
Abstract
Corticosterone (CORT) is a kind of corticosteroid produced by cortex of adrenal glands. Hypothalamic–pituitary–adrenal (HPA) axis hyperfunction leads to excessive CORT, which is associated with depression. Few studies have investigated the role of CORT in voltage-gated ion channels and its upstream signaling pathway in central nervous system. In this study, we investigated the mechanism of excessive CORT resulting in brain impairment on voltage-gated ion channels, and its upstream signaling effectors in hippocampal CA1 neurons. The action potential (AP) and voltage-gated potassium currents were determined by using whole-cell patch-clamp. Insulin and CORT improved the neuronal excitability. Independent effects existed in transient potassium channel (IA) and delay rectifier potassium channel (IK). The inhibition of potassium currents, IA in our experiment, could increase neuronal excitability. CORT led to the excitotoxicity of hippocampal neurons via phosphatidylinositol 3 kinase (PI3K)-mediated insulin-signaling pathway. Therefore, the stimulation of excessive CORT induces excitotoxicity of hippocampal neurons and sensitivity of potassium channels via PI3K-mediated insulin-signaling pathway, which indicates one possible way of depression treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The primary physiological function of glucocorticoids (cortisol in humans and corticosterone in rodents) (Grinevich et al. 2012) is to mediate the specificity and magnitude of neural, behavioral and hormonal responses to stress (Lucassen et al. 2014). The impaired feedback regulation of hypothalamic-pituitary- adrenal (HPA) axis results in the increased level of corticosterone (CORT) (Buhl et al. 2010; Lee et al. 2015). Short-term and long-term of excessive CORT have the profound influence on physiological and neurobehavioral functions during adolescent development (Kinlein et al. 2017).
Insulin signaling has been studied extensively in peripheral organ systems and central nervous system (CNS) (Chiu and Cline 2010; Dai et al. 2014). Particularly, it is reported that insulin signaling plays various roles in CNS, including the regulation of neuronal survival, synaptic plasticity, cognition, and memory (Chiu et al. 2008). Insulin resistance can be induced by CORT treatment, including the reduction of associated intracellular pathways and insulin receptor (IR) activation (Buhl et al. 2010; Dobarro et al. 2013; Solas et al. 2013). Chronic CORT treatment can alter gene expression of insulin-signaling pathway, resulting in the impairment of learning and memory function (Osmanovic et al. 2010). In addition to responding through gene-mediated signaling pathways, CORT can also rapidly and reversibly change nongenomic hippocampal signaling within minutes after stress-induced elevation of corticosteroid level (Karst et al. 2005). Short-term CORT administration and peripheral insulin resistance can damage insulin signaling in rat hippocampus (Piroli et al. 2007).
Ion channels are vital to the activation of action potential (AP) and the maintaining of resting membrane potential. AP is a fundamental property of hippocampal neurons, which mainly reflects the excitability of cell membrane. It has been demonstrated that toxic impairments could lead to neuronal excitotoxicity by virtue of modifying AP (Chen et al. 2013). Voltage-gated potassium ion (K+) channels have been demonstrated similar characteristics in pancreatic β-cells, smooth muscle cells and central neurons (Teramoto 2006). K+ channels affect resting membrane potential, influence repolarization and depolarization, and alter waveform and frequency of AP.
Depression is a common debilitating mood disorder and becomes the second disease contributing to the disability worldwide (Cai et al. 2015). Some studies showed that CORT was associated with cognitive impairments and behavior changes, such as depression (Aurand et al. 2016; Grinevich et al. 2012; Lee et al. 2015). CORT suggested a possibility of exploring the mechanism of antidepressants (Aurand et al. 2016). Several effective antidepressant drugs and therapies that regulate cortisol secretion have shown promise in clinical trials (Johnson et al. 2006). However, the definite changes of excessive CORT in voltage-gated ion channels in CNS and its upstream signaling effectors remain unclear. In this study, we constructed an in vitro model of excessive CORT (Karst et al. 2005) to explore its impact on voltage-gated ion channels and insulin-signaling pathway in hippocampal CA1 pyramidal neurons. We reported that the stimulation of excessive CORT induced excitotoxicity of hippocampal neurons and sensitivity of potassium channels via phosphatidylinositol 3 kinase (PI3K)-mediated insulin-signaling pathway, which played an important role in cognitive diseases.
Material and methods
Slice preparation
The experimental protocol was approved by the local Ethical Committee of Nankai University. Male Wistar rats (10-14d on postnatal days) were used in the experiment (Yin et al. 2017). They were purchased from the Laboratory Animal Center, Academy of Military Medical Science of People’s Liberation Army, and temporarily fed in the animal house of Medicine School, Nankai University. After the rats were decapitated, their brains were quickly removed and immersed in an ice-cold and oxygenated (95% O2 and 5% CO2) dissection buffer, containing the following (in mM): 220 sucrose, 2.5 KCl, 6 MgCl2, 1 CaCl2, 1.23 NaH2PO4, 26 NaHCO3 and 10 Glucose, and pH 7.4. Subsequently, hippocampal slices (400 μm thick) were cut using a vibratome (VT1000S, Leica, Germany). They were placed in a water bath kettle and incubated with artificial cerebrospinal fluid (ACSF) containing the following (in mM):125 NaCl, 25 NaHCO3, 1.25 NaH2PO4, 1.25 KCl, 1.5 MgCl2, 2.0 CaCl2 and 10 Glucose, and pH 7.4 (saturated with 95% O2–5% CO2). Then, slices were used for whole-cell patch-clamp recording and Western blot assay.
Drug application
The final concentrations of CORT and insulin were 0.1 μM and 0.5 μM, respectively. CORT was dissolved in ethanol. The concentration of ethanol was less than 0.009% (Karst et al. 2005). Transient outward potassium current (IA) and delay rectifier potassium current (IK) were two types of voltage-dependent potassium channels. IA and IK were separated by 25 mM tetraethylammonium chloride (TEA-Cl) and 3 mM 4-Aminopyrodine (4-AP) (Chen et al. 2014; Liu et al. 2014). Tetrodotoxin (TTX, 1 μM) and CdCl2 (0.2 mM) blocked sodium and calcium channels, respectively. TTX was purchased from the Research Institute of the Aquatic Products of Hebei (China). TEA-Cl, 4-AP, CdCl2, EGTA, HEPES and ATP-Na2 were obtained from Sigma (USA), and other reagents were of A.R. grade.
Whole-cell patch-clamp recording
Whole-cell patch-clamp recordings obtained from the CA1 hippocampal neurons using a patch-clamp amplifier (EPC-10, HEKA, Germany). It was viewed with an upright microscope (BX51WI, Olympus, Japan) and visualized on a television monitor connected to a low-light-sensitive CCD camera (710 M, DVC, USA). After transferred into a glass-bottomed recording chamber, slices were submerged in 1 ml oxygenated ACSF solution. The patch electrodes were made into opening tips of 1-2 μm by a vertical puller (PIP5, HEKA, Germany). Then, electrodes with 4–8 MΩ electrical resistance were filled with standard solution, containing the following (in mM): 140 KCl, 10 HEPES, 10 EGTA, 2 MgCl2·6H2O and 2 ATP-Na2, and pH 7.4. All cells were held at −70 mV when slow and fast capacitance compensation was automatically performed.
Western blot assay
Hippocampal slices were used for Western blot assay. Slices were incubated in ACSF for 1 h before treatment with CORT and insulin. Preparation of tissue lysates has been described in our previous studies (Xu et al. 2012). Equal protein loading was separated by SDS-PAGE, and it was transferred to polyvinylidene-difluoride (PVDF) membranes. Non-specific binding sites on membranes were incubated with blocking buffer, which was 5% fat-free milk powder in Tris-buffered saline including 5% Tween 20 (TBST) for 1 h at room temperature. Membranes were incubated in the following antibodies: primary antibodies (anti-PI3K, 1:2000 dilution, CST; anti-Akt, 1:2000 dilution, CST; anti-p-Akt, 1:2000 dilution, CST; anti-β-actin, 1:2000 dilution, Abcam) overnight at 4 °C and horseradish peroxidase-conjugated secondary antibodies (anti-rabbit IgG, 1:2500 dilution, Invitrogen) for 1 h at room temperature. Blots was detected by a chemiluminescent imaging system (Tanon 5500, Tanon Science & Technology Co., Ltd., China). Ser473 of p-Akt was tested. β-actin was served as an internal control on the same Western blots. The experiments repeated at least three times.
Data acquisition and analysis
Whole-cell patch-clamp data were acquired with a patch-clamp amplifier (EPC-10, HEKA, Germany) and analyzed by Clampfit 10.5, Origin 9 and SPSS 22. Western blot assay data were detected by a chemiluminescent imaging system (Tanon 5500, Tanon Science & Technology Co., Ltd., China) and analyzed by Photoshop CS6, Origin 9 and SPSS 22. Paired sample t test was applied in vehicle and vehicle-insulin groups, CORT and CORT-insulin groups. The activated currents were converted to conductance (G) using the formula: G = I/(Vm - Vr), where Vr was reversal potential. The current-voltage curves, activation curves and steady-state inactivation curves were respectively fitted with Boltzmann function: I = 1/{1 + exp[(Vm – Vh1)/k]}, G/Gmax = 1/{1 + exp[(Vm – Vh2)/k]} and I/Imax = 1/{1 + exp[(Vm – Vh3)/k]}. Vh1 was the voltage of half-maximal activation. Vh2 was the voltage of half-maximal conductance. Vh3 was the voltage of half-maximal inactivation. k was a slope factor. The recovery time of curves was fitted with a mono-exponential function: I/I max = A{1 - exp[Δt/τ]}, where I was current amplitude at time of Δt, A was the coefficient of amplitude and τ was the time constant.
The values were expressed as means ± SEM. p < 0.05 was recognized as significant difference.
Results
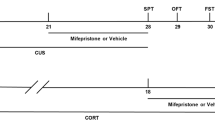
As shown in Fig. 1, there were four groups in our experiment. Before recording, the vehicle group received no treatment and the CORT group was pretreated with CORT for 5 min. The recordings of vehicle and CORT groups were performed 5 min after the establishment of recording pattern. The recordings of vehicle-insulin and CORT -insulin groups were performed 5 min after insulin treatment.

The schematic of four groups with different treatments. -5 min represented the time before the establishment of recording pattern, defined as 0 min. Drugs in the ovals added at the appointed time. Rectangles represented group and recording of electrophysiological indexes at a particular time. The concentration of ethanol was less than 0.009%. cort = corticosterone; ins = insulin
Effects of corticosterone on the excitability of hippocampal neurons
AP is used for describing the property of excitability of hippocampal pyramidal neurons. The frequency of AP was evoked by a 50 pA depolarizing current injection, 500-ms maintained (Fig. 2a). After insulin application, the frequency of AP was significantly increased by 15.50 ± 5.80% (Fig. 2c, p < 0.05, t = −2.67, paired sample t test; n = 6; % of vehicle). However, after pretreatment with CORT, the frequency of AP was not influenced by insulin (Fig. 2c, p > 0.05, t = −0.70, paired sample t test; n = 6; % of CORT). There was a marked increase in the frequency of AP (Fig. 2e, p < 0.01,t = −3.20, independence sample t test; n = 6) induced by CORT itself. The results indicated that insulin or CORT could increase the excitability of neurons. However, insulin had no effect on the AP frequency increased by CORT.

Comparison of firing frequency and half-width of AP before and after application of drugs in four groups. (a, b) A recording example before and after application of drugs in four groups. The evoking firings, depolarizing current (500 ms, 50 pA), stimulated the neurons to acquire the distinction of insulin sensitivity in four groups. AP was evoked by 5 ms brief depolarizing current pulses in four groups. (c, d) Comparison of firing frequency and half-width in four groups. (e) The contrast of firing frequency and half-width between vehicle and cort groups. Data were presented as means ± SEM. * p < 0.05, ** p < 0.01. cort = corticosterone
The half-width of AP was elicited by a 5-ms depolarizing current pulse of 100 pA (Fig. 2b). After treatment with insulin, the half-width of AP increased by 10.06 ± 3.65% (Fig. 2d, p < 0.05, t = −2.76, paired sample t test; n = 6; % of vehicle). After pretreatment with CORT, insulin increased the half-width by 14.37 ± 2.8% (Fig. 2d, p < 0.01, t = −5.12, paired sample t test; n = 6; % of CORT). CORT itself showed no effect on the half-width of AP (Fig. 2e, p > 0.05, t = −1.21, independence sample t test; n = 6). The results implied that insulin had an evident influence on the half-width of AP. CORT showed no effect on the half-width of AP and no mutual interference with insulin.
Effects of corticosterone on the I-V changes of IA and IK
To record IA, the holding potential was −70 mV, and the current traces of IA were evoked by using an 80 ms constant depolarizing pulse from −50 to +90 mV in increments of 10 mV (Fig. 3a). IK was obtained using a 300 ms constant depolarizing pulse by a similar pulse protocol (Fig. 3b). The current-voltage curves of IA and IK in four groups were fitted well with the Boltzmann equation: I = 1/{1 + exp[(Vm–Vh)/k]}. The detailed parameter list was showed in Table 1. After insulin application, IA currents were decreased significantly (Fig. 3c, p < 0.05, t = −3.10, paired sample t test; n = 6; % of vehicle) while IK currents had no significant change (Fig. 3d, p > 0.05, t = −0.14, paired t test; n = 8; % of vehicle) at different command potentials. After pretreatment with CORT, the effect of insulin on IA currents was abolished (Fig. 3c, p > 0.05, t = −0.10, paired t test; n = 6; % of CORT), and the effect of insulin on IK currents had no significant change (Fig. 3d, p > 0.05, t = −2.14, paired sample t test; n = 6; % of CORT). CORT alone had no effect on IA and I K currents (Fig.S1a, b). It showed that insulin could effectively reduce the amplitudes of IA currents. After pretreatment with CORT, however, insulin had no effect on the amplitudes of IA currents.

Effects of corticosterone on the I-V relationship of IA and IK. (a, b) A recording example before and after application of drugs in four groups. IA and IK were obtained by 80 and 300 ms depolarizing pulses, respectively, from a command potential of −50 to +90 mV in increments of 10 mV, and the holding potential was −70 and − 50 mV, respectively. Comparison of the effects of corticosterone on current-voltage activation kinetics curves of IA (c) and IK (d) in four groups. The steady-state activation kinetics curves of IA (e) and IK (f). The current-voltage curves were fitted with a Boltzmann equation. Utilized the eq. G = I/(Vm - Vr), amplitudes of IA and IK currents were converted into conductance and fitted with a Boltzmann function. Data were presented as means ± SEM. * p < 0.05, ** p < 0.01. cort = corticosterone
Effects of corticosterone on the activation kinetics of IA and IK
The activation kinetics curves of IA and IK in four groups were visible (Fig. 3e, Fig. 3f), which were fitted well with the Boltzmann equation: G/Gmax = 1/{1 + exp[(Vm–Vh)/k]}. It showed that after the addition of insulin for 5 min, the activation curves of IA and IK showed no visible alteration. Moreover, after pretreatment with CORT, there were no significant change in the activation curves of IA and IK with insulin application. The detailed parameter list was shown in Table 2. The CORT alone had no influence on the activation curves of IA and IK (Fig.S1c, Fig.S1d).
Effects of corticosterone on the inactivation kinetics of IA
To obtain the steady-state inactivation kinetics, neurons were under the parameter: being at the holding potential of −70 mV, elicited with an 80 ms test pulse to +50 mV, proceeded by 80 ms prepulse to potentials between −110 and + 10 mV of currents (Fig.4a). The amplitudes for IA currents were normalized as I/I max, and the curves were fitted up with the Boltzmann equation: I/Imax = 1/{1 + exp [(Vm – Vh)/k]. The parameter of inactivation curves was displayed in Table 3. It showed that insulin significantly reduced the Vh and increased the slope factor of IA inactivation curves (Fig.4c). With CORT pretreatment, there was no further change following insulin administration. CORT alone showed no effect on the steady-state inactivation (Fig.S1e).

The steady-state inactivation kinetics curves of IA and recovery from inactivation curves kinetics of IA. (a) Normalized steady-state inactivation of IA before and after application of drugs. Currents were elicited with a 80 ms test pulse to +50 mV proceeded by 80 ms prepulse to potentials between −110 and + 10 mV. Steady-state inactivation of IA was normalized as I/Imax and fitted with a Boltzmann function. (c) Effects of corticosterone on IA steady-state inactivation kinetics curves. (b) The recovery from inactivation of IA was well fitted with a mono-exponential function. (d) Effects of corticosterone on the recovery from inactivation of IA. Data were presented as means ± SEM. cort = corticosterone
Effects of corticosterone on the recovery from inactivation of IA
To explore the kinetics of recovery from activation, neurons were under the holding potential of −70 mV and applied with an 80 ms conditioning depolarizing pulse of +50 mV that completely inactivated the transient outward potassium channels. Then neurons were treated with an 80 ms test pulse of +50 mV that was following a series of 80 mV intervals varying from 10 to 265 ms (Fig. 4b). The I2/I1 ratio signified the recovery from inactivation, in which I1 was the peak value of IA evoked by the conditioning pulse, and that of I2 was evoked by the test pulse. The recovery from inactivation of I2/I1 vs. the interval of the 80 mV was well fitted up with a mono-exponential function: I/Imax = A + B exp(−t/τ). It showed that insulin did not influence the recovery from inactivation of IA (Fig.4d). With CORT pretreatment, there were no further change following insulin administration. CORT alone did not make significant change in the recovery from inactivation (Fig.S1f). τ, the parameter of recovery from inactivation of IA, listed in Table 4.
Corticosterone alleviated the activation of insulin-signaling pathway induced by insulin
As we know, the activation of insulin-signaling pathway can upregulate the protein expression of Kv4.2 (Yao et al. 2012), which is the main α-subunit of the transient potassium channels. Insulin activates the IR tyrosine kinase, which phosphorylates and recruits different substrate adaptors. PI3K plays a major role in insulin function and mainly phosphorylates the downstream target protein, Akt. PI3K and Akt were determined by Western blot assay. The expression of PI3K was significantly increased in the vehicle and vehicle-insulin groups (Fig.5b, p < 0.05, t = −5.05, paired sample t test; n = 3; % of vehicle) while no significant difference exists between CORT and CORT-insulin groups (Fig.5c, p > 0.05, t = −0.61, paired sample t test; n = 3; % of CORT). We also examined the possible downstream proteins, Akt. It showed that insulin treatment significantly increased the phosphorylation of Akt compared to that of vehicle group (Fig.5d, p < 0.05, t = 4.53, paired sample t test; n = 3), and the phosphorylation of Akt was increased in the CORT and CORT-insulin groups (Fig.5e, p < 0.05, t = −4.44, paired sample t test; n = 3). CORT alone did not change the expression of PI3K (Fig.5f, p > 0.05, t = −0.36, independence sample t test; n = 3) and the phosphorylation of Akt (Fig.5g, p > 0.05, t = −0.66, independence sample t test; n = 3). These results demonstrated that insulin possibly inhibited transient potassium currents via the activation of PI3K signaling pathway, and CORT alleviated insulin-induced inhibition of transient potassium currents by affecting the activation of PI3K/Akt signaling pathway.

Effects of corticosterone on PI3K/Akt of CA1 neurons. (a) The representative immunoreactive bands of PI3K (80 kDa), p-Akt (60 kDa) and β-actin (43 kDa) of four groups. (b, c) Quantitative analysis of PI3K/β-actin in four groups. (d, e) Quantitative analysis of Akt/p-Akt in four groups. (f, g) Quantitative analysis of PI3K/β-actin and Akt/p-Akt between vehicle and cort groups.* p < 0.05, ** p < 0.01. (n = 3)
Discussion
Stress can induce activation of HPA axis and lead to excessive corticosteroids released into the systemic bloodstream (Russell et al. 2018). Excessive CORT, resulting from the disorder of HPA axis, is vital to the progression of depression (Grinevich et al. 2012; Johnson et al. 2006; Lee et al. 2015; Wieczorek et al. 2015). Chronic CORT treatment induces cellular changes of hippocampus, which can be prevented by antidepressants (Buhl et al. 2010; Murray et al. 2008). Neuroscientists have put forward a principal hypothesis that depression could result from excitotoxic damage to neurons in hippocampus (Kudryashova 2015), which causes cognitive impairments and behavior changes. Exposure to CORT results in cognitive deficits in neurogenesis and synaptic plasticity (Dobarro et al. 2013; Stranahan et al. 2008) or insulin resistant with decreased IR phosphorylation (Solas et al. 2013). In this study, we reported that the stimulation of excessive CORT induced excitotoxicity of hippocampal neurons and sensitivity of potassium channels by PI3K-mediated insulin-signaling pathway,which played an important role in neuronal development and maturation.
AP is the basic characteristic reflecting neuronal excitability on CNS, which is regulated by ion channels in membrane. Toxic impairment or activation of signaling pathways can modify the excitability of neurons (Joseph and Turrigiano 2017; Larimore et al. 2017), which ultimately leads to neuronal excitotoxicity (Chen et al. 2013). We observed that insulin and CORT could increase the excitability of CA1 pyramidal neurons. The increased frequency, induced by CORT, was not increased further by insulin treatment (Fig.2). These results suggest that CORT has mutual effect against insulin-induced excitability.
Many toxins and drugs aim at ion channels of cell membrane. K+ channels are important to repolarization and hyperpolarization, which determine the excitability of neurons. Transient potassium channel (IA) and delay rectifier potassium channel (IK) are two main types of neuronal K+ currents. They can be activated by depolarizing voltage gradually (Schroder et al. 2000). IA plays an important role in repetitive firing of AP while IK is associated with the process of repolarization (Zhang and McBain 1995). In the present study, insulin effectively restrained IA while insulin had no effect on IA with CORT pretreatment (Fig.3). It suggests that CORT alters IA to produce excitotoxicity on neurons and induces insulin resistant. The block of potassium currents, IA in our study, could increase the neuronal excitability. Short-term modulation of IA can arise from a rapid mechanism due to changes in voltage-gating properties or intracellular trafficking of the channel proteins (Yao et al. 2013). We observed that CORT applied acutely to the bath solution significantly increased neuronal excitability without modification of IA activation and inactivation properties, indicating that the mechanism of action of CORT involved long-term effects. Therefore, we explored its upstream signaling mechanism to explain this phenomenon.
The dysregulation of PI3K/Akt pathway is implicated in a number of human diseases including cancer, diabetes, cardiovascular disease and neurological diseases. Insulin enhanced IR phosphorylation and activated potassium channels (Yasui et al. 2008). Chronic CORT inactivated the IR-mediated pathway, including the Akt/GSK-3β pathway, and resulted in the cognitive deficits (Solas et al. 2013). In the present study, we observed that insulin inhibited transient potassium currents via the activation of PI3K-mediated signaling pathway, and CORT possibly alleviated insulin-induced inhibition of transient potassium currents by affecting the activation of PI3K/Akt signaling pathway. However, CORT could not inhibit the activation of downstream signaling effectors of PI3K signaling in hippocampal neurons (Fig. 5). It confirmed that Akt was the important intermediate medium of multiple signaling pathways. A pathway-like picture was to describe the results (Fig. 6).

A pathway-like figure of conclusion description. Insulin could cause changes of AP, K-currents and protein levels. However, all these effects were abolished with CORT pretreatment. The mechanism would rely on the PI3K-mediated insulin-signaling pathway. Cort = corticosterone; ins = insulin; IR = insulin receptor
The connection between stress and depression was drawn from observations of elevated cortisol levels in depressed patients (Gold et al. 2015; Ulloa et al. 2010), which was one of the most robust findings in biological psychiatry (Scott and Dinan 1998). Cognitive impairments in hippocampal function was related to cortisol levels in depressed patients (Brown et al. 2004). The acute increase of glucocorticoid could effectively induce depression (Xiao et al. 2017). In this study, we found that CORT regulated the excitability of hippocampal neurons and the sensitivity of potassium channels through insulin-signaling pathway. It implied a possible mechanism of depression induced by elevated CORT. However, the deeper mechanism of potassium channel activity in depression process remained unknown. Other ion channels may have interferences in potassium channels, such as sodium channels. Due to all these possibilities, further studies need to explore the deeper mechanism of depression, ion channels and insulin-signaling pathway.
Conclusions
Corticosterone induces the neuronal excitotoxicity and possibly alleviates insulin-induced inhibition of transient potassium currents by affecting the activation of PI3K/Akt signaling pathway. Insulin activates insulin-signaling pathway, and the downstream effectors involves potassium channels. Therefore, insulin-signaling pathway is a possible pathway that CORT leads to insulin resistant and depression. Thus, the CORT → insulin resistant → PI3K/Akt → IA axis may play an important role in neuronal development and depression treatment.
References
Aurand J-M et al (2016) Antidepressant-like activity of red wine phenolic extracts in repeated corticosterone-induced depression mice via BDNF/TrkB/CREB signaling pathway. BIO Web of Conferences 7:04009. https://doi.org/10.1051/bioconf/20160704009
Brown ES, Varghese FP, McEwen BS (2004) Association of depression with medical illness: does cortisol play a role? Biol Psychiatry 55:1–9. https://doi.org/10.1016/s0006-3223(03)00473-6
Buhl ES et al (2010) Treatment with an SSRI antidepressant restores hippocampo-hypothalamic corticosteroid feedback and reverses insulin resistance in low-birth-weight rats. Am J Phys Endocrinol Metab 298:E920–E929. https://doi.org/10.1152/ajpendo.00606.2009
Cai S, Huang S, Hao W (2015) New hypothesis and treatment targets of depression: an integrated view of key findings. Neurosci Bull 31:61–74. https://doi.org/10.1007/s12264-014-1486-4
Chen T, Yang J, Ren G, Yang Z, Zhang T (2013) Multi-walled carbon nanotube increases the excitability of hippocampal CA1 neurons through inhibition of potassium channels in rat's brain slices. Toxicol Lett 217:121–128. https://doi.org/10.1016/j.toxlet.2012.12.013
Chen L, Dai J, Wang Z, Zhang H, Huang Y, Zhao Y (2014) Ginseng Total Saponins reverse corticosterone-induced changes in depression-like behavior and hippocampal plasticity-related proteins by interfering with GSK-3 beta -CREB signaling pathway Evid Based Complement Alternat Med : eCAM 2014:506735 https://doi.org/10.1155/2014/506735
Chiu SL, Cline HT (2010) Insulin receptor signaling in the development of neuronal structure and function. Neural Dev 5:18. https://doi.org/10.1186/1749-8104-5-7
Chiu SL, Chen CM, Cline HT (2008) Insulin receptor signaling regulates synapse number, dendritic plasticity, and circuit function in vivo. Neuron 58:708–719. https://doi.org/10.1016/j.neuron.2008.04.014
Dai W, Yang J, Chen T, Yang Z (2014) Protective effects of bexarotene against amyloid-beta25-35-induced dysfunction in hippocampal neurons through the insulin signaling pathway. Neurodegener Dis 14:77–84. https://doi.org/10.1159/000358397
Dobarro M, Orejana L, Aguirre N, Ramirez MJ (2013) Propranolol reduces cognitive deficits, amyloid beta levels, tau phosphorylation and insulin resistance in response to chronic corticosterone administration. Int J Neuropsychopharmacol 16:1351–1360. https://doi.org/10.1017/s1461145712001393
Gold PW, Machado-Vieira R, Pavlatou MG (2015) Clinical and biochemical manifestations of depression: relation to the neurobiology of stress. Neural Plasticity 2015:1–11. https://doi.org/10.1155/2015/581976
Grinevich V, Seeburg PH, Schwarz MK, Jezova D (2012) Homer 1 – a new player linking the hypothalamic-pituitary-adrenal axis activity to depression and anxiety. Endocr Regul 46:153–159. https://doi.org/10.4149/endo_2012_03_153
Johnson SA, Fournier NM, Kalynchuk LE (2006) Effect of different doses of corticosterone on depression-like behavior and HPA axis responses to a novel stressor. Behav Brain Res 168:280–288. https://doi.org/10.1016/j.bbr.2005.11.019
Joseph A, Turrigiano GG (2017) All for one but not one for all: excitatory synaptic scaling and intrinsic excitability are Coregulated by CaMKIV, whereas inhibitory synaptic scaling is under independent control. J Neurosci 37:6778–6785. https://doi.org/10.1523/jneurosci.0618-17.2017
Karst H, Berger S, Turiault M, Tronche F, Schutz G, Joels M (2005) Mineralocorticoid receptors are indispensable for nongenomic modulation of hippocampal glutamate transmission by corticosterone. Proc Natl Acad Sci U S A 102:19204–19207. https://doi.org/10.1073/pnas.0507572102
Kinlein SA, Shahanoor Z, Romeo RD, Karatsoreos IN (2017) Chronic corticosterone treatment during adolescence has significant effects on metabolism and skeletal development in male C57BL6/N mice. Endocrinology 158:2239–2254. https://doi.org/10.1210/en.2017-00208
Kudryashova IV (2015) Neurodegenerative changes in depression: excitotoxicity or a deficit of trophic factors? Neurochem J 9:1–7. https://doi.org/10.1134/s1819712415010043
Larimore J et al (2017) Dysbindin deficiency modifies the expression of GABA neuron and ion permeation transcripts in the developing Hippocampus. Front Genet 8:14. https://doi.org/10.3389/fgene.2017.00028
Lee B, Sur B, Shim I, Lee H, Hahm DH (2015) Angelica gigas ameliorate depression-like symptoms in rats following chronic corticosterone injection. BMC Complement Altern Med 15:210. https://doi.org/10.1186/s12906-015-0746-9
Liu DH et al (2014) Voltage dependent Potassium Channel remodeling in murine intestinal smooth muscle hypertrophy induced by partial obstruction. PLoS One 9:12. https://doi.org/10.1371/journal.pone.0086109
Lucassen PJ et al (2014) Neuropathology of stress. Acta Neuropathol 127:109–135. https://doi.org/10.1007/s00401-013-1223-5
Murray F, Smith DW, Hutson PH (2008) Chronic low dose corticosterone exposure decreased hippocampal cell proliferation, volume and induced anxiety and depression like behaviours in mice. Eur J Pharmacol 583:115–127. https://doi.org/10.1016/j.ejphar.2008.01.014
Osmanovic J, Plaschke K, Salkovic-Petrisic M, Gruenblatt E, Riederer P, Hoyer S (2010) Chronic exogenous corticosterone administration generates an insulin-resistant brain state in rats stress-the international. Journal on the Biology of Stress 13:123–131. https://doi.org/10.3109/10253890903080379
Piroli GG, Grillo CA, Reznikov LR, Adams S, McEwen BS, Charron MJ, Reagan LP (2007) Corticosterone impairs insulin-stimulated translocation of GLUT4 in the rat hippocampus. Neuroendocrinology 85:71–80. https://doi.org/10.1159/000101694
Russell AL, Tasker JG, Lucion AB, Fiedler J, Munhoz CD, Wu TJ, Deak T (2018) Factors promoting vulnerability to dysregulated stress reactivity and stress-related disease. J Neuroendocrinol:e12641. https://doi.org/10.1111/jne.12641
Schroder W, Hinterkeuser S, Seifert G, Schramm J, Jabs R, Wilkin GP, Steinhauser C (2000) Functional and molecular properties of human astrocytes in acute hippocampal slices obtained from patients with temporal lobe epilepsy. Epilepsia 41:S181–S184. https://doi.org/10.1111/j.1528-1157.2000.tb01578.x
Scott LV, Dinan TG (1998) Vasopressin and the regulation of hypothalamic-pituitary-adrenal axis function: implications for the pathophysiology of depression. Life Sci 62:1985–1998
Solas M, Gerenu G, Gil-Bea FJ, Ramirez MJ (2013) Mineralocorticoid receptor activation induces insulin resistance through c-Jun N-terminal kinases in response to chronic corticosterone: cognitive implications. J Neuroendocrinol 25:350–356. https://doi.org/10.1111/jne.12006
Stranahan AM, Arumugam TV, Cutler RG, Lee K, Egan JM, Mattson MP (2008) Diabetes impairs hippocampal function through glucocorticoid-mediated effects on new and mature neurons. Nat Neurosci 11:309–317. https://doi.org/10.1038/nn2055
Teramoto N (2006) Physiological roles of ATP-sensitive K+ channels in smooth muscle. J Physiol 572:617–624. https://doi.org/10.1113/jphysiol.2006.105973
Ulloa JL et al (2010) Comparison of the antidepressant sertraline on differential depression-like behaviors elicited by restraint stress and repeated corticosterone administration. Pharmacol Biochem Behav 97:213–221. https://doi.org/10.1016/j.pbb.2010.08.001
Wieczorek L, Fish EW, O'Leary-Moore SK, Parnell SE, Sulik KK (2015) Hypothalamic-pituitary-adrenal axis and behavioral dysfunction following early binge-like prenatal alcohol exposure in mice. Alcohol 49:207–217. https://doi.org/10.1016/j.alcohol.2015.01.005
Xiao X, Zhang H, Wang H, Li Q, Zhang T (2017) Neuroprotective effect of amantadine on corticosterone-induced abnormal glutamatergic synaptic transmission of CA3-CA1 pathway in rat's hippocampal slices 71 https://doi.org/10.1002/syn.22010
Xu P, Xu J, Li Z, Yang Z (2012) Expression of TRPC6 in renal cortex and hippocampus of mouse during postnatal development. PLoS One 7:e38503. https://doi.org/10.1371/journal.pone.0038503
Yao JJ, Gao XF, Chow CW, Zhan XQ, Hu CL, Mei YA (2012) Neuritin activates insulin receptor pathway to up-regulate Kv4.2-mediated transient outward K+ current in rat cerebellar granule neurons. J Biol Chem 287:41534–41545. https://doi.org/10.1074/jbc.M112.390260
Yao JJ, Sun J, Zhao QR, Wang CY, Mei YA (2013) Neuregulin-1/ErbB4 signaling regulates Kv4.2-mediated transient outward K+ current through the Akt/mTOR pathway. Am J Physiol Cell Physiol 305:C197–C206. https://doi.org/10.1152/ajpcell.00041.2013
Yasui S et al (2008) Insulin activates ATP-sensitive potassium channels via phosphatidylinositol 3-kinase in cultured vascular smooth muscle cells. J Vasc Res 45:233–243. https://doi.org/10.1159/000112545
Yin H, Wang H, Zhang H, Gao N, Zhang T, Yang Z (2017) Resveratrol attenuates Aβ-induced early hippocampal neuron excitability impairment via recovery of function of potassium channels. Neurotox Res. https://doi.org/10.1007/s12640-017-9726-9
Zhang L, McBain CJ (1995) Potassium conductances underlying repolarization and after-hyperpolarization in rat CA1 hippocampal interneurones. J Physiol 488(Pt 3):661–672
Acknowledgements
This study was supported by grants from the National Natural Science Foundation of China (81571804, 81771979).
Author information
Authors and Affiliations
Contributions
Zhuo Yang, Qingqing Xia and Hui Wang conceived the study and designed the experiments. Qingqing Xia performed all experiments and data analysis, wrote the manuscript and generated the figures. Hongqiang Yin provided the guidance of technique and data analysis. All authors have read and approved the manuscript.
Corresponding author
Ethics declarations
All animal experiments were approved by the Animal Research Ethics Committee, School of Medicine, Nankai University and were performed in accordance with the Animal Management Rules of the Ministry of Health of the People’s Republic of China.
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
ESM 1
(DOCX 212 kb)
Rights and permissions
About this article

Cite this article
Xia, Q., Wang, H., Yin, H. et al. Excessive corticosterone induces excitotoxicity of hippocampal neurons and sensitivity of potassium channels via insulin-signaling pathway. Metab Brain Dis 34, 119–128 (2019). https://doi.org/10.1007/s11011-018-0326-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11011-018-0326-z