
Abstract
Maple syrup urine disease (MSUD) is an autosomal recessive disorder affecting branched-chain amino acids (BCAAs) metabolism and caused by a defect in the thiamine-dependent enzyme branched chain α-ketoacid dehydrogenase (BCKD) with subsequent accumulation of BCAAs and corresponding branched-chain keto acids (BCKAs) metabolites. Presently, at least 4 genes of BCKDHA, BCKDHB, DLD and DBT have been reported to cause MSUD. Furthermore, more than 265 mutations have been identified as the cause across different populations worldwide. Some studies have reported the data of gene mutations in Chinese people with MSUD. In this study, we present clinical characteristics and mutational analyses in five Chinese Han child with MSUD, which had been screened out by tandem mass spectrometry detection of amino acids in blood samples. High-throughput sequencing, Sanger sequence and real-time qualitative PCR were performed to detect and verify the genetic mutations. Six different novel genetic variants were validated in BCKDHB gene and BCKDHA gene, including c.523 T > C, c.659delA, c.550delT, c.863G > A and two gross deletions. Interestingly, 3 cases had identical mutation of BCKDHB gene (c.659delA). We predicted the pathogenicity and analyzed the clinical characteristics. The identification of these mutations in this study further expands the mutation spectrum of MSUD and contributes to prenatal molecular diagnosis of MSUD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Maple syrup urine disease (MSUD, OMIM #248600) is an autosomal recessive disorder characterized by a deficiency of the thiamine-dependent enzyme branched chain α-ketoacid dehydrogenase (BCKD)which could result in the accumulation of branched-chain amino acids (BCAAs), including leucine, isoleucine, and valine (Burrage et al. 2014). The BCAAs and corresponding branched-chain keto acids (BCKA) metabolites accumulated in brain, causing a range of clinical presentation, especially neurological damage, such as anorexia, apnoea,stereotyped movements, and so on (Zeltner et al. 2014). MSUD was first described by Menkes et al. in 1954 (Menkes et al. 1954). The current prevalence was reported about 1 in 185,000 worldwide (Lee et al. 2008) and much higher in some ethnic and racial crowds, such as the Mennonites (up to 1 in 358 live births) and Iran (38.6% in consanguineous marriage) (Puffenberger 2003; Abiri et al. 2017), but the incidence in Chinese population has not yet been reported.
Tandem mass spectrometry (MS/MS) is widely used in neonatal screening in china which enables to diagnose MSUD (Lin et al. 2013). The results of tandem mass spectrometry also provide reference for MSUD gene mutation detection. To date, at least 4 genes of BCKDHA, BCKDHB, DLD and DBT have been reported (Abiri et al. 2017) to be the causative gene for MSUD, furthermore, there have been 88 BCKDHA, 85 BCKDHB, 21 DLD and 71 DBT mutations listed in the human gene mutation database (HGMD). Some reports have also presented some new mutation of genes for MSUD in China (Li et al. 2015; Yang et al. 2012; Hou and Hwang 2014; Wang et al. 2011; Guo et al. 2015; Wang et al. 2012). Genetic testing for families at-risk of MSUD is very helpful in Prenatal Diagnosis (PND) and Preimplantation Genetic Diagnosis (PGD).
In the present study, we present clinical characteristics and mutational analyses in five Han Chinese patients with MSUD which had been identified by liquid chromatography-tandem mass spectrometry (LC-MS/MS) for amino acids detection in blood samples and gas chromatography mass spectrometry (GC/MS) for organic acidurias screening in urine samples as well as High throughput sequencing for gene mutation testing. We found 6 novel mutations in BCKDHA and BCKDHB genes including 2 in BCKDHA and 4 in BCKDHB in 5 patients from Shandong province of North China.
Material and methods
Compliance with ethical standards
The work was approved by Medical Ethics Committee of Qilu Children’s Hospital of Shandong University. The parents of the patients gave their written informed consent before clinical and laboratory examinations. All the relevant regulations and institutional polices were followed strictly with the Declaration of Helsinki. The patients’ information was anonymized prior to submission.
Patients
Five children (3 males and 2 females) from 5 unrelated Chinese families were diagnosed as MSUD at neonatal intensive care unit and rehabilitation department of the Qilu Children’s Hospital of Shandong University screened from 5432 infants by LC/MS-MS and GC/MS between May 2011 and May 2014. The parents of the patients were healthy and non-consanguineous. Blood samples were obtained from the patients and their parents in accordance with informed consent in the study.
Routine tests and metabolic analysis
Routine physical examination and biochemical laboratory tests were performed for liver and renal function, glucose, ammonia, lactic acid and blood gas analysis, and so on by using blood, urine and cerebrospinal fluid. Cranial MRI was conducted except for one patient (case 1) who was too serious to do it.
The levels of blood amino acids including valine (val), leucine (Leu), and isoleucine (Ile) in dried blood spots were measured by liquid chromatography-tandem mass spectrometry (LC-MS/MS) using an Applied Biosystems API 3200 analyzer (ABSCIEX, Foster City, USA) and the ChemoView software(ABSCIEX, Foster City, USA). Meanwhile, the levels of organic acidurias were measured by gas chromatography mass spectrometry (GC/MS) using a GCMS-QP2010 analyzer (Shimadzu, Tokyo, Japan) and the Inborn Errors of Metabolism Screening System software(Shimadzu, Tokyo, Japan) .
Targeted captured sequencing
Peripheral blood samples were collected from the patients and their parents for DNA extraction using a DNA extraction kit (TIANamp Blood Genomic DNA Purification Kit; Tiangen Biotech, Beijing, China). Genomic DNA was purified according to manufacturer’s instructions (concentration = 100~150 ng/μl; OD260/OD280 = 1.7~1.9).
Whole exome targeted capture sequencing was applied for the mutation screening of the suspicious patients. A total of 712 genes associated with inherited metabolic diseases were selected and their coding regions were captured using a custom exome enrichment kit (Agilent, Santa Clara, USA). Fifteen microgram of DNA from the proband was used to generate index libraries (average size 350-450 bp, including adapter sequences) for HiSeq2000 sequencer (Illumina, San diego, USA). Sequencing was carried out with 90 cycles per read. The obtained mean exome coverage was more than 98%, with more than 99% of variants accuracy.
Validation of genetic variants
Sanger sequencing and quantitative PCR (qPCR) were then utilized to validate the potentially pathogenic variants of point mutations and gross deletions in the patients with designed specific primers. The primer sets were designed to target different fragments within the deleted regions using an online primer designing tool-Primer 3 (http://primer3.ut.ee/). The primers were then synthesized by Shanghai Invitrogen Biotechnology Company (Shanghai, China) (Table 1).
Six mutations, such as c.508C > T, c.659delA, c.523 T > C, c.93_103dup11, c.550delT and c.863G > A were amplified with routine procedures. The PCR amplification products were purified and sequenced using an ABI Prism 3700 automated sequencer (Applied Biosystems, Foster City, CA). Three gross deletions in case 2, 3 and 5 were quantitative amplified according to the manufacturer’s recommendations on a real-time PCR system (LightCycler 480 II, Roche, Foster City, USA). Copy number variations were determined based on the ratio of copies of the deletion fragment to a reference gene (GAPDH). Genomic DNA samples from normal male and female individuals were used simultaneously as two control samples. Each qPCR was carried out in triplicate with the SYBR Premix Ex Taq II PCR reagent kit (TakaRa Bio, Dalian, China) according to the manufacturer’s protocol.
Bioinformatic analysis of mutation
Homologous protein sequence alignments and bioinformatic analysis of the mutations were conducted. Potential effects of the mutations on function were assessed using the following databases: UCSC Genome Bioinformatics (http://genome.ucsc.edu/) (comparing with the reference sequence of BCKDHA gene and BCKDHB gene); Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php) (comparing with an integrated set of variants); 1000 Genomes Project (www.1000genomes.org) and ExAC(http://exac.broadinstitute.org/about). PolyPhen-2 database (http://genetics.bwh.harvard.edu/pph2/index.shtml) and SIFT sequence database (http://sift.jcvi.org/www/SIFT_seq_submit2.html) were used to predict the pathogenicity of missense alterations. ClustalX software and OMIM (http://www.ncbi.nlm.nih.gov/omim/limits) were used to obtain the multiple sequence alignments. Generation of protein plots was performed using Swiss-bdb Viewer4.0.
Result
Clinical characteristics and laboratory tests
Five index cases with MSUD were identified among 5432 by high risk infants screening. All the five index cases presented with increased Leucine (Leu), Ileucine (Ile), and Leu + Ile/Phe (phenylalanine) ratio in dried blood spots, and 4 patients (case 1, 3, 4 and 5) have elevated valine (Val) (Table 2). So they were highly suspected to suffer from maple syrup urine disease.
Clinical data of five index cases were collected (Table 2). All patients aged less than 10 days at the first onset. The main clinical manifestations included irritability, feeding difficulties, abnormal muscle tension, seizure, and so on. Among them, case 1 and case 2 presented hypertonia, while other patients (case 3, 4 and 5) exhibited acid-base imbalance including respiratory acidosis, respiratory alkalosis and metabolic acidosis. Blood biochemical index showed that blood ammonia is mostly increasing, but the blood lactic acid is normal which is different from the previous report (Yang et al. 2012). Liver enzymes including glutamic-oxalacetic transaminase and glutamic-pyruvic transaminase were normal, and glutamyl transpetidase was high, but amylase was decreased in four patiens (case 1, 2, 4 and 5). Cranial MRI showed widespread abnormal signals in four patients (case 2, 3, 4 and 5), while the case 1 was too serious to do MRI (Fig. 1, Table 2). Case 2 was found to have severe anemia with hemoglobin 78 g/L at 1-month-10-day old which was considered to be from persistent feeding difficulty in his first month. After the treatment for MSUD, the hemoglobin was normal, 120 g/L, at 3-year-3 months old.

Magnetic resonance imaging of brain in case 3, 4, 5. All of them are accord with the manifestation of inherited metabolic encephalopathy. Widespread abnormal signals were found in the brain including bilateral brain bridge, midbrain, pons, medulla, cerebellar vermis, thalamus, corona radiata, semioval center, bilateral globus pallidus, internal capsule. A. axial T2-weighted images (T2 W1); B. axial T1-weighted images (T1 W1); C. diffusion weighted imaging (DWI); D. Fluid attented inversion recovery (FLAIR)
Genetic analysis
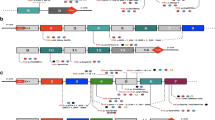
Whole exome targeted capture sequencing for inherited metabolic diseases were applied to detect the mutations in patients. Sanger sequencing and quantitative PCR (qPCR) were utilized for verification. Eight different mutations were identified in BCKDHB and BCKDHA with compound heterozygosis. Of which, 6 were novel mutation, including c.523 T > C, c.659delA, c.550delT, c.863G > A and two gross deletions (see Table 3 and Fig. 2).


Identification of mutations by NGS, Sanger sequencing and qPCR for case 1–5. A. Compound heterozygous BCKDHB point mutations in case 1 inherited from her father and mother. B. Compound heterozygous BCKDHB mutations (deletion and point) in case 2 inherited from his father and mother. C. Homozygous BCKDHB point mutation (c.659delA) in case 3 inherited from his father but mother was not available. D. Compound heterozygous BCKDHB mutations (deletion and duplication) in case 4 inherited from his father and mother. E. Compound heterozygous BCKDHA mutations (deletion and point) in case 5 inherited from his father and mother. The gross deletions in case 2 and 5 were detected by SYBR qPCR assays. The numbers on the y-coordinate refer to the ratio of deleted region versus GAPDH copies. Generally the normal copies is 0.7 < 2-ΔΔCt <1.3 and 0.3 < 2-ΔΔCt <0.6 is loss of heterozygosity. P: proband; F: father; M: mother
None of these mutations was observed in databases and the patients’ healthy family members. The mutations of c.550delT and c.659delA could result in frameshift mutation in coding sequence of BCKDHB, in which, the 229th codon became premature stop codon in the same way, thus, the length of the encoded peptide chain was reduced from 392 to 229 amino acid residues (p.G209LfsX1 and p.S184PfsX1 respectively). The two point mutations, c.863G > A in BCKDHA and c.523 T > C in BCKDHB, are highly conservative in seven species sequence analysis and were predicted to be pathogenic by polyphen-2 (Fig. 3). In addition, the crystallographic structure of the human E1 tetramer of the BCKD complex predicted by Swiss-PDB Viewer 4.0 indicated the two missense mutations might affect E1 function (Fig. 4). Finally, the two point mutations in the study were regarded as likely pathogenic.

In silico analysis of the novel point mutations in BCKDHA and BCKDHB. A. The point mutations c.863G > A (p.G288D) in BCKDHA gene and c.523 T > C (p.F175 L) in BCKDHB gene are highly conservative in seven species. B. p.F175 L mutation is predicted to be probably damaging with a score of 0.995 (sensitivity 0.68; specificity: 0.97); p.G288D mutation is predicted to be probably damaging with a score of 1.000 (sensitivity 0.00; specificity: 1.00)

3D–structure of wide-type and mutant type BCKDHA and BCKDHB. A. 3D–structure of E1α and E1β. E1α (green), E1β(blue). The red arrows marked the locations of F175 and G288. B. 3D structures of E1α bearing wild-type or mutated G288-α. The mutation D288-α change the side strand structure. C. 3D structures of E1β bearing wild-type or mutated F175-β. The mutation L175-β loss the benzene ring structure
Treatment and follow up
In the acute phase, the best treatment was invasive interventions such as peritoneal dialysis, hemodiafiltration or hemodialysis. But our four patients (case 1, 3, 4 and 5) were all decompensation, which they could not tolerate the general anesthesia and dialysis. Therefore, the prompt and aggressive treatment was carried out to reduce leucine levels, and the basal prescription was a high-rate glucose infusion (12.5%) to stimulate insulin secretion and suppress protein catabolism, levocarnitine (100 mg/kg daily) to increase the plasma free carnitine and decrease malondialdehyde and protect the neuron, as well as thiamine (100 mg daily), BCAA-free medical formula (6%), and mannitol (20%, 1.25–2.5 ml/kg) to prevent the encephaledema. Sadly, the treatment did not take effect and the four patients died from severe metabolic crisis and cerebral injury though the liver transplantation was recommended.
The case 2 was not in the severe acute phase when he was diagnosed as MSUD and the good effect was received after using the basal prescription same as that for other 4 patients. When the plasma BCAA levels was normal, the long-term treatment was maintained using dietary modifications, such as protein restriction and BCAA-free medical formula, and periodic monitoring by MS/MS and GC/MS. Thiamine was not prescribed in the long-term treatment in which the proband was ineffective by initial experimental thiamine administration. After the follow up of 3-year-3-month, he showed slight mental and language developmental delay.
Since the MUSD was diagnosed by MS/MS and GC/MS, We have carried out the thiamine administration in all cases 100 mg daily. However, case 1, 4 and 5 were dead from severe metabolic crisis soon, and case 2 and 3 were ineffective.
Discussion
The branched-chain amino acids (BCAAs) including valine, isoleucine and leucine are essential amino acids for hydrophobic side chains. BCAAs comprise about 20–40% in most dietary proteins. In the body, BCAAs undergo ammonia stripping by branched chain aminotransferase (BCAT) to their respective α-ketoacids, besides α-ketoacids was catalyzed oxidative decarboxylation by the branched-chain ketoacid dehydrogenase complex (BCKDC), in order to carry out catabolism (Burrage et al. 2014). BCKDC, a highly conserved and large catalytic complex (4.5 MDa), consists of a heterodimeric E1 decarboxylase component (E1a and E1b subunits which were encoded by BCKDHA(NC_000019.10) and BCKDHB(NC_000006.12)), E2 transacylase component with 24 identical subunits which was encoded by DBT gene (NC_000001.11), and a homodimeric E3 component encoded by DLD gene (NC_000007.14)(Lu et al. 2009; Abiri et al. 2016). Any one subunit change of BCKDC could affect its catalytic function, which is likely to cause MSUD. Otherwise, the change of regulatory enzyme to BCKDC, PPM1K encoded by PPM1K which activates BCKDC by dephosphorylation of the E1α subunit (Oyarzabal et al. 2013) could also cause MSUD.
MSUD is a heterogeneous disorder with variable clinical severity. Based on the clinical features and residual activity of BCKDC, MUSD was classified into five types(Abiri et al. 2016; Taylor et al. 1978), including the classic type with 0–2% enzyme activity and neonatal presentation, the intermediate type with 3–30% enzyme activity and onset at any age, the intermittent type with 5–20% enzyme activity and similarly presenting at any age, the thiamin- responsive type with 2–40% activity and being the late onset form, and dihydrolipoamide dehydrogenase deficiency (E3 − deficiency) type with 8–20% activity and usually infancy manifestation. According to the onset age and the severity of illness, case 1, 3, 4 and 5 were classified as classic MSUD, and case 2 is intermediate MSUD.
At present, MSUD can be screened out by MS/MS testing, but the specific genetic causes of MSUD have to be further tested by molecular techniques which provided valuable methods for diagnosis of the patients and carriers as well as the chance for the treatment. Different molecular techniques can be applicable, such as Sanger sequencing, real-time qPCR, STR linkage analysis in the past (Abiri et al. 2017), and high-throughput sequencing at present.
High-throughput sequencing has been increasingly accessible in the clinical laboratory with the advantages of rapid, accurate, and relatively low cost. Target region capture sequencing was developed mainly to enrich and sequence specific regions of particular interest, such as specific exons or entire exomes on the basis of High-throughput sequencing (Stoddard et al. 2014), offering a cost-effective solution for investigating genetic mutations of inherited metabolic diseases. In this study, we used a panel of target region capture sequencing containing 712 genes associated with inherited metabolic diseases in which four genes of BCKDHA, BCKDHB, DLD and DBT related to maple diabetes were included. Two patients (case 2 and 5) were identified to have the gross deletions spanning several exons in BCKDHB and BCKDHA, individually, which were further validated by qPCR. Our results showed that the advantages of using both techniques of high throughput sequencing and qPCR in detecting the gross deletion.
In recent years, some cases with MSUD have been found in the Chinese population by the promotion of MS/MS screening technology (Lin et al. 2013), and some molecular data was obtained showing that point mutations were most common in Chinese patients with MSUD(Li et al. 2015; Yang et al. 2012). We summarized the reports about MSUD in Chinese population, and the variants happened in BCKDHA, BCKDHB and DBT genes. However, in this study, small deletion/insertion and gross deletion are the common mutations, and the cases with BCKDHB mutations were in the majority, which were different from other reports. Meanwhile, the novel mutation c.659delA in BCKDHB gene was found in two patients (case 1 and 3) demonstrating that c.659delA mutation was more common mutation in this cohort.
Basically, there are three therapeutic strategies for MSUD (Burrage et al. 2014).First, the prompt and aggressive treatment is to remove the toxic substances in body and symptomatic treatment, especially in the acute phase, the best treatments was invasive interventions such as including peritoneal dialysis, hemodiafiltration or hemodialysis, a high-rate glucose infusion to stimulate insulin secretion and suppress protein catabolism, levocarnitine which could increase the plasma free carnitine and decrease malondialdehyde to protect the neuron (Ribas et al. 2014), mannitol to prevent the encephaledema. Second, protein restriction and BCAA-free medical formula limit the source, especially for the long-term treatment. The last is that increases BCKDC activity via tissue or cell transplantation, such as liver transplantation (Miryounesi et al. 2015; Al-Shamsi et al. 2016). Unfortunately, 4 patients in our study died from rapid deterioration of the illness. Case 2 benefited from a relative later onset, in-time discovery and early therapy. Moreover, he had the compound heterozygous mutations c.523 T > C and ex.1–7 del*, meanwhile the BCKDHB protein with the point mutation c.523 T > C possibly remains very little residual activity combining clinical characteristics with in silico analysis.
In summary, we present the clinical characteristics and mutation analysis of 5 patients with MUSD, which had been identified by LC-MS/MS, GC-MS screening and high-throughput sequencing. Seven novel mutations were found in BCKDHA and BCKDHB genes. The identification of these mutations in this study further expands the spectrum of known gene mutations and contributes to the genotype-phenotype correlation and prenatal molecular diagnosis of MSUD.
References
Abiri M, Karamzadeh R, Karimipoor M, Ghadami S, Alaei MR, Bagheri SD, Bagherian H, Setoodeh A, Noori-Daloii MR, Sirous Z (2016) Identification of six novel mutations in Iranian patients with maple syrup urine disease and their in silico analysis. Mutat Res 786:34–40. https://doi.org/10.1016/j.mrfmmm.2016.01.005
Abiri M, Karamzadeh R, Mojbafan M, Alaei MR, Jodaki A, Safi M, Kianfar S, Bandehi Sarhaddi A, Noori-Daloii MR, Karimipoor M, Zeinali S (2017) In silico analysis of novel mutations in maple syrup urine disease patients from Iran. Metab Brain Dis 32(1):105–113. https://doi.org/10.1007/s11011-016-9867-1
Al-Shamsi A, Baker A, Dhawan A, Hertecant J (2016) Acute metabolic crises in maple syrup urine disease after liver transplantation from a related heterozygous living donor. JIMD Rep 30:59–62. https://doi.org/10.1007/8904_2016_532
Burrage LC, Nagamani SC, Campeau PM, Lee BH (2014) Branched-chain amino acid metabolism: from rare Mendelian diseases to more common disorders. Hum Mol Genet 23(R1):R1–R8. https://doi.org/10.1093/hmg/ddu123
Guo Y, Liming L, Jiang L (2015) Two novel compound heterozygous mutations in the BCKDHB gene that cause the intermittent form of maple syrup urine disease. Metab Brain Dis 30(6):1395–1400. https://doi.org/10.1007/s11011-015-9711-z
Hou JW, Hwang TL (2014) Different gene preferences of maple syrup urine disease in the aboriginal tribes of Taiwan. Pediatr Neonatol 55(3):213–217. https://doi.org/10.1016/j.pedneo.2013.09.009
Lee JY, Chiong MA, Estrada SC, Cutiongco-De la Paz EM, Silao CL, Padilla CD (2008) Maple syrup urine disease (MSUD)--clinical profile of 47 Filipino patients. J Inherit Metab Dis 31(Suppl 2):S281–S285
Li X, Ding Y, Liu Y, Ma Y, Song J, Wang Q, Li M, Qin Y, Yang Y (2015) Eleven novel mutations of the BCKDHA, BCKDHB and DBT genes associated with maple syrup urine disease in the Chinese population: report on eight cases. Eur J Med Genet 58(11):617–623. https://doi.org/10.1016/j.ejmg.2015.10.002
Lin N, Ye J, Qiu W, Han L, Zhang H, Gu X (2013) Application of liquid chromatography -tandem mass spectrometry in the diagnosis and follow-up of maple syrup urine disease in a Chinese population. J Pediatr Endocrinol Metab 26(5–6):433–439. https://doi.org/10.1515/jpem-2012-0343
Lu G, Sun H, She P, Youn JY, Warburton S, Ping P, Vondriska TM, Cai H, Lynch CJ, Wang Y (2009) Protein phosphatase 2Cm is a critical regulator of branched-chain amino acid catabolism in mice and cultured cells. J Clin Invest 119(6):1678–1687. https://doi.org/10.1172/JCI38151
Menkes JH, Hurst PL, Craig JM (1954) A new syndrome: progressive familial infantile cerebral dysfunction associated with an unusual urinary substance. Pediatrics 14(5):462–467
Miryounesi M, Ghafouri-Fard S, Goodarzi H, Fardaei M (2015) A new missense mutation in the BCKDHB gene causes the classic form of maple syrup urine disease (MSUD). J Pediatr Endocrinol Metab 28(5–6):673–675. https://doi.org/10.1515/jpem-2014-0341
Oyarzabal A, Martínez-Pardo M, Merinero B, Navarrete R, Desviat LR, Ugarte M, Rodríguez-Pombo P (2013) A novel regulatory defect in the branched-chain α-keto acid dehydrogenase complex due to a mutation in the PPM1K gene causes a mild variant phenotype of maple syrup urine disease. Hum Mutat 34(2):355–362. https://doi.org/10.1002/humu.22242
Puffenberger EG (2003) Genetic heritage of the old order Mennonites of southeastern Pennsylvania. Am J Med Genet C: Semin Med Genet 121C(1):18–31. https://doi.org/10.1002/ajmg.c.20003
Ribas GS, Vargas CR, Wajner M (2014) L-carnitine supplementation as a potential antioxidant therapy for inherited neurometabolic disorders. Gene 533(2):469–476. https://doi.org/10.1016/j.gene.2013.10.017
Stoddard JL, Niemela JE, Fleisher TA, Rosenzweig SD (2014) Targeted NGS: a cost-effective approach to molecular diagnosis of PIDs. Front Immunol 5:531
Taylor J, Robinson BH, Sherwood WG (1978) A defect in branched-chain amino acid metabolism in a patient with congenital lactic acidosis due to dihydrolipoyl dehydrogenase deficiency. Pediatr Res 12(1):60–62. https://doi.org/10.1203/00006450-197801000-00018
Wang J, Liu H, Chen G, Tsuei SH, Yu T, Fu Q (2011) Identification of two novel BCKDHA mutations in a Chinese patient with maple syrup urine disease. J Pediatr Endocrinol Metab 24(9–10):827–829
Wang YP, Qi ML, Li TT, Zhao YJ (2012) Two novel mutations in the BCKDHB gene (R170H, Q346R) cause the classic form of maple syrup urine disease (MSUD). Gene 498(1):112–115. https://doi.org/10.1016/j.gene.2012.01.082
Yang N, Han L, Gu X, Ye J, Qiu W, Zhang H, Gong Z, Zhang Y (2012) Analysis of gene mutations in Chinese patients with maple syrup urine disease. Mol Genet Metab 106(4):412–418. https://doi.org/10.1016/j.ymgme.2012.05.023
Zeltner NA, Huemer M, Baumgartner MR, Landolt MA (2014) Quality of life, psychological adjustment, and adaptive functioning of patients with intoxication-type inborn errors of metabolism - a systematic review. Orphanet J Rare Dis 9(1):159. https://doi.org/10.1186/s13023-014-0159-8
Acknowledgments
This work was financially supported by The Natural Science Training Foundation of Shandong Province (ZR2014HP051) and Jinan Excellent Science and Technology Innovation Team Project (20150515). The authors are grateful to the patients and their parents for their contribution to the study. We would like to thank SinoPath (Beijing) Medical Laboratory for technical support.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflicts of interests
There are no conflicts of interest.
Rights and permissions
About this article

Cite this article
Li, X., Yang, Y., Gao, Q. et al. Clinical characteristics and mutation analysis of five Chinese patients with maple syrup urine disease. Metab Brain Dis 33, 741–751 (2018). https://doi.org/10.1007/s11011-017-0168-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11011-017-0168-0