
Abstract
Sustained elevation of corticosterone (CORT) is one of the common causes of aging and major depression disorder. However, the role of elevated CORT in late life depression (LLD) has not been elucidated. In this study, 18-month-old female rats were subjected to bilateral adrenalectomy or sham surgery. Their CORT levels in plasma were adjusted by CORT replacement and the rats were divided into high-level CORT (H-CORT), low-level CORT (L-CORT), and Sham group. We showed that L-CORT rats displayed attenuated depressive symptoms and memory defects in behavioral tests as compared with Sham or H-CORT rats. Furthermore, we showed that glutamatergic transmission was enhanced in L-CORT rats, evidenced by enhanced population spike amplitude (PSA) recorded from the dentate gyrus of hippocampus in vivo and increased glutamate release from hippocampal synaptosomes caused by high frequency stimulation or CORT exposure. Intracerebroventricular injection of an enzymatic glutamate scavenger system, glutamic-pyruvic transmine (GPT, 1 μM), significantly increased the PSA in Sham rats, suggesting that extracelluar accumulation of glutamate might be the culprit of impaired glutamatergic transmission, which was dependent on the uptake by Glt-1 in astrocytes. We revealed that hippocampal Glt-1 expression level in the L-CORT rats was much higher than in Sham and H-CORT rats. In a gradient neuron–astrocyte coculture, we found that the expression of Glt-1 was decreased with the increase of neural percentage, suggesting that impairment of Glt-1 might result from the high level of CORT contributed neural damage. In sham rats, administration of DHK that inhibited Glt-1 activity induced significant LLD symptoms, whereas administration of RIL that promoted glutamate uptake significantly attenuated LLD. All of these results suggest that glutamatergic transmission impairment is one of important pathogenesis in LLD induced by high level of CORT, which provide promising clues for the treatment of LLD.
Similar content being viewed by others


Introduction
Major depressive disorder (MDD) has been estimated to be the second most prevalent public health problem by 2030, especially in aged populations. As many as 10% of individuals over 65 years old in primary care settings suffer from major depression and dysthymic disorder [1], which are characterized by anhedonia and cognitive impairment [2, 3]. However, the available antidepressants commonly cause more side effects in aged patients than in young adults, including QT prolongation [4] and hypertension [1], indicating that traditional antidepressants are not suitable for the treatment of late life depression (LLD).
Dysfunction of glutamatergic neurotransmission is increasingly considered to be a core feature of stress-related mental illness, which occurs predominantly within the confines of a tripartite synapse and involves basal and activated presynaptic glutamate release, postsynaptic receptor trafficking and function, and transporter-mediated uptake and recycling of glutamate through the glutamate/glutamine cycle. It has been demonstrated that several regulatory processes within the synapse are sensitive to stress and elevated corticosterone (CORT) [5]. For example, acute exposure to stress or administration of CORT rapidly induces glutamate release in the hippocampus [6], and 100 nM CORT significantly increases the frequency of excitatory postsynaptic potentials in CA1 pyramidal neurons [7]. Approximately 30% of aged rats experienced basal CORT hypersecretion, which was correlated with cognitive impairment and hippocampal atrophy [8]. Artificially lowering the concentration of CORT during middle age significantly reduced the development of both memory dysfunction and hippocampal atrophy [9, 10]. However, the relationship between glutamatergic transmission and CORT levels in late life is still unclear.
Substantial evidence suggests that the hippocampus is involved in the pathogenesis of LLD [11, 12]. First, LLD is clinically recognized as a stress-sensitive illness [13], and the hippocampus is one of the brain regions involved in the negative feedback of the HPA axis [14, 15]. Second, LLD is always accompanied by impairments in cognitive function, which are heavily dependent on the function of the hippocampus [16]. Third, the hippocampus is one of several regions involved in emotional modulation [11]. Therefore, the hippocampus was considered the region of interest in the present study.
According to the basal level of CORT in plasma, 18-month-old rats were separated into two subpopulations: high-level CORT (H-CORT) animals and normal animals in which the CORT concentration was not increased (N-CORT). The glutamatergic system was investigated by electrophysiological, molecular and behavioral approaches in these aged rats. Based on our results, we propose that low-level CORT (L-CORT) ameliorates LLD and increases glutamatergic transmission and that the Glt-1 modulator may be a promising candidate agent for the treatment of LLD.
Materials and methods
Animals
Eighteen-month-old female Sprague–Dawley rats weighing 430–550 g were purchased from Vital River Laboratories, Beijing, China. All animals were maintained under a 12 h light/dark cycle with ad libitum access to food and water. Animals were habituated to the housing conditions for 5 days before the beginning of the experimental procedures. All procedures were conducted in accordance with the guidelines established by the National Institutes of Health for the Care and Use of Laboratory Animals and approved by the Animal Care Committee of the Peking Union Medical College and Chinese Academy of Medical Sciences.
Adrenalectomy (ADX) and corticosterone replacement
Rats underwent either bilateral ADX (n = 150) or SHAM surgery (n = 30) between 8 and 10 a.m. as described previously [17]. ADX rats were given 0.9% NaCl after surgery to compensate for the loss of salt. Some SHAM and ADX rats were examined 3 days after surgery to verify that ADX suppressed plasma CORT levels. All remaining ADX rats (n = 115) received CORT replacement treatment, which mimics the circadian rhythm of circulating adrenal steroids (ADX/CORT). This treatment consisted of (i) s.c. implantation of a CORT pellet (40 mg) to mimic the basal level of CORT in the diurnal cycle [18] and (ii) addition of CORT to the drinking water at night to mimic the nocturnal rise in CORT (at concentrations of 25 μg/mL (L-CORT) and 250 μg/mL (H-CORT) in 0.9% NaCl with 0.15% ethanol) [19]. Tail vein blood from SHAM and ADX/CORT rats was collected for the analysis of plasma CORT via ELISA (Cayman, UK).
Late life depression determination
Morris water maze (MWM)
The rats participated in the MWM test twice when they were 18 months old and 26 months old (after ADX). The water maze apparatus (Chinese Academy of Medical Sciences, China) consisted of a black circular pool (180 cm diameter, 75 cm high) divided into four equally spaced quadrants filled with water to a depth of 40 cm (22 ± 1 °C). A black Plexiglas goal platform submerged 1.0 cm below the surface of the water was hidden in a constant position in the center of one quadrant during the training period and then removed at the time of the probe task. To monitor the rat swimming parameters, a closed-circuit television camera was mounted onto the ceiling directly above the center of the pool and linked to a computer-based image analyzer [20].
The memory training test consisted of two consecutively repeated trials per day for a period of 4 days. Each rat was placed into the water facing the wall of the tank at the starting point and allowed to swim until they found the hidden platform located in the maze. During each trial, each rat was allowed 60 s to find the hidden platform. If the rat remained in the water after 60 s, the rat was gently guided to and placed on the platform. It was then allowed to stay on the platform for 10 s before a similar second trial was initiated. After the trials, the rat was returned to its cage and placed under a 60 W lamp to allow it to dry and warm up for 5 min. The time required for the rat to reach the platform (escape latency) was recorded. On the 5th day, the rats were subjected to a probe trial session in which the platform was removed from the pool, and they were allowed to swim for 60 s to search for it. A record was kept of the swimming time in the quadrant of the pool where the platform was previously located.
Forced swim test (FST)
FST was performed twice at 18 months and 26 months of age as described previously [21]. Briefly, animals were placed in a cylindrical tank (60 cm height, 30 cm diameter) filled with tap water (25 °C) to a depth of 45 cm for 6 min. Their performance was recorded by a video camera on top of the swimming tank. Immobility was defined as floating or a lack of active movement other than that necessary to keep the nose above the water. The immobility time in the last 4 min was used to evaluate the depression symptoms of rats.
Open field test (OPT)
Locomotive activities were measured by the OPT for a 5 min period when the rats were 18 months old and 26 months old. Briefly [22], the open field (120 cm × 120 cm) was divided into nine subfields (40 cm × 40 cm) of equal size. Animals were released into the central subfield, and their activities were recorded by a video camera on top of the open field. The crossing counts and rearing counts were determined to measure the rat locomotive activities. The field was cleaned with 70% alcohol after each recording session.
Electrophysiological recording
Recordings of evoked potentials were conducted as previously described [23]. Briefly, rats were anesthetized with urethane carbamate (1.5 g/kg, ip) before being fixed on an SR-6N stereotaxic apparatus (Narishige, Japan). Two holes were sequentially drilled at 3.8 and 7.5 mm posterior to the bregma and 2.5 and 4.2 mm lateral to the midline to allow the placement of a monopolar recording electrode and a bipolar stimulating electrode, respectively. The recording electrode was placed in the granular cell layer of the dentate gyrus at a depth of 3.0–3.5 mm, and the stimulating electrode was sunk into the perforant path to a depth of 3.0–3.5 mm. The synaptic responses were monitored with a VC-11 memory oscilloscope (Nihon Kohden, Japan). The population spike amplitude (PSA) was employed to assess the excitation level of the granular cell population in the dentate gyrus. An evoked response was generated in the granular cell layer by stimulating the perforant path at a low frequency (0.033 Hz) with single constant current pulses (150 μs in duration) triggered by an SEN-7203 electrical stimulator (Nihon Kohden, Japan) through an SS-202J isolator (Nihon Kohden, Japan). To determine the input–output responses, PSA was elicited at a series of different stimulation intensities (0.1–1.0 mA) with the same duration as the pulses. After determining the input/output curve, the baseline responses were evoked by a stimulus with an intensity that produced 20% of the maximal PSA. To measure the paired pulse ratio (PPR), PSA was evoked by a series of stimulation intervals (0.01–1.00 s) with the same intensity [24]. To observe the synaptic response to high frequency stimulation, theta-burst stimulation (TBS, 5 trains of bursts at 100 Hz, 200 ms intervals; repeated for 10 cycles at intervals of 200 ms) [25, 26] was introduced after obtaining a stable baseline of PSA for 30 min. After TBS, PSA was recorded over the next 60 min to determine the long-term potentiation (LTP).
To directly investigate the effect of CORT on the synaptic response, CORT (100 nM) [7] was delivered into the brain via an outer guide cannula placed into the intracerebroventricular area (0.8 mm posterior to the bregma and 1.8 mm lateral to the midline at a 3.0–3.5 mm depth from the cerebral surface). PSA was measured for the following 60 min to determine the LTP. In addition, mineralocorticoid receptor (MR) antagonists (RU28318) and glucocorticoid receptor (GR) antagonists (RU486) were preadministered for 20 min to block the respective receptor activity, and CORT (100 nM) was then delivered into the brain by the same method. PSA was analyzed over the following 60 min to determine the roles of these receptors during this process.
To observe the role of the impairment of the glutamate/glutamine cycle in glutamatergic neurotransmission, L-glutamine (1 μM) or glutamic-pyruvic transaminase (GPT, 1 μM) was preadministered for 20 min to restore the imbalance between glutamate and glutamine. Then, TBS was conducted as previously described, and PSA was recorded over the next 60 min to reveal the variations in glutamatergic transmission.
Glutamate release from synaptosomes
Hippocampal synaptosomes were prepared with a discontinuous Percoll gradient procedure according to the method of Dunkley et al. [27, 28] with minor modifications. Briefly, the hippocampus was quickly removed after animals were decapitated and then homogenized in 10 volumes of 0.32 M sucrose (pH 7.4) in a glass/Teflon tissue grinder (clearance 0.25 mm). The homogenate was centrifuged at 1000 × g, and the supernatant was stratified on a discontinuous Percoll gradient (6%, 10%, and 20% v/v in Tris-buffered sucrose) and centrifuged at 33,500 × g for 5 min. The layer between 10% and 20% Percoll (synaptosomal fraction) was collected and washed by centrifugation.
To determine glutamate release, the synaptosomes were resuspended in physiological medium and incubated at 37 °C for 15 min with gentle shaking in a 95% O2 and 5% CO2 atmosphere [29]. The physiological medium was composed of (in mM): 125 NaCl, 3 KCl, 1.2 MgSO4, 1.2 CaCl2, 1 NaH2PO4, 22 NaHCO3, and 10 glucose. At the end of the incubation period, aliquots of the synaptosomal suspensions (500 μg of protein) were layered on microporous filters at the bottom of a set of parallel superfusion chambers maintained at 37 °C. Superfusion was started at a rate of 0.5 mL/min with physiological medium aerated with 95% O2 and 5% CO2. After 36 min of superfusion, the samples were collected according to the following scheme: the 36–37, 37–38, and 38–39 min fractions were collected as the basal outflow content, and the 39–40, 40–41, 41–42, and 42–43 min fractions were collected to measure stimulus-evoked release. A 1 min flush of 15 mM KCl or 200 nM CORT was applied at t = 39 min after the basal sample had been collected. An equimolar concentration of NaCl was administered at t = 44 after the evoked released glutamate was collected. The collected fractions were analyzed for their endogenous glutamate content. The endogenous amino acid release was expressed as ng per milligram of protein. The stimulus-evoked overflow was estimated by subtracting the transmitter content of the basal outflow from that of the release-evoked sample collected each min during and after the depolarization pulse. Appropriate controls were always run in parallel.
For glutamate uptake measurement, the protein concentration of the synaptosomes [26] was adjusted to 20 μg/mL in HEPES-buffered saline (HBS) [30]. Briefly, 1 μM D-aspartate and 0.25 μCi/assay D-[3H] aspartate (50 nM) were added to the HBS that contained the synaptosomes. Uptake assays were performed for 4 min at room temperature and terminated by filtration on a Packard 96-well Unifilter (Whatman GF/B) plate, followed by two washes with ice-cold buffer. Microscint 20 (Perkin Elmer, US) was added directly to the filter plates for the determination of the [3H] retained on the filters using a Packard Top Count Adapter. Uptake was determined to be linear for up to a 5 min incubation period, and the data were corrected to represent the sodium-dependent uptake.
Glutamate and glutamine determination in the hippocampus
Endogenous glutamate was measured by HPLC analysis as described by Bonanno with minor modifications [28]. After precolumn derivatization with o-phthalaldehyde, separation was performed on a C18 reverse-phase chromatographic column (10 mm × 4.6 mm, 3 μm at 30 °C; Chrompack, Middleburg, The Netherlands) coupled with fluorometric detection (excitation wavelength, 350 nm; emission wavelength, 450 nm). Buffers and the gradient program were as follows: solvent A, 0.1 M sodium acetate (pH 5.8)/methanol, 80:20; solvent B, 0.1 M sodium acetate (pH 5.8)/methanol, 20:80; solvent C, sodium acetate (pH 6.0)/methanol, 80:20; gradient program, 100% C for 4 min from the initiation of the program; 90% A and 10% B for 1 min; isocratic flow, 2 min; 78% A and 22% B for 2 min; isocratic flow, 6 min; 66% A and 34% B for 3 min; 42% A and 58% B for 1 min; 100% B for 1 min; isocratic flow, 2 min; 100% C for 3 min; flow rate, 0.9 mL/min. Homoserine was used as the internal standard in the present study.
Electrical transmission microscopy detection of neurotransmitter vesicles
Processing and EM were conducted as previously described [31]. Briefly, anesthetized animals were perfused, and the hippocampus was separated. The hippocampal sections were postfixed with 2.5% glutaraldehyde for 2 h, washed with 0.1 M PBS, and then exposed to 1% osmium tetraoxide for 2 h. After several subsequent washes with water, the tissues were dehydrated with an alcohol gradient and embedded in epon resin. Randomly selected ultrathin sections were stained with uranyl acetate and lead citrate and examined using a transmission electron microscope (H-7650, HITACHI, Tokyo, Japan). Only nonperforated asymmetric (excitatory) synapses with well-defined presynaptic compartments containing at least 3 vesicles, a clear synaptic cleft and PSD were examined. Then, at a magnification of 100,000×, the number of neurotransmitter vesicles was randomly measured with MetaMorph software. Docked vesicles were defined as vesicles located within 10 nm of the active zone membrane (n = 6 animals).
Glt-1 expression and its modulation by neural activity
Astrocytes were prepared as described previously with minor modifications [32]. Cortices were dissected from P1-3 rat pups in DMEM (Invitrogen, San Diego, CA, USA) and digested with 0.1% w/v trypsin. The tissue was triturated and suspended after centrifugation in astrocyte culture medium (DMEM containing 10% fetal calf serum and 1% pen/strep). Cells were plated in T-75 flasks at a density of two cortices per flask at 37 °C in 5% CO2. The culture medium was changed every 3rd day. After 8–10 days, the cultures were shaken for 12–16 h at 250 rpm on an orbital shaker (HZQ-X100, China), followed by incubation in culture medium containing 20 μM cytosine-1-β-D-arabinofuranoside (Sigma-Aldrich, St. Louis, MO, USA) for 2–3 days to deplete the precursor cells. To directly determine the effect of CORT or glutamate on the expression of Glt-1 in astrocytes, astrocytes were replated in 6-well plates (100,000/well) and exposed to CORT (200 nM) or glutamate (1, 10 mM) for 5 days, and the expression of Glt-1 was determined by Western blotting.
E18 hippocampal neurons were cultured as described previously [33]. Hippocampal neurons were isolated from E18 rat embryos in Hanks’ balanced salt solution with 0.1% w/v glucose and 10 mM 4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid (HEPES) buffer, followed by digestion with 0.25% w/v trypsin (Invitrogen, San Diego, CA, USA). The triturated neurons were then centrifuged (120 × g for 10 min) and resuspended in neural basal medium with 2% v/v B27 supplement (both Invitrogen, San Diego, CA, USA). To observe the effect on neural activities, neurons (10 DIV) were cultured with CORT (200 nM) for 5 days, and their expression of synaptic markers (PSD95, synapsin I, NR2A and NR2B) were investigated by Western blotting.
To generate the gradient neuron–astrocyte cultural system, cell culture inserts (30 mm diameter, Millipore, Temecula, CA, USA) with a pore size of 0.4 μm were employed for the cultivation of astrocytes separately from neurons. Briefly, hippocampal neurons, which were isolated as described above, were placed in cell culture inserts at a gradient density of 0, 25,000, 50,000, or 75,000 cells per insert. Astrocytes were removed from the flasks through enzymatic digestion (0.25% (w/v) trypsin in MEM), and the cells were replated in 6-well plates at concentrations of 100,000, 75,000, 50,000, and 25,000 cells/well. After 2 days, the medium was replaced with neural basal medium, and the inserts were placed into 6-well plates with hippocampal neurons (DIV 12 days). The proportion of astrocytes among neurons was ~100%, 75%, 50%, or 25% in this coculture system. CORT (200 nM) was added to this system for 5 days to investigate the effect of neuron impairment on the expression of Glt-1. In addition to hippocampal neurons, midbrain neurons were extracted at E14 as described previously and cultured with astrocytes in the same manner.
Western blotting analysis
Hippocampal neurons or astrocytes were homogenized in lysis buffer. Protein concentrations were determined by a bicinchoninic acid protein assay. A total of 15 μg of protein for each sample was separated by SDS-PAGE and transferred to a PVDF membrane (Millipore, Temecula, CA, USA). The membrane was blocked with 3% BSA and incubated with primary antibody overnight at 4 °C (anti-Glt-1, CST, Danvers, MA, USA, 1:500; anti-PSD-95, synapsin I, NR2A, and NR2B, Santa Cruz, CA, USA; anti-β-actin, Sigma, 1:10,000), which was followed by incubation with horseradish peroxidase (HRP)-conjugated secondary antibody (1:5000; KPL, Gaithersburg, MD, USA). The protein bands were detected using enhanced chemiluminescence. Densitometric analysis of the immunoreactivity of each protein was conducted using Gel-Pro Analyzer software (Media Cybernetics).
DHK-induced LLD in SHAM animals
Thirty rats (20 SHAM animals and 10 H-CORT animals) were anesthetized using sodium pentobarbital (60 mg/kg, i.p.). The rats were placed into a stereotaxic apparatus, and a craniotomy was performed. A small hole was drilled through the skull using the following coordinates: 0.9 mm anterior/posterior and 1.5 mm medial/lateral relative to the bregma according to Paxinos and Watson’s coordinates. A 52 mm cannula constructed from PE-20 tubing was inserted into this opening and slowly lowered into the brain so that the tip of the cannula resided within the lateral ventricle. Three small holes were also drilled into the skull for the placement of three anchor screws (size 0–80 × 1/8 inch in length). A dental cement stage was then constructed around each cannula and allowed to harden, thereby securing the cannula in place [34]. Animals were divided into three groups: the SHAM + PBS (vehicle) group, the SHAM + DHK (DHK, Glt-1 antagonist) group and the H-CORT + PBS (H-CORT) group. Three days after implanting the cannula, the rats received i.c.v. injections (10 μL) of DHK (2.5 μg/μL) or PBS for 5 consecutive days. Starting on day 6, the MWM, FST, and OPT were performed as previously described to evaluate the role of Glt-1 in LLD.
RIL attenuated LLD in H-CORT animals
Thirty rats (20 H-CORT rats and 10 L-CORT rats) were used to observe the role of Glt-1 activity in the treatment of LLD. The poor activity of Glt-1 was improved by oral administration of RIL (5 mg/kg). H-CORT rats were divided into a vehicle (double-distilled water) group and RIL group. L-CORT animals were used as a positive control. After 21 days of administration, the MWM, FST and OPT were performed as previously described to evaluate the role of Glt-1 in LLD.
Statistical analysis
Data are presented as the mean ± SEM. Raw data were statistically analyzed with Graph Pad Prism 7.0. Escape latency in MWM and electrophysiological recordings were analyzed using two-way analysis of variance (ANOVA) with repeated measures followed by Newman–Keuls post hoc test. The other data were analyzed using one-way ANOVA followed by Newman–Keuls post hoc test. A value of P < 0.05 was considered statistically significant.
Results
Late life depression was attenuated in L-CORT animals
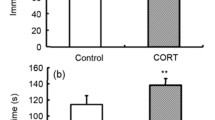
To observe the impact of CORT on LLD, cognitive, emotional, and locomotive activities were investigated twice in rats at 18 months old and 26 months old, and 3-month-old female rats were employed as control animals. After the determination of CORT levels in plasma, the rats underwent bilateral ADX or SHAM surgery followed by CORT replacement treatment. According to the concentration of CORT that was obtained, rats were divided into the H-CORT group (ADX + 250 μg/mL CORT replacement), L-CORT group (ADX + 25 μg/mL CORT replacement), and SHAM group (SHAM + 0 μg/mL CORT replacement) (Fig. 1a). Cognitive function was observed with the MWM. In the navigation training, SHAM and H-CORT animals showed a prolonged escape latency in the 2nd MWM compared to that in the 1st MWM (P < 0.05, Fig. 1b, c), and L-CORT animals showed similar performance in both MWM tests (P > 0.05, Fig. 1d). In the second MWM, a significant decrease in escape latency was found in the L-CORT group on the 4th day compared to that in the H-CORT and SHAM groups (P < 0.05, Fig. 1e). The probe trial showed trends similar to those of the navigation trial (Fig. 1f). The retention time within the target quadrant was significantly decreased in the 2nd MWM compared with the 1st MWM in both the SHAM (16.2 ± 5.3 s vs. 29.6 ± 6.9 s, respectively, P < 0.05) and H-CORT (13.3 ± 7.3 s vs. 25.3 ± 5.4 s, respectively, P < 0.05) groups. L-CORT animals did not show notable changes during either MWM. Moreover, L-CORT animals spent more time in the target quadrant than SHAM and H-CORT animals during the 2nd MWM, indicating that L-CORT attenuates cognitive function decline in aged female rats, which is consistent with data previously presented by Landfield.

a The concentration of CORT in rat plasma at 18 months (before surgery) and 26 months (before behavioral tests) of age. Plasma was obtained from tail vein blood, and CORT was determined by ELISA tests. ##P < 0.01 vs. SHAM animals at 26 months old, **P < 0.01 vs. H-CORT animals at 26 months old. b–d L-CORT inhibited the aging-induced prolongation of the escape latency in the MWM. MWM tests were conducted twice, when the rats were 18 months (1st MWM) and 26 months of age (2nd MWM). For each MWM, a navigation trial (60 s) was conducted for 4 successive days, and the escape latency was analyzed by two-way ANOVA. *P < 0.05 vs. 1st MWM. e L-CORT animals showed a shortened escape latency in the 2nd MWM. #P < 0.05 vs. SHAM animals, *P < 0.05 vs. H-CORT animals. f L-CORT increased the retention time in the target quadrant in the probe trial of the 2nd MWM. The probe trial was performed on day 5 of each MWM, and the retention time in the target quadrant was analyzed by one-way ANOVA. #P < 0.05 vs. SHAM animals, *P < 0.05 vs. H-CORT animals. g L-CORT decreased the immobility time in the FST. The FST was also conducted twice after each MWM. The immobility time in the last 4 min was analyzed, #P < 0.05 vs. SHAM animals in the 2nd FST. *P < 0.05 vs. H-CORT animals in the 2nd FST. h, i L-CORT improved locomotive activities in the OPT. Locomotive activities were assessed by determining the crossing counts and rearing counts in the 5 min trial, #P < 0.05 vs. SHAM animals in the 2nd OPT. *P < 0.05 vs. H-CORT animals in the 2nd OPT.
The FST was a useful screen of the antidepressant efficacy of a treatment. In the present study, we observed the effect of CORT on the emotional variation of rats. As shown in Fig. 1g, H-CORT animals showed increased immobility time in the 2nd FST compared with that in the 1st FST (93.7 ± 31.4 s vs. 177.7 ± 30.2 s, P < 0.05). In the 2nd FST, the L-CORT rats (78.5 ± 20.3 s) exhibited a significantly reduced immobility time compared with the SHAM (128.7 ± 25.2 s) and H-CORT (177.7 ± 30.2 s) animals, indicating that L-CORT reduced depressive behavior in the aged animals.
The results of the OPT, which are expressed as the mean values of the rearing and crossing counts during the 6-min test, revealed the rat locomotive activity. Figure 1h, i show that there was a significant decrease in locomotive activity in the SHAM and H-CORT animals between the 1st OPT and 2nd OPT, as demonstrated by the decrease in the crossing count (167.47 ± 26.17 vs. 95.05 ± 17.04 in the SHAM animals, respectively, P < 0.05; 159.49 ± 31.48 vs. 53.97 ± 28.19 in the H-CORT animals, respectively, P < 0.05) and the rearing count (17.40 ± 5.73 vs. 21.48 ± 5.58 in the SHAM animals, respectively, P > 0.05; 11.23 ± 4.61 vs. 23.57 ± 6.49 in the H-CORT animals, respectively, P < 0.05). The 2nd OPT results indicated that the H-CORT rats had a significantly lower motor activity performance than the L-CORT rats, indicating that L-CORT improved locomotive activity in late life. Taken together, these results suggest that L-CORT significantly attenuated LLD.
Glutamatergic transmission was improved in L-CORT animals
To examine the impact of CORT on glutamatergic transmission, synaptic responses in the perforant pathway were measured in anesthetized rats. First, input/output curves were generated according to the variation in PSA evoked by a series of stimuli with different intensities (from 0 to 1.0 mA). As shown in Fig. 2a, the PSA was markedly improved in the L-CORT animals (0.62 ± 0.06 mV at 1.0 mA, P < 0.01) compared with that in the H-CORT animals (0.22 ± 0.05 mV at 1.0 mA). The PPR, which is sensitive to presynaptic glutamate release, was measured at 1 mA after different stimulus intervals. The L-CORT animals showed a significantly increased PPR at intervals of 0.025, 0.05, 0.1, and 0.2 s compared with the H-CORT animals (Fig. 2b). All of these results suggest that presynaptic function was impaired at high CORT levels, while lower CORT levels could improve the presynaptic efficacy, although there was no difference in the PPR when the interval was prolonged to 0.2 s.

a L-CORT increased the slope of the input/output curve in the perforant path. PSA was used to evaluate synaptic activity. The left image shows the summarized input/output curve in response to a series of stimulation intensities. The right picture contains representative images of different synaptic responses at 0.6, 0.8, and 1.0 mA. b PPR was improved by L-CORT treatment. The plot of the PPR evoked by double pulses at various intervals (0.025–0.2 s) is shown. c L-CORT enhanced the PSA, which was significantly triggered by TBS. TBS was performed after obtaining a stable baseline for 30 min, and the PSA was then analyzed by two-way ANOVA for the next 60 min. d L-CORT improved the PSA after icv CORT delivery. CORT (100 nM) was administered after a stable baseline was recorded, and the PSA was then analyzed for the following 60 min. e Inactivation of MRs abolished the improvement of the PSA induced by CORT delivery. An MR antagonist (RU28318) or GR inhibitor (RU486) was administered 20 min prior to CORT delivery as previously described, and the PSA was analyzed for the following 60 min by two-way ANOVA. **P < 0.01 vs. SHAM animals, #P < 0.05; ##P < 0.01 vs. H-CORT animals. n = 6.
To further investigate glutamatergic transmission in these animals, TBS was used to evoke LTP. The PSA was recorded during the first 30 min as the baseline, and neurons in the perforant path were depolarized by TBS. As shown in Fig. 2c, the PSA was increased significantly in the L-CORT animals compared with that in the H-CORT animals within the first 20 min, indicating that L-CORT attenuated the synaptic response evoked by TBS, although it failed to evoke LTP. The H-CORT animals did not show notable changes during this process.
To observe the sensitivity to CORT, the PSA was directly measured after CORT exposure. As shown in Fig. 2d, CORT exposure did not affect the PSA in the H-CORT animals, while the L-CORT animals showed a remarkable increase in the PSA after 60 min, indicating that L-CORT maintained sensitivity to CORT, while H-CORT impaired it.
CORT has two receptors, MR and GR. To further confirm which one is involved in synaptic transmission after CORT exposure, RU28318 (MR antagonist) and RU486 (GR antagonist) were introduced in this study. As shown in Fig. 2e, no difference was observed in the enhancement of synaptic transmission between the L-CORT and L-CORT + RU486 animals, indicating that GR was not involved in this process. However, the action of CORT was significantly blocked by RU28318, suggesting that MR plays an important role in the evoked synaptic response.
Glutamate release from synaptosomes was improved in L-CORT rats
Hippocampal synaptosomes were prepared from aged rats to investigate glutamate release under different conditions. First, 15 mM KCl was employed to induce synaptosomal membrane depolarization, which mimicked stimulation at 100 Hz. The amount of released glutamate is shown in Fig. 3a. L-CORT synaptosomes (2.76 ± 0.19 ng/mg protein) were released more quickly than SHAM (1.66 ± 0.28 ng/mg protein) and H-CORT (0.61 ± 0.28 ng/mg protein) synaptosomes in the first min. Over time, the release velocity decreased. After 4 min of exposure, the release velocity decreased to the resting level. Then, CORT (100 nM) was added to the synaptosomes to trigger glutamate release. As shown in Fig. 3b, the L-CORT group (2.34 ± 0.29 ng/mg protein) released more glutamate in the first min than the SHAM (0.94 ± 0.24 ng/mg protein) and H-CORT (0.43 ± 0.19 ng/mg protein) groups, and the release rate decreased to that of the resting state after 2 min. Figure 3c shows the amount of glutamate release stimulated by KCl or CORT. In addition, MR played an important role during glutamate release induced by CORT exposure (Fig. 3g). Taken together, these results suggest that glutamate release from the L-CORT animals was greater than that from the SHAM and H-CORT animals.

a L-CORT increased the velocity of glutamate release triggered by KCl (15 mM). Synaptosomes were prepared as previously mentioned. After the basal outflow content was collected, 15 mM KCl was added, and the medium was collected for the next 4 min to determine the changes in the velocity of glutamate release. Glutamate content was determined by the HPLC system combined with a fluorescence detector (excitation wavelength: 340 nm; emission wavelength: 450 nm). b L-CORT increased the velocity of glutamate release triggered by CORT exposure (100 nM). c L-CORT significantly increased the amount of released glutamate. d, e L-CORT increased the number of neurotransmitter vesicles in the excitatory synapse, as measured by ETM. Hippocampal fractions were prepared to observe the neurotransmitter vesicles in the excitatory synapse, and the vesicles were counted by Metaphor 7.0. e shows the representative images for each group, and the analysis is shown in (e, f). **P < 0.01 vs. SHAM animals, #P < 0.05; ##P < 0.01 vs. H-CORT animals. n = 6. g The inactivation of MR abolished the improvement of glutamate release induced by CORT exposure. An MR antagonist (RU28318) or GR inhibitor (RU486) was administered 20 min prior to CORT addition, and the glutamate released over the following 4 min was then analyzed by two-way ANOVA. $$P < 0.01; $$$P < 0.001 vs. RU28318.
To investigate the reason for the reduction of glutamate release from the SHAM and H-CORT animals, ETM was employed to observe the number of neurotransmitter vesicles in the readily released pool in excitatory synapses located near the synaptosomal membrane to control the rapid release of glutamate. As shown in Fig. 3d, hippocampal synapses in the L-CORT animals contained many small and bright vesicles in the readily released pool, indicating that there was a large quantity of glutamate waiting to be released. The SHAM and H-CORT synapses contained fewer vesicles than the L-CORT synapses. One-way ANOVA showed that L-CORT synapses had a significantly increased density of vesicles. When compared with that in the SHAM or H-CORT animals (P < 0.01), the vesicle density in the active zone was also remarkably increased in the L-CORT animals (Fig. 3e, f), which provided a rational explanation for the decrease in glutamate release in SHAM and H-CORT animals.
Impairment of the glutamate/glutamine cycle was reduced in L-CORT animals
The glutamate/glutamine cycle plays an important role in the homeostasis of glutamatergic transmission. To reveal the cause of decreases in the number of glutamatergic vesicles in the SHAM and H-CORT animals, the glutamate and glutamine contents were determined by a colorimetric method. As shown in Fig. 4a, a one-way ANOVA indicated there was increased glutamate (5.72 ± 0.97 nmol/mg) and glutamine (2.67 ± 0.18 nmol/mg, P < 0.05, P < 0.01) content in the L-CORT rat hippocampus compared with that in the SHAM (glutamate: 3.84 ± 0.73 nmol/mg; glutamine: 1.53 ± 0.08 nmol/mg) and H-CORT (glutamate: 2.86 ± 0.60 nmol/mg; glutamine: 0.66 ± 0.13 nmol/mg) rat hippocampus. Based on the variation in glutamate and glutamine, the L-CORT animals exhibited glutamatergic homeostasis, which was reflected as a lower ratio between glutamate and glutamine (red stars). This ratio was also elevated with an increase in CORT (Fig. 4b).

a L-CORT increased the concentration of glutamate and glutamine in the hippocampus. Glutamate and glutamine content in the hippocampus were separated by the HPLC system and monitored using a fluorescence detector at an excitation wavelength of 340 nm and an emission wavelength of 450 nm. b The relationship between the CORT concentration and the Glu/Gln ratio in the hippocampus is shown. The CORT level in the hippocampus was measured by ELISA according to a standard protocol. c, d L-glutamine supplementation did not significantly change synaptic activity. L-glutamine (1 μM) was delivered into the lateral ventricle of the SHAM and H-CORT animals prior to TBS for 20 min, and the PSA was measured for the following 60 min. e, f Inactivation of extracellular glutamate attenuated synaptic activity in SHAM animals (e) but abolished synaptic activity in L-CORT animals (f). Extracellular glutamate was inactivated by the delivery of GPT before TBS was performed, and the PSA was determined as previously described. *P < 0.05 vs. vehicle animals. g The glutamate uptake capacity was improved in L-CORT animals. Hippocampal synaptosomes were prepared in KHBS for 30 min, and the initial rate of glutamate uptake was then determined at various concentrations by a liquid scintillation counter. Substrate levels reflect the sum of the endogenous and exogenous glutamate at the start of uptake determination. **P < 0.01, ***P < 0.001 vs. H-CORT group, #P < 0.05, ##P < 0.01 vs. SHAM group. n = 3.
The homeostasis of the glutamate system is disrupted in LLD, which is characterized by excessive glutamate production or a lack of glutamine. To test the role of the lack of glutamine in glutamatergic transmission in the SHAM and H-CORT animals, L-glutamine was administered to the hippocampus by icv. Two-way ANOVA indicated that L-glutamine delivery did not have a notable effect on the PSA in the SHAM and H-CORT animals (Fig. 4c, d). To evaluate the role of an elevated extracellular glutamate concentration, an enzymatic glutamate scavenger system (glutamic-pyruvic transaminase, GPT) was used to reduce extracellular glutamate levels. As shown in Fig. 4e, f, GPT had no effect on the PSA before TBS. However, it significantly increased the PSA in SHAM animals under TBS compared with that in the vehicle-pretreated animals (Fig. 4e) but failed to increase the PSA in L-CORT animals (Fig. 4f). All of these results suggest that excess glutamate, not glutamine deficiency, is the key cause of impaired glutamatergic transmission in aged rats.
Because there is no specific enzyme for the degradation of glutamate, the clearance of glutamate relies on the transporter activity. To observe glutamate transporter activity, [3H]-glutamate was incubated with hippocampal synaptosomes. Figure 4g indicates that more glutamate was taken up by the L-CORT synaptosomes than by the SHAM and H-CORT rat synaptosomes. All of these results suggest that poor glutamate uptake may be the key mechanism of impaired glutamate transmission in aged rats.
Expression of Glt-1 was suppressed by CORT and contributed to neural impairment
Glt-1 is the main executor of forebrain glutamate uptake in the adult CNS, especially in the hippocampus. Figure 5a shows that the expression of Glt-1 in the L-CORT hippocampus (2.66 ± 0.37) was much higher than that in the SHAM (1.00 ± 0.15) and H-CORT (0.49 ± 0.18) hippocampus. To directly observe the effect of CORT on the expression of Glt-1, primary astrocytes were exposed to CORT (200 nM) for 5 days. One-way ANOVA revealed that the Glt-1 level was not significantly affected (Fig. 5b). However, hippocampal neurons suffered serious damage when cultured in the same conditions, as shown by the loss of synaptic markers and decreased glutamate release and expression of its receptor (Fig. 5c, d), suggesting that hippocampal neurons were more vulnerable to CORT than astrocytes. Therefore, we speculated that the decrease in Glt-1 may result from the contribution of CORT to neural impairment.

a Total hippocampal protein was extracted by RIPA buffer, and Glt-1 expression was detected by a specific antibody (upper lane). The relative density of the bands was analyzed by one-way ANOVA (lower lane). *P < 0.05; **P < 0.01 vs. SHAM animals, ##P < 0.01 vs. H-CORT animals. b The expression of Glt-1 was not affected by CORT exposure in astrocytes. Astrocytes were exposed to CORT at a concentration of 200 nM for 1, 3, and 5 days, and the cell lysates were used to determine Glt-1 expression by Western blotting. The upper lane contains the representative bands, and the bottom lane shows the normalized density analysis performed with one-way ANOVA. c Glutamate release was significantly suppressed by CORT exposure in hippocampal synaptosomes. Synaptosomes were isolated after exposure for 1, 3, and 5 days by the discontinuous Percoll gradient procedure. Glutamate release was triggered by CORT (200 nM) as previously described. *P < 0.05; **P < 0.01 vs. D1. d CORT exposure impaired the expression of synaptic markers and glutamate receptors in hippocampal neurons. The left column contains representative images of the synaptic marker (synapsin I), postsynaptic marker (PSD95), and glutamate receptors (NR2A, NR2B), which were labeled using specific antibodies, and β-actin was employed as a loading control. The relative density of these bands was analyzed using one-way ANOVA (right lane). *P < 0.05; **P < 0.01 vs. D1; #P < 0.05 vs. D3. e The expression of Glt-1 was decreased with the increase in the hippocampal neuron percentage in the gradient neuron–astrocyte coculture. Different ratios of neurons and astrocytes were cocultured in a separate manner (insert culture system). CORT (200 nM) was added to this system for 5 days, and then, Glt-1 in the astrocytes was measured by Western blotting. The upper row contains representative images, and the bottom row shows the normalized analysis with a one-way ANOVA. *P < 0.05; **P < 0.01 vs. 0% neurons. f The expression of Glt-1 did not change with the increase in midbrain neurons in the gradient neuron–astrocyte coculture. The upper row contains a representative image, and the bottom row shows the normalized analysis performed with one-way ANOVA. g Glutamate significantly impaired the expression of Glt-1 in astrocytes. Astrocytes were cultured with high concentrations of glutamate (1, 10 mM) for 5 days, and then Glt-1 expression was examined in the same manner as in (e, f). *P < 0.05; **P < 0.01 vs. 0 mM glutamate.
To verify our speculation, a gradient neuron–astrocyte coculture was established. As shown in Fig. 5e, with the increase in the hippocampal neuron percentage, the expression of Glt-1 decreased gradually. Interestingly, dopaminergic neurons failed to affect the expression of Glt-1 when they were cocultured with astrocytes under the same concentration of CORT (Fig. 5f). Moreover, long-term exposure of glutamate to astrocytes significantly decreased the level of Glt-1 (Fig. 5g). All of these results suggest that the decrease in Glt-1 may result from the contribution of CORT to neural impairment in the hippocampus.
Enhancement of glutamate uptake attenuated LLD
RIL, a drug currently used to slow the progression of amyotrophic lateral sclerosis, is believed to increase the expression of Glt-1 and to facilitate astrocytic glutamate uptake. DHK has been shown to block the clearance of glutamate. Both of these drugs were used to examine the role of Glt-1 in LLD. Administration of DHK to the SHAM animals significantly increased LLD symptoms compared with those in vehicle-treated animals. These effects were reflected by impaired cognition, decreased locomotive activity and increased helplessness. In the MWM, DHK significantly prolonged the escape latency (P < 0.05, Fig. 6a) in the navigation trials and shortened the retention time (P < 0.05, Fig. 6b) in the target quadrant in the probe trial compared with the results observed for vehicle-treated animals. DHK treatment increased the immobility time in the FST compared with vehicle treatment in the SHAM animals (Fig. 6c). Furthermore, DHK notably decreased the crossing count (Fig. 6d) and rearing count (Fig. 6e) compared with those in vehicle-treated animals in the OPT. The H-CORT animals were used as positive controls in this study. In summary, impairment of Glt-1 activity induced the occurrence of LLD.

a–e Inhibition of Glt-1 activity induced LLD in the SHAM animals. DHK, a Glt-1 antagonist, was delivered by icv every other day to block Glt-1 activity. The vehicle-treated and H-CORT animals received vehicle administration in the same manner. The MWM was conducted as previously described after three administrations of DHK. DHK treatment prolonged the escape latency (a) and decreased the retention time in the target quadrant (b) in the SHAM animals. After the MWM, the FST was performed the next day, and DHK delivery increased immobility time in the FST (c). The OPT was conducted after the FST, and DHK administration decreased locomotive activity, as shown by the crossing (d) and rearing counts (e). The H-CORT animals were used as a positive control. f–j Enhanced Glt-1 activity attenuated LLD in the H-CORT animals. RIL (Glt-1 activity enhancer) was orally administered for 21 days, and behavioral tests were performed as previously described. In the MWM, RIL administration decreased the escape latency (f) and increased the retention time in the target quadrant (g). In the FST, RIL treatment significantly shortened the immobility time (h) in the FST and increased locomotive activity in the OPT (i, j). The L-CORT animals were used as a positive control. *P < 0.05; **P < 0.01 vs. corresponding vehicle-treated animals (n = 8–10).
However, RIL treatment significantly attenuated cognitive function, as evidenced by a decrease in the escape latency (Fig. 6f) and an increase in the retention time (Fig. 6g) in the target quadrant of the MWM in the H-CORT animals. FST showed that RIL significantly decreased the immobility time compared with that observed for the vehicle-treated animals (Fig. 6h). Furthermore, RIL improved locomotive activity in the OPT, as shown by an increase in the crossing count (Fig. 6i) and rearing count (Fig. 6j). Taken together, these results suggest that increased Glt-1 function attenuated LLD.
Discussion
In the present study, we showed that LLD was attenuated by reducing the level of CORT in female rats via enhanced glutamatergic neurotransmission. The core feature of glutamatergic transmission in LLD is a deficiency in glutamate uptake, which is due to Glt-1. The results of the gradient neuron–astrocyte coculture suggested that the expression of Glt-1 was downregulated with the increase in the number of hippocampal neurons, indicating that impairment of Glt-1 induced by H-CORT contributed to neuron impairment.
As previously mentioned, MDD is always accompanied by hyperactivation of the HPA axis and poor cognitive function [35, 36]. Decreasing the level of CORT could improve the spatial memory capacity of aged rats [9], suggesting that there is a close relationship between CORT and discrete spatial function [37,38,39]. Our MWM results led to similar conclusions regarding the modulation of CORT levels in late life. Locomotive activity, as indicated by the OPT, indicated that L-CORT rescued the impairment of exploratory behaviors, as evidenced by the increases in the crossing and rearing counts. In addition, the L-CORT animals exhibited reduced helplessness compared to the H-CORT and SHAM animals in the FST. These behavioral changes suggest that H-CORT induced LLD and that L-CORT alleviated LLD according to multiple lines of evidence.
The dysfunction of glutamatergic neurotransmission is considered a core feature of stress-related mental illness [5]. This dysfunction is broadly distributed throughout the central nervous system, especially in the hippocampus. Models of short- and long-term plasticity, such as the PPR and LTP, can be readily induced in the hippocampus [40]. LTP, which is characterized by a persistent increase in synaptic potential, is considered a cellular model of cognition [41]. PPR, which is characterized by an increase in synaptic transmission potential, is regarded as a readout of presynaptic function [24]. In the present study, the H-CORT animals exhibited worse short-term plasticity than the SHAM animals, as shown by the decrease in the I/O curve and the impairment of the PPR. L-CORT significantly reversed these changes, indicating that L-CORT could act to prevent the impairment of the glutamatergic synaptic response observed in LLD. In addition to short-term plasticity, TBS was used to induce LTP after the baseline result was recorded, which showed that synaptic transmission failed to be improved by TBS in the H-CORT and SHAM animals. L-CORT significantly attenuated synaptic transmission. An electrophysiological evaluation suggested that there was a notable decrease in the impairment of glutamatergic neurotransmission in LLD and that L-CORT could significantly reverse this impairment.
Converging lines of evidence suggest that the acute administration of CORT rapidly increases glutamate release [6] and induces LTP [7] in adult animals. Adrenalectomy attenuates stress-induced elevations in extracellular glutamate concentrations in the hippocampus [42]. In the present study, the delivery of CORT did not significantly increase the PSA in the SHAM animals, indicating that there was a poor synaptic response to elevated CORT levels. Moreover, similar findings were observed in the H-CORT animals. Unlike the SHAM and H-CORT animals, CORT exposure induced a significant increase in PSA in the L-CORT animals, indicating that decreases in CORT could maintain CORT sensitivity in late life.
Microdialysis demonstrated an increase (above 68%) in basal glutamate release in the hippocampus of 24-month-old rats compared with that in young rats [43]. However, the glutamate distribution was heterogeneous in the extracellular matrix, and the released glutamate may have contacted receptors or transporters on neighboring terminals [5]. Therefore, microdialysis analysis cannot be interpreted to reflect neurotransmitter release. To determine whether there was excessive or inadequate glutamate release in these animals, hippocampal synaptosomes were isolated to observe the release of glutamate induced by KCl. H-CORT synaptosomes showed reduced glutamate release compared with that in the SHAM groups, suggesting that there was a reduction in glutamate release. To confirm whether abundant glutamate-containing vesicles were readily released, the number of neurotransmitter vesicles in the excitatory synapses of the hippocampus was investigated by ETM. The results showed that H-CORT animals had fewer vesicles that were ready to be released than the SHAM animals, suggesting that excessive rapid release of glutamate is impossible in H-CORT animals. L-CORT synaptosomes released more glutamate at higher rates than the SHAM and H-CORT synaptosomes, indicating that decreased levels of CORT preserve presynaptic functions during the aging process. In addition, glutamate release was also triggered by CORT exposure. H-CORT synaptosomes released lower levels of glutamate at a slower velocity compared to SHAM synaptosomes, and L-CORT significantly reversed this release. All of these results suggest that L-CORT could ameliorate presynaptic function and maintain CORT sensitivity in LLD.
Two types of CORT receptors are enriched in the hippocampus: MRs and GRs. Previous studies have demonstrated that high-affinity, rapidly activated MRs are implicated in the onset of stress responses, whereas low-affinity, progressively activated GRs are involved in the termination of stress reactions [44, 45]. The presence of membrane MR-like signaling can mediate the fast nongenomic actions of CORT, which manifest as the reversible increase in presynaptic glutamate release and facilitate LTP in hippocampal neurons. A mechanistic study showed that MR was involved in the surface trafficking of the AMPA receptor and the distribution and dynamics of the NMDA receptor. In this study, we demonstrated that increased synaptic transmission (triggered by CORT delivery) was disrupted when MR capability was blocked in L-CORT animals. GR was not involved in this process. Further studies have shown that glutamate release also relies on the activation of MR, suggesting that MR plays an important role in the improvement of glutamatergic transmission induced by CORT.
Decreased release of glutamate and vesicles from synaptosomes indicated that there may be an inadequate quantity of glutamate in the neurons, which is replenished mainly by the glutamate/glutamine cycle [46, 47]. In this pathway, glutamate is taken up by glia and converted to glutamine by glutamine synthetase. Glutamine is then released to the extracellular fluid, where it is taken up by neurons and converted back into glutamate by the action of phosphate-activated glutaminase [48]. It has been shown that a decreased glutamate/glutamine ratio may mediate the efficacy of cytidine in treating bipolar depression [49]. In this study, we demonstrated that H-CORT animals experienced a greater increase in the ratio of Glu/Gln than L-CORT rats, while L-CORT decreased the ratio significantly.
However, increased glutamate levels were found in the medial prefrontal cortex in patients with postpartum depression [50], and glutamine deficiency in the prefrontal cortex increases depressive-like behavior in male mice [51]. To identify whether glutamate or glutamine is the cause of LLD, further studies were conducted. First, L-glutamine was supplemented to observe the variations in synaptic transmission. However, the PSA showed no remarkable changes. Second, increases in glutamate were inactivated by GPT, and the PSA was improved significantly in the H-CORT animals but was decreased in the L-CORT animals, suggesting that increased extracellular glutamate levels are the root cause of LLD. Excitatory synaptic transmission relies on the glutamate level in the synaptic cleft, which is responsible for the activation of glutamate receptors on the postsynaptic density, contributing to the influx of positive ions and promoting synaptic transmission. However, it was found that elimination of glutamate increased the PSA in the SHAM and H-CORT groups in this study, which seems to contradict the results of the previous study. It was found that the distribution of glutamate is heterogeneous, which is manifested by the fact that the highest level of glutamate in the brain is located in the presynapse, followed by the synaptic cleft, and the concentration of extrasynaptic glutamate is very low. Extrasynaptic glutamate elevation is not only unrelated to PSA but also activates extrasynaptic NMDA receptors, which then exert neurotoxicity. In this study, we assumed that the increase in the PSA induced by the elimination of glutamate may result from the decrease in extrasynaptic glutamate levels, which needs further investigation.
Because there is no specific enzyme for the degradation of glutamate in vivo, the clearance of glutamate relies on transporter activity to maintain glutamate/glutamine cycle homeostasis [52, 53]. In this study, we found that the capacity for glutamate uptake in H-CORT synaptosomes was less than that of the SHAM group and that L-CORT significantly restored glutamate uptake. All of these results suggest that glutamate uptake is insufficient in LLD animals and that glutamate uptake activity can be increased by lowering CORT levels.
Astrocytes regulate glutamate levels by removing glutamate via the glutamate transporter (EAAT1-5) [54]. In the rat hippocampus, the predominant glutamate transporters are Glt-1 (EAAT2), which shapes synaptic transmission by limiting the leakage of glutamate into extrasynaptic receptors or glutamate spillover into neighboring synapses [52, 55, 56]. Chronic restraint stress upregulates GLT-1 mRNA and protein expression in the adult hippocampus [57]. However, we found that there was reduced Glt-1 expression in the H-CORT hippocampus, and a larger quantity of glutamate was accumulated in the extracellular space, which induced LLD. Lowering CORT levels restored Glt-1 expression, which maintained stable glutamatergic neurotransmission and improved LLD.
Unlike hippocampal granule neurons and microglia, astrocytes are resistant to CORT-induced apoptosis due to their reduced production of reactive oxygen species (ROS) and their increased buffering capacity against the cytotoxic actions of ROS [58, 59]. To directly determine the effect of CORT on the expression of Glt-1, primary astrocytes were exposed to CORT for 5 days. No gliosis was found (data not shown), and the activity of glutamate transporters was not significantly changed, indicating that the decrease in Glt-1 was independent of CORT impairment. Consistent with previous studies, we found that hippocampal neurons endured substantial damage after long-term exposure to CORT, including synaptic loss and glutamate receptor dysfunction. These distinct results suggest that CORT mediates the interaction between astrocytes and neurons in the hippocampus in a way that is unclear.
A 5-day gradient hippocampal neuron–astrocyte coculture under CORT exposure was used to further investigate the interaction between astrocytes and neurons. The expression of Glt-1 decreased with an increase in the hippocampal neuron percentage. To exclude the nonspecific role of glutamate, a dopaminergic neuron–astrocyte coculture was generated under the same exposure to glutamate. Interestingly, there was no significant change in the expression of glutamate transporters. All of these results suggest that the accumulation of extracellular glutamate over a long duration decreases Glt-1 expression.
To further investigate the role of Glt-1 deficiency in LLD, its activity was regulated by pharmacological methods in SHAM animals. We tested whether LLD-like signs could be induced by dihydrokainic acid (DHK, an inhibitor of Glt-1) [53] via behavioral tests, including the MWM, FST, and OPT. We found that DHK impaired performance in the MWM in a depressive-like manner, which could reflect reduced cognitive function or an increased immobility time (helplessness). All of these behavioral changes suggest that the inactivation of Glt-1 leads to LLD. Glt-1 was elevated by Riluzole (RIL, a drug that elevates Glt-1 expression [60] and facilitates glutamate uptake [46, 61, 62]) in LLD animals. The behavioral tests showed that RIL significantly attenuated depressive signs compared with those observed in the vehicle-treated animals, as shown by the increase in locomotive movement and improvement of cognitive function. Two sets of comprehensive behavioral experiments found that Glt-1 activity plays an important role in the incidence and treatment of LLD.
Taken together, these data suggest that the dysfunction of glutamatergic neurotransmission in LLD may result from deficient Glt-1 activity, which directly contributes to the accumulation of extracellular glutamate, directly contributing to the overactivation of glutamatergic receptors, especially extrasynaptic NMDA receptors, which are considered the main sources of excitatory neurotoxicity. Moreover, the poor uptake of glutamate contributed to the reduced production of glutamine, which is an important substrate for neuron survival. Moreover, the decrease in glutamine production led to poor neural viability and the deficiency of excitatory neurotransmission, resulting in enhancement of Glt-1 activity and a significant anti-LLD effect, which provides promising clues for the treatment of LLD.
References
Unutzer J. Clinical practice. Late-life depression. N Engl J Med. 2007;357:2269–76.
Unutzer J, Katon W, Callahan CM, Williams JW Jr, Hunkeler E, Harpole L, et al. Collaborative care management of late-life depression in the primary care setting: a randomized controlled trial. JAMA. 2002;288:2836–45.
Byers AL, Yaffe K. Depression and risk of developing dementia. Nat Rev Neurol. 2011;7:323–31.
Marcum ZA, Vande Griend JP, Linnebur SA. FDA drug safety communications: a narrative review and clinical considerations for older adults. Am J Geriatr Pharmacother. 2012;10:264–71.
Popoli M, Yan Z, McEwen BS, Sanacora G. The stressed synapse: the impact of stress and glucocorticoids on glutamate transmission. Nat Rev Neurosci. 2012;13:22–37.
Venero C, Borrell J. Rapid glucocorticoid effects on excitatory amino acid levels in the hippocampus: a microdialysis study in freely moving rats. Eur J Neurosci. 1999;11:2465–73.
Karst H, Berger S, Turiault M, Tronche F, Schutz G, Joels M. Mineralocorticoid receptors are indispensable for nongenomic modulation of hippocampal glutamate transmission by corticosterone. Proc Natl Acad Sci USA. 2005;102:19204–7.
Issa AM, Rowe W, Gauthier S, Meaney MJ. Hypothalamic-pituitary-adrenal activity in aged, cognitively impaired and cognitively unimpaired rats. J Neurosci. 1990;10:3247–54.
Landfield PW, Baskin RK, Pitler TA. Brain aging correlates: retardation by hormonal-pharmacological treatments. Science. 1981;214:581–4.
Lupien SJ, McEwen BS, Gunnar MR, Heim C. Effects of stress throughout the lifespan on the brain, behaviour and cognition. Nat Rev Neurosci. 2009;10:434–45.
MacQueen G, Frodl T. The hippocampus in major depression: evidence for the convergence of the bench and bedside in psychiatric research? Mol Psychiatry. 2011;16:252–64.
Sexton CE, Le Masurier M, Allan CL, Jenkinson M, McDermott L, Kalu UG, et al. Magnetic resonance imaging in late-life depression: vascular and glucocorticoid cascade hypotheses. Br J Psychiatry. 2012;201:46–51.
Hegel MT, Unutzer J, Tang L, Arean PA, Katon W, Noel PH, et al. Impact of comorbid panic and posttraumatic stress disorder on outcomes of collaborative care for late-life depression in primary care. Am J Geriatr Psychiatry. 2005;13:48–58.
Bishop NA, Lu T, Yankner BA. Neural mechanisms of ageing and cognitive decline. Nature. 2010;464:529–35.
Tasker JG, Herman JP. Mechanisms of rapid glucocorticoid feedback inhibition of the hypothalamic-pituitary-adrenal axis. Stress. 2011;14:398–406.
Tsopelas C, Stewart R, Savva GM, Brayne C, Ince P, Thomas A, et al. Neuropathological correlates of late-life depression in older people. Br J Psychiatry. 2011;198:109–14.
Marinelli M, Piazza PV, Deroche V, Maccari S, Le Moal M, Simon H. Corticosterone circadian secretion differentially facilitates dopamine-mediated psychomotor effect of cocaine and morphine. J Neurosci. 1994;14:2724–31.
Meyer JS, Micco DJ, Stephenson BS, Krey LC, McEwen BS. Subcutaneous implantation method for chronic glucocorticoid replacement therapy. Physiol Behav. 1979;22:867–70.
Eisch AJ, Barrot M, Schad CA, Self DW, Nestler EJ. Opiates inhibit neurogenesis in the adult rat hippocampus. Proc Natl Acad Sci USA. 2000;97:7579–84.
Xue W, Hu JF, Yuan YH, Sun JD, Li BY, Zhang DM, et al. Polygalasaponin XXXII from Polygala tenuifolia root improves hippocampal-dependent learning and memory. Acta Pharmacol Sin. 2009;30:1211–9.
Crochemore C, Lu J, Wu Y, Liposits Z, Sousa N, Holsboer F, et al. Direct targeting of hippocampal neurons for apoptosis by glucocorticoids is reversible by mineralocorticoid receptor activation. Mol Psychiatry. 2005;10:790–8.
Shever’ova VM. Influence of chronic emotional stress on the behaviour in “open field” test of rats with different level of motor activity. Fiziol Zh. 2011;57:94–105.
Wang XY, Zhang JT. NO mediates ginsenoside Rg1-induced long-term potentiation in anesthetized rats. Acta Pharmacol Sin. 2001;22:1099–102.
Yuen EY, Wei J, Liu W, Zhong P, Li X, Yan Z. Repeated stress causes cognitive impairment by suppressing glutamate receptor expression and function in prefrontal cortex. Neuron. 2012;73:962–77.
Sierra-Mercado D, Dieguez D Jr., Barea-Rodriguez EJ. Brief novelty exposure facilitates dentate gyrus LTP in aged rats. Hippocampus. 2008;18:835–43.
Chen Y, Kramar EA, Chen LY, Babayan AH, Andres AL, Gall CM, et al. Impairment of synaptic plasticity by the stress mediator CRH involves selective destruction of thin dendritic spines via RhoA signaling. Mol Psychiatry. 2012;18:485–96.
Dunkley PR, Jarvie PE, Heath JW, Kidd GJ, Rostas JA. A rapid method for isolation of synaptosomes on Percoll gradients. Brain Res. 1986;372:115–29.
Bonanno G, Giambelli R, Raiteri L, Tiraboschi E, Zappettini S, Musazzi L, et al. Chronic antidepressants reduce depolarization-evoked glutamate release and protein interactions favoring formation of SNARE complex in hippocampus. J Neurosci. 2005;25:3270–9.
Raiteri L, Raiteri M. Synaptosomes still viable after 25 years of superfusion. Neurochem Res. 2000;25:1265–74.
Dunlop J, Eliasof S, Stack G, McIlvain HB, Greenfield A, Kowal D, et al. WAY-855 (3-amino-tricyclo[2.2.1.02.6]heptane-1,3-dicarboxylic acid): a novel, EAAT2-preferring, nonsubstrate inhibitor of high-affinity glutamate uptake. Br J Pharmacol. 2003;140:839–46.
Jasinska M, Siucinska E, Glazewski S, Pyza E, Kossut M. Characterization and plasticity of the double synapse spines in the barrel cortex of the mouse. Acta Neurobiol Exp (Wars). 2006;66:99–104.
Chen PS, Peng GS, Li G, Yang S, Wu X, Wang CC, et al. Valproate protects dopaminergic neurons in midbrain neuron/glia cultures by stimulating the release of neurotrophic factors from astrocytes. Mol Psychiatry. 2006;11:1116–25.
Kaech S, Banker G. Culturing hippocampal neurons. Nat Protoc. 2006;1:2406–15.
Wang X, Xu H, Rothman RB. Intracerebroventricular administration of anti-endothelin-1 IgG selectively upregulates endothelin-A and kappa opioid receptors. Neuroscience. 2004;129:751–6.
Aune T, Stiles TC. The effects of depression and stressful life events on the development and maintenance of syndromal social anxiety: sex and age differences. J Clin Child Adolesc Psychol. 2009;38:501–12.
Kessler RC. The effects of stressful life events on depression. Annu Rev Psychol. 1997;48:191–214.
Mizoguchi K, Ishige A, Takeda S, Aburada M, Tabira T. Endogenous glucocorticoids are essential for maintaining prefrontal cortical cognitive function. J Neurosci. 2004;24:5492–9.
Yau JL, Seckl JR. Local amplification of glucocorticoids in the aging brain and impaired spatial memory. Front Aging Neurosci. 2012;4:24.
Atsak P, Hauer D, Campolongo P, Schelling G, McGaugh JL, Roozendaal B. Glucocorticoids interact with the hippocampal endocannabinoid system in impairing retrieval of contextual fear memory. Proc Natl Acad Sci USA. 2012;109:3504–9.
Cazakoff BN, Howland JG. Acute stress disrupts paired pulse facilitation and long-term potentiation in rat dorsal hippocampus through activation of glucocorticoid receptors. Hippocampus. 2010;20:1327–31.
Whitlock JR, Heynen AJ, Shuler MG, Bear MF. Learning induces long-term potentiation in the hippocampus. Science. 2006;313:1093–7.
Lowy MT, Gault L, Yamamoto BK. Adrenalectomy attenuates stress-induced elevations in extracellular glutamate concentrations in the hippocampus. J Neurochem. 1993;61:1957–60.
Segovia G, Porras A, Del Arco A, Mora F. Glutamatergic neurotransmission in aging: a critical perspective. Mech Ageing Dev. 2001;122:1–29.
de Kloet ER, Joels M, Holsboer F. Stress and the brain: from adaptation to disease. Nat Rev Neurosci. 2005;6:463–75.
Yuen EY, Liu W, Karatsoreos IN, Ren Y, Feng J, McEwen BS, et al. Mechanisms for acute stress-induced enhancement of glutamatergic transmission and working memory. Mol Psychiatry. 2011;16:156–70.
Banasr M, Chowdhury GM, Terwilliger R, Newton SS, Duman RS, Behar KL, et al. Glial pathology in an animal model of depression: reversal of stress-induced cellular, metabolic and behavioral deficits by the glutamate-modulating drug riluzole. Mol Psychiatry. 2010;15:501–11.
Yang J, Shen J. In vivo evidence for reduced cortical glutamate-glutamine cycling in rats treated with the antidepressant/antipanic drug phenelzine. Neuroscience. 2005;135:927–37.
Shen J, Petersen KF, Behar KL, Brown P, Nixon TW, Mason GF, et al. Determination of the rate of the glutamate/glutamine cycle in the human brain by in vivo 13C NMR. Proc Natl Acad Sci USA. 1999;96:8235–40.
Yoon SJ, Lyoo IK, Haws C, Kim TS, Cohen BM, Renshaw PF. Decreased glutamate/glutamine levels may mediate cytidine’s efficacy in treating bipolar depression: a longitudinal proton magnetic resonance spectroscopy study. Neuropsychopharmacology. 2009;34:1810–8.
McEwen AM, Burgess DT, Hanstock CC, Seres P, Khalili P, Newman SC, et al. Increased glutamate levels in the medial prefrontal cortex in patients with postpartum depression. Neuropsychopharmacology. 2012;37:2428–35.
Lee Y, Son H, Kim G, Kim S, Lee DH, Roh GS, et al. Glutamine deficiency in the prefrontal cortex increases depressive-like behaviours in male mice. J Psychiatry Neurosci. 2012;37:120024.
John CS, Smith KL, Van’t Veer A, Gompf HS, Carlezon WA Jr, Cohen BM, et al. Blockade of astrocytic glutamate uptake in the prefrontal cortex induces anhedonia. Neuropsychopharmacology. 2012;37:2467–75.
Bechtholt-Gompf AJ, Walther HV, Adams MA, Carlezon WA Jr, Ongur D, Cohen BM. Blockade of astrocytic glutamate uptake in rats induces signs of anhedonia and impaired spatial memory. Neuropsychopharmacology. 2010;35:2049–59.
Potier B, Billard JM, Riviere S, Sinet PM, Denis I, Champeil-Potokar G, et al. Reduction in glutamate uptake is associated with extrasynaptic NMDA and metabotropic glutamate receptor activation at the hippocampal CA1 synapse of aged rats. Aging Cell. 2010;9:722–35.
Autry AE, Grillo CA, Piroli GG, Rothstein JD, McEwen BS, Reagan LP. Glucocorticoid regulation of GLT-1 glutamate transporter isoform expression in the rat hippocampus. Neuroendocrinology. 2006;83:371–9.
Rothstein JD, Martin L, Levey AI, Dykes-Hoberg M, Jin L, Wu D, et al. Localization of neuronal and glial glutamate transporters. Neuron. 1994;13:713–25.
Reagan LP, Rosell DR, Wood GE, Spedding M, Munoz C, Rothstein J, et al. Chronic restraint stress up-regulates GLT-1 mRNA and protein expression in the rat hippocampus: reversal by tianeptine. Proc Natl Acad Sci USA. 2004;101:2179–84.
Yu S, Yang S, Holsboer F, Sousa N, Almeida OF. Glucocorticoid regulation of astrocytic fate and function. PloS ONE. 2011;6:e22419.
Jacobsson J, Persson M, Hansson E, Ronnback L. Corticosterone inhibits expression of the microglial glutamate transporter GLT-1 in vitro. Neuroscience. 2006;139:475–83.
Carbone M, Duty S, Rattray M. Riluzole elevates GLT-1 activity and levels in striatal astrocytes. Neurochem Int. 2012;60:31–8.
Fumagalli E, Funicello M, Rauen T, Gobbi M, Mennini T. Riluzole enhances the activity of glutamate transporters GLAST, GLT1 and EAAC1. Eur J Pharmacol. 2008;578:171–6.
Frizzo ME, Dall’Onder LP, Dalcin KB, Souza DO. Riluzole enhances glutamate uptake in rat astrocyte cultures. Cell Mol Neurobiol. 2004;24:123–8.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81973499, 81960729, 81873026, 81730096, and 81773924), the Natural Science Foundation of Beijing (7192135), the CAMS Innovation Fund for Medical Sciences (CIFMS) (2016-I2 M-1-004), and The Opening Program of the Shanxi Key Laboratory of Chinese Medicine Encephalopathy (CME-OP-2017001).
Author information
Authors and Affiliations
Contributions
SFC and NHC designed research; SFC, ZZ, XZ, WBH, BY, LYC, HYH and ZZW performed research; WBH and BY contributed new analytical tools and reagents; SFC, ZZ and NHC analyzed data; SFC wrote the paper.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Rights and permissions
About this article

Cite this article
Chu, Sf., Zhang, Z., Zhou, X. et al. Low corticosterone levels attenuate late life depression and enhance glutamatergic neurotransmission in female rats. Acta Pharmacol Sin 42, 848–860 (2021). https://doi.org/10.1038/s41401-020-00536-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41401-020-00536-w
- Springer Nature Singapore Pte Ltd.
Keywords
This article is cited by
-
Clarification of the molecular mechanisms underlying glyphosate-induced major depressive disorder: a network toxicology approach
Annals of General Psychiatry (2024)
-
Itaconate inhibits corticosterone-induced necroptosis and neuroinflammation via up-regulating menin in HT22 cells
Journal of Physiology and Biochemistry (2024)
-
Botulinum neurotoxin A ameliorates depressive-like behavior in a reserpine-induced Parkinson’s disease mouse model via suppressing hippocampal microglial engulfment and neuroinflammation
Acta Pharmacologica Sinica (2023)
-
New monoamine antidepressant, hypidone hydrochloride (YL-0919), enhances the excitability of medial prefrontal cortex in mice via a neural disinhibition mechanism
Acta Pharmacologica Sinica (2022)