID, that exerts its main pharmacodynamic effect by inhibiting COX-2. Our experimental results showed that compared with the control group, the expression of COX-2 was decreased in the IBP group but cause no seizures. And the expression of COX-2 was lower in the IBP + PTZ group than that in the PTZ group. According to the literature, inhibition of COX-2 reduces the expression of NLRP3 and its downstream inflammatory effectors [12, 13]. Our experimental results also proved that compared with the PTZ group, the expression of NLRP3, caspase-1 and IL-18 decreased in the IBP + PTZ group. Moreover, our current results suggested that IBP improved behavior and reduced EEG epileptiform activity in PTZ-induced rats. And the hippocampal neuronal damage was reduced in the IBP + PTZ group. Thus, inhibition of the COX-2/NLRP3/IL-18 pathway may contribute to the antiepileptic and neuroprotective effects of IBP.
Neuronal hyperexcitability is an important pathological basis of seizures [37]. Studies have shown that hippocampal hyperexcitability exists even before recurrent spontaneous seizures begin [38]. The hyperexcitability of hippocampal CA1 was found in the epileptic models induced by kainic acid [39] and pilocarpine [40]. Our study results are consistent with the mentioned above studies that the frequency of action potentials was increased, and the latency and duration of action potentials were significantly decreased in hippocampal CA1 neurons from PTZ-induced epileptic rats.
It has been reported that neuronal depolarization and hyperexcitability may be related to the increase of extracellular glutamate levels [41]. N-methyl-d-aspartate (NMDA)-receptor is a subtype of ionic glutamate receptor and its activation induces burst discharges in neurons. IBP has been reported to regulate the function of NMDA receptors [42, 43]. The change of K+ and Ca2+ ion channels is also an important link affecting the excitability of neurons. It has been demonstrated that the T-type Ca2 + channel is related to burst firing in CA1 hippocampal pyramidal neurons in the animal epilepsy model [44]. There is evidence that IBP exerts a neuroprotective effect by inhibiting mitochondrial Ca2+ overload [15]. IBP also inhibits cardiac Na+ and Ca2+ channels [45]. Our experimental results showed that the latency and duration of action potentials and rheobase were significantly higher in the IBP group than them in the control group. Compared with the PTZ group, the rheobase, the latency and duration of action potentials were significantly increased, and the frequency of action potentials was significantly decreased from the IBP + PTZ group rats. The above results are evidence that IBP can reduce seizures by affecting the neuronal excitability in PTZ-induced epileptic rats. However, the specific mechanism of IBP on neuronal excitability, including the effects on NMDA and GABA receptors and various ion channels, need to be further studied.
In conclusion, IBP can reduce seizure scores and hippocampal neuronal damage and affect the excitability of hippocampal neurons in PTZ induced rats through COX-2/NLRP3/IL-18 pathway. These effects of IBP may be the basis of its antiepileptic and neuroprotective actions.
References
Rana A, Musto AE (2018) The role of inflammation in the development of epilepsy. J Neuroinflamm 15(1):144
Mazarati AM, Lewis ML, Pittman QJ (2017) Neurobehavioral comorbidities of epilepsy: role of inflammation. Epilepsia 58(Suppl 3):48–56
Malik A, Kanneganti TD (2017) Inflammasome activation and assembly at a glance. J Cell Sci 130(23):3955–3963
Rathinam VAK, Fitzgerald KA (2016) Inflammasome complexes: emerging mechanisms and effector functions. Cell 165(4):792–800
Shao BZ, Xu ZQ, Han BZ et al (2015) NLRP3 inflammasome and its inhibitors: a review. Front Pharmacol 6:262
Han X, Sun S, Sun Y et al (2019) Small molecule-driven NLRP3 inflammation inhibition via interplay between ubiquitination and autophagy: implications for Parkinson disease. Autophagy 15(11):1860–1881
Li L, Ismael S, Nasoohi S et al (2019) Thioredoxin-interacting protein (TXNIP) associated NLRP3 inflammasome activation in human Alzheimer’s disease brain. J Alzheimers Dis 68(1):255–265
Gugliandolo A, Giacoppo S, Bramanti P et al (2018) NLRP3 inflammasome activation in a transgenic amyotrophic lateral sclerosis model. Inflammation 41(1):93–103
Rong S, Wan D, Fan Y et al (2019) Amentoflavone affects epileptogenesis and exerts neuroprotective effects by inhibiting NLRP3 inflammasome. Front Pharmacol 10:856
Lai ZZ, Yang HL, Ha SY et al (2019) Cyclooxygenase-2 in endometriosis. Int J Biol Sci 15(13):2783–2797
Rojas A, Jiang J, Ganesh T et al (2014) Cyclooxygenase-2 in epilepsy. Epilepsia 55(1):17–25
Hua KF, Chou JC, Ka SM et al (2015) Cyclooxygenase-2 regulates NLRP3 inflammasome-derived IL-1β production. J Cell Physiol 230(4):863–874
Xue Z, Zhang Z, Liu H et al (2019) lincRNA-Cox2 regulates NLRP3 inflammasome and autophagy mediated neuroinflammation. Cell Death Differ 26(1):130–145
Peng J, Wu S, Guo C et al (2019) Effect of ibuprofen on autophagy of astrocytes during pentylenetetrazol-induced epilepsy and its significance: an experimental study. Neurochem Res 44(11):2566–2576
Sanz-Blasco S, Calvo-Rodriguez M, Caballero E et al (2018) Is it all said for NSAIDs in Alzheimer’s disease? Role of mitochondrial calcium uptake. Curr Alzheimer Res 15(6):504–510
Wixey JA, Sukumar KR, Pretorius R et al (2019) Ibuprofen treatment reduces the neuroinflammatory response and associated neuronal and white matter impairment in the growth restricted newborn. Front Physiol 10:541
Orlando BJ, Lucido MJ, Malkowski MG (2015) The structure of ibuprofen bound to cyclooxygenase-2. J Struct Biol 189(1):62–66
Gao B, Wu Y, Yang YJ et al (2018) Sinomenine exerts anticonvulsant profile and neuroprotective activity in pentylenetetrazole kindled rats: involvement of inhibition of NLRP1 inflammasome. J Neuroinflamm 15(1):152
Wallenstein MC (1991) Attenuation of epileptogenesis by nonsteroidal anti-inflammatory drugs in the rat. Neuropharmacology 30(6):657–663
Chindo BA, Schröder H, Becker A (2015) Methanol extract of Ficus platyphylla ameliorates seizure severity, cognitive deficit and neuronal cell loss in pentylenetetrazole-kindled mice. Phytomedicine 22(1):86–93
Zhu X, Shen K, Bai Y et al (2016) NADPH oxidase activation is required for pentylenetetrazole kindling-induced hippocampal autophagy. Free Radic Biol Med 94:230–242
Altwegg-Boussac T, Schramm AE, Ballestero J et al (2017) Cortical neurons and networks are dormant but fully responsive during isoelectric brain state. Brain 140(9):2381–2398
Zhang X, Wu Q, Zhang Q et al (2017) Resveratrol attenuates early brain injury after experimental subarachnoid hemorrhage via inhibition of NLRP3 inflammasome activation. Front Neurosci 11:611
Si J, Wang S, Liu N et al (2017) Anticonvulsant effect of exogenous β-hydroxybutyrate on kainic acid-induced epilepsy. Exp Ther Med 14(1):765–770
Xie Y, Yu N, Chen Y et al (2017) HMGB1 regulates P-glycoprotein expression in status epilepticus rat brains via the RAGE/NF-κB signaling pathway. Mol Med Rep 16(2):1691–1700
Scorza CA, Marques MJG, Gomes da Silva S et al (2018) Status epilepticus does not induce acute brain inflammatory response in the Amazon rodent Proechimys, an animal model resistant to epileptogenesis. Neurosci Lett 668:169–173
Ozaki E, Campbell M, Doyle SL (2015) Targeting the NLRP3 inflammasome in chronic inflammatory diseases: current perspectives. J Inflamm Res 8:15–27
Zendedel A, Johann S, Mehrabi S et al (2016) Activation and regulation of NLRP3 inflammasome by intrathecal application of SDF-1a in a spinal cord injury model. Mol Neurobiol 53(5):3063–3075
Zhang D, Wang X, Wang B et al (2017) Adiponectin regulates contextual fear extinction and intrinsic excitability of dentate gyrus granule neurons through AdipoR2 receptors. Mol Psychiatry 22(7):1044–1055
Shen K, Mao Q, Yin X et al (2018) NLRP3 inflammasome activation leads to epileptic neuronal apoptosis. Curr Neurovasc Res 15(4):276–281
Zhu X, Liu J, Huang S et al (2019) Neuroprotective effects of isoliquiritigenin against cognitive impairment via suppression of synaptic dysfunction, neuronal injury, and neuroinflammation in rats with kainic acid-induced seizures. Int Immunopharmacol 72:358–366
Serdar M, Kempe K, Rizazad M et al (2019) Early pro-inflammatory microglia activation after inflammation-sensitized hypoxic-ischemic brain injury in neonatal rats. Front Cell Neurosci 13:237
Yatsiv I, Morganti-Kossmann MC, Perez D et al (2002) Elevated intracranial IL-18 in humans and mice after traumatic brain injury and evidence of neuroprotective effects of IL-18-binding protein after experimental closed head injury. J Cereb Blood Flow Metab 22(8):971–978
Sil S, Ghosh T (2016) Role of cox-2 mediated neuroinflammation on the neurodegeneration and cognitive impairments in colchicine induced rat model of Alzheimer’s disease. J Neuroimmunol 291:115–124
Vezzani A, Friedman A, Dingledine RJ (2013) The role of inflammation in epileptogenesis. Neuropharmacology 69:16–24
Serrano GE, Lelutiu N, Rojas A et al (2011) Ablation of cyclooxygenase-2 in forebrain neurons is neuroprotective and dampens brain inflammation after status epilepticus. J Neurosci 31(42):14850–14860
Staley K (2015) Molecular mechanisms of epilepsy. Nat Neurosci 18(3):367–372
Navidhamidi M, Ghasemi M, Mehranfard N (2017) Epilepsy-associated alterations in hippocampal excitability. Rev Neurosci 28(3):307–334
Wu K, Leung LS (2003) Increased dendritic excitability in hippocampal ca1 in vivo in the kainic acid model of temporal lobe epilepsy: a study using current source density analysis. Neuroscience 116(2):599–616
Cossart R, Dinocourt C, Hirsch JC et al (2001) Dendritic but not somatic GABAergic inhibition is decreased in experimental epilepsy. Nat Neurosci 4(1):52–62
Unichenko P, Yang JW, Luhmann HJ et al (2015) Glutamatergic system controls synchronization of spontaneous neuronal activity in the murine neonatal entorhinal cortex. Pflugers Arch 467(7):1565–1575
Ozturk Bilgin O, Kumbul Doguc D, Altuntas I, Sutcu R et al (2013) Effects of subchronic treatment with ibuprofen and nimesulide on spatial memory and NMDAR subunits expression in aged rats. Iran J Pharm Res 12(4):877–885
Márquez Loza A, Elias V, Wong CP et al (2017) Effects of ibuprofen on cognition and NMDA receptor subunit expression across aging. Neuroscience 344:276–292
Çarçak N, Ali I, Powell K et al (2019) Ca 3.2 T-type calcium channel mutation influences kindling-induced thalamic neuronal firing patterns in genetic absence epilepsy rats from Strasbourg. Epilepsia 60(7):1378–1386
Yang ZF, Wang HW, Zheng YQ et al (2008) Possible arrhythmiogenic mechanism produced by ibuprofen. Acta Pharmacol Sin 29(4):421–429
Acknowledgements
This work was supported by grants from National Natural Science Foundation of China (to Shuhua Wu, No. 81772637), Shandong Medical and Health Technology Development Plan (to Zhongbo Hu, No. 2017WS553), Binzhou Medical University Science and Technology Plan Project (to Chong Guo, No. BY2015KJ13).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical Approval
All protocols were approved by the Institutional Animal Ethical Committee of Binzhou Medical University Hospital (China), and experiments were performed in accordance to the CPCSEA guidelines for ethical use of animals.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Liu, R., Wu, S., Guo, C. et al. Ibuprofen Exerts Antiepileptic and Neuroprotective Effects in the Rat Model of Pentylenetetrazol-Induced Epilepsy via the COX-2/NLRP3/IL-18 Pathway. Neurochem Res 45, 2516–2526 (2020). https://doi.org/10.1007/s11064-020-03109-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11064-020-03109-9