
Abstract
Maple Syrup Urine Disease (MSUD) is a rare autosomal recessive disorder of branched-chain amino acid (BCAA) metabolism. The disease is mainly caused by mutations either in the BCKDHA, BCKDHB, DBT or DLD genes encoding components of the E1α, E1β, E2 and E3 subunits of branched-chain α-keto acid dehydrogenase complex (BCKDC), respectively. BCKDC is a mitochondrial enzyme which is responsible for the normal breakdown of BCAA. The rate of consanguineous marriage in Iran is 38.6 %, so the prevalence of autosomal recessive disorders is higher in comparison to other countries. Consanguinity increases the chance of the presence of pathogenic mutations in a homoallelic state. This phenomenon has made homozygosity mapping a powerful tool for finding the probable causative gene in heterogeneous disorders like IEM (Inborn Errors of Metabolism). In this study, two sets of multiplex polymorphic STR (Short Tandem Repeat) markers linked to the above-mentioned genes were selected to identify the probable pathogenic gene in the studied families. The families who showed a homozygous haplotype for the STR markers of the BCKDHB gene were subsequently sequenced. Four novel mutations including c.633 + 1G > A, c.988G > A, c.833_834insCAC, and a homozygous deletion of whole exon 3 c. (274 + 1_275–1) _(343 + 1_344–1), as well as one recently reported (c. 508G > T) mutation have been identified. Interestingly, three families shared a common haplotype structure along with the c. 508G > T mutation. Also, four other families revealed another similar haplotype with c.988G > A mutation. Founder effect can be a suggestive mechanism for the disease. Additionally, structural models of MSUD mutations have been performed to predict the pathogenesis of the newly identified variants.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Maple Syrup Urine Disease (MSUD; OMIM #248,600) is a rare metabolic disease of branched-chain amino acid metabolism. Accumulation of leucine, isoleucine and valine and the corresponding branched-chain keto acids (BCKA) metabolites induce ketoacidosis, developmental disturbances, neurological impairment and coma and may be fatal if untreated (Korein et al. 1994; Yoshino et al. 1999). Therefore, early detection and disease management are crucial for disease outcomes.
MSUD is a heterogeneous disorder with variable clinical severity. The disease is classified into five types based on the amount of branched-chain alpha-keto acid dehydrogenase (BCKD) activity. The first type is the classic which is the most severe with 0–2 % enzyme activity and neonatal presentation. The second is the intermediate form with 3–30 % enzyme activity and can present at any age. The third one is intermittent with 5–20 % activity of the enzyme and can present from infancy to adulthood. The fourth type is thiamin-responsive with 2–40 % activity and is the late onset form. The last type is dihydrolipoamide dehydrogenase deficiency (E3−deficiency) that usually present during infancy. The enzyme activity is about 8–20 % (Shaw 2014).
The disease mainly results because of the impaired activity of BCKDC (BCKD; EC 1.2.4.4), the second enzyme in the catabolic pathway of branched chain amino acids. BCKDC is a member of the highly conserved mitochondrial α-ketoacid dehydrogenase complexes. It is composed of three catalytic subunits: E1 (EC 1.2.4.4.), E2 (EC 2.3.1.168) and E3 (EC 1.8.1.4.) (Harris et al. 2004). The E1 subunit consists of a heterotetramer of α2β2 which functions as a BCKA dehydrogenase/decarboxylase (Chuang 2001).
The E1β subunit which is encoded by the BCKDHB gene is located at the 6q14 chromosomal position and includes 11 exons. It encodes for a 1.4 kb mRNA which is translated into 342 amino acid residues.
The E2 is a dihydrolipoyl transacylase that exists as a homo-24-mer and is the functional core of the complex (Chuang 2001). E3 is a dihydrolipoamide dehydrogenase and functions as a dihydrolipoyl dehydrogenase and is encoded by the DBT gene. Two regulatory enzymes (i.e. a branched-chain ketoacid dehydrogenase kinase (BCKDK) and protein phosphatase 2Cm (PPM1K or PP2Cm) regulate the activity of the complex (Lu et al. 2009; Reed et al. 1985). Defects in any part of the complex may lead to the disease state.
One of the considerable demographic features of the Iranian population is the elevated rate of consanguineous marriage. Unfortunately, the exact prevalence of MSUD is not clearly known in Iran, but it is expected to have a higher rate due to consanguineous marriage, than of the estimate of 1 in 185,000 affected infants (Chuang 2001) worldwide. This population structure is very suitable for homozygosity mapping studies because it increases the possibility of finding disease-causing mutations in blocks of homozygosity.
Genetic testing in MSUD is very helpful in Prenatal Diagnosis (PND) and Preimplantation Genetic Diagnosis (PGD) for at-risk families. It also facilitates molecular screening of patients to yield epidemiological data for implantation of community-based carrier testing in populations.
Homozygosity mapping was done with the help of STR (Short Tandem Repeats) markers to the above four mentioned genes to quickly identify the responsible gene in our MSUD patients.
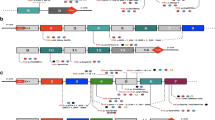
The present survey aims to show a BCAA profile, and a clinical and molecular genetics study of families who showed homozygosity for the markers of in and around the BCKDHB gene. Haplotype analysis of four families with the same structure revealed the same mutation c.988G > A (p. Glu330Lys) (Fig. 1a). Haplotype B (Fig. 1b) was seen among three other families with a c. 508G > T (p. Arg170 Cys) mutation. Investigation of the remaining families revealed an insertion of 3 nucleotides.

a The common haplotype structure is seen in the families with E330K mutation. b The common haplotype of the three families with R170 C mutation
c.833_834insCAC (p. Gly278_Thr279insThr), a splice site mutation c.633 + 1G > A and a homozygous deletion of exon-3. Computational analyzes were performed to see the impact of the novel mutations at the protein level.
Material and methods
Case descriptions
Twenty-one unrelated families were referred to our center by endocrine and metabolic specialists. The affected member of the family showed clinical presentations of MSUD and elevated levels of BCAAs and allo-isoleucine in the plasma and abnormal urine organic acids. Parents of all patients participating in this study had consanguinity. All patients studied here suffer from the classic form of the disease which is the most severe type. Genetic counseling was conducted and participants signed the consent form. The study was approved by the Ethical Committee of the Tehran University of Medical Sciences. Clinical features and the BCAA profile of each patient are presented in Table 1.
Molecular genetic study
Five ml peripheral blood samples were taken from the patients and their parents in tubes containing EDTA as an anticoagulant. DNA was extracted from the blood samples by salting out method (Miller et al. 1988).
Suitable short tandem repeat (STR) markers flanking the BCKDHA, BCKDHB, DBT and DLD genes were selected using Tandem Repeat Finder (TRF) and Sequence-based Estimation of Repeat Variability (SERV) (Benson 1999; Legendre et al. 2007). An attempt was made to select markers with higher mean heterozygosity in the sample population. The other very important criterion in marker selection was the length of the repeats. Markers with 4–6 nucleotide repeats were preferred. We used six STR markers flanking each of the genes responsible for MSUD. These markers were mixed in two multiplex sets of 12 STRs. Primers for microsatellite markers were fluorescently labeled (Applied Biosystems) and primer sequences are available upon request. Fragment analysis was performed on an ABI 3130 Genetic Analyzer (Life Technologies, LT). The Genotypes of each individual were ascertained using Gene Mapper software. Then the haplotype for each family was drawn.
After finding the probably mutated gene, the entire coding region and intron-exon boundaries of the BCKDHB gene were amplified from genomic DNA. Primers are available upon request. PCR amplification was performed based on the protocol reported previously (Foroozani et al. 2015).
PCR products were directly sequenced using BigDye Terminator kit (Thermo Fisher Scientific, Life Technologies, USA, TS) according to the manufacturer’s protocol. The samples were run on an ABI3130XL Genetic Analyzer at the KBC (Kawsar Biotech Company) facility. Sequences were compared with human genomic and cDNA sequences of the BCKDHB gene with the accession number (NC_000006.11, NM_183050.2).
Nomenclature of the novel mutations was performed according to the recommendations of Human Genome Variation Society (Den Dunnen and Antonarakis 2000).
In the case of novel missense mutations, Amplification Refractory Mutation System (ARMS) was performed on 50 healthy subjects from the same ethnicity to check the presence of mutant or normal form of the allele.
Computational analysis
The X-ray crystal structure of E1b (PDB entry: 2J9F) was used as the initial conformation for computational analysis. Modeller 9.13 was used for repairing missing residues (Eswar et al. 2006). Generation of p. Gly278_Thr279insThr mutant structure was performed with Modeller 9.13 (Eswar et al. 2006). Hydrogen was added using reduce 3.23 for the mutant of p. Glu330Lys, p. Arg170Cys, p. Gly278_Thr279insThr) and the wild-type complexes. Then, structures were prepared for energy minimization using the psfgen plugin of VMD (Humphrey et al. 1996).
All complex systems were minimized to 1000 steps by the conjugate gradient algorithm using NAMD software package to remove bad contacts (Phillips et al. 2005). The hydrogen bonds and salt bridges were measured by H Bonds and Salt Bridges plugin of VMD, respectively.
Structural impact of exon-3 deletion on BCKDHB was analyzed by generating the mutant conformation from the crystal structure (PDB entry: 2J9F) by I-TASSER web server (Roy et al. 2010; Yang et al. 2015; Yang and Zhang 2015; Zhang 2008). The Best proposed model was selected based on C-score and estimated TM-score for further analysis. Then, the wild type and mutant complexes were superimposed and secondary structure distributions were assessed by Timeline plugin of VMD (Humphrey et al. 1996). VMD was used for creating molecular graphics (Humphrey et al. 1996). Further analysis was accomplished by R.
Results
The study included twenty-one families with clinical diagnosis of MSUD. Ten patients showed homozygous haplotypes for markers flanking the BCKDHB gene, six for BCKDHA, two for DBT and no family was homozygous for the markers flanking the DLD gene. The remaining three families did not show homozygosity to any of the mentioned genes. All ten patients with homozygous haplotype block of the BCKDHB gene had the classic form of the disease which is the most severe form.
Investigation of all exons and exon/intron boundaries of the BCKDHB gene allowed successful identification of the disease-causing mutations in the investigated families. The identified mutations in the patients were homozygous as expected by the homozygote haplotype results. Parents were heterozygous for the identified mutations (Table 1).
Analysis of ten families with the homozygote haplotype of the BCKDHB gene revealed four families with the same haplotype structure (Fig. 1a) and further analysis showed the same mutation (c.988G > A). Three of the other families showed the B haplotype with the same mutation of c.508G > T (Fig. 1b). The last three families showed a different haplotype and also a different mutation including a splice site mutation (c.633 + 1G > A), an insertion (C.833_834insCAC) and homozygous deletion of exon-3 of the gene c. (274 + 1_275–1) _(343 + 1_344–1).
The relative locations of the mutations of p. Gly278_Thr279insThr, p. Glu330Lys and Arg170Cys in E1b heterodimer are shown in Fig. 2. As shown in this figure, Gly278_Thr279insThr and p. Glu330Lys mutations are located near the end of the alpha helix and beta sheets secondary structures of the β subunit of the BCKDC. The p. Arg170Cys mutation is located at the interface of the β, β’ and α subunits close to the metal ion and cofactor binding site. To elucidate the individual structural impact of these mutations on conformational stability of E1b, hydrogen bonds, and salt bridges were calculated in mutant complexes compared to the wild type after energy minimization. All the mutations resulted in redisposition of hydrogen bonds, mostly in the β and α subunits (Figs. 3 and 4).

a 3D structural representation of the E1b heterotetramer: E1α (white); E1β (blue); E1α′ (green) and E1β′ (pink). The mutated residues Arg170Cys, Gly278_Thr279insThr and Glu330Lys are indicated in orange, yellow and red, respectively. Magnesium and potassium ions are presented in light and dark brown, respectively. Thiamin diphosphates (ThDPs) are shown in black. For detailed description see “Results and Discussion”

The structural illustration of the mutated region related to the Arg170Cys mutation. a Arginine 170 situation with exclusive hydrogen bonds and salt bridges in the wild-type conformation. b Cysteine 170 position with exclusive hydrogen bonds in the mutant conformation. Red loop corresponds to the residues of 206 to 211 of the E1α subunit. Mutated residue is colored yellow. E1α, E1β, E1α′, and E1β′ are shown in white, blue, green and pink, respectively. Thiamin diphosphates (ThDPs) are colored black. Oxygen, nitrogen, sulfur and carbon atoms are in red, blue, yellow and cyan, respectively. Hydrogen bonds and salt bridges are visualized by black and green dash-lines, respectively. The magnesium and potassium ions are presented in light and dark brown, respectively. For detailed descriptions see “Results and Discussion”

The structural illustration of the mutated region related to the Gly278_Thr279insThr mutation. a The position of the mutation before the insertion of Threonine with exclusive hydrogen bonds in the wild-type conformation. b The location of the inserted Threonine between Gly278 and Thr279 with exclusive hydrogen bonds in the mutant conformation. Mutated residue is colored yellow. E1β, E1α′, and E1β′ are shown in blue, green and pink, respectively. Oxygen, nitrogen, sulfur and carbon atoms are in red, blue, yellow and cyan, respectively. Hydrogen bonds are indicated by black dash-lines. For detailed descriptions see “Results and Discussion”
Figure 3a, b display the exclusive hydrogen bonds and salt bridges in wild-type and p. Arg170Cys mutant conformations, respectively. As shown in Fig. 3, the p. Arg170Cys mutation affected not only the intra-subunit, but also inter-subunit interactions between α and β subunits. In particular, substitution of Arginine by Cysteine resulted in the elimination of salt bridges (green dash lines) between Arg170 and Glu163 inside the β subunit. Moreover, the hydrogen bond between Ser206-α and Glu163-β was also removed. The rearrangement of these bonds might have an influence on the stability of adjacent loops due to the close distance to the potassium ion. This situation might also affect the ThDP (Thiamin Diphosphate) binding site.
The superimposition of wild type and mutant complexes with their exclusive hydrogen bonds is shown in Figs. 4a, b, respectively. As shown in these figures, insertion of Threonine between Glycine 278 and Threonine 279 results in the alteration of loop structure between sheet A and the following helix. This insertion retreated sheets B and C in comparison to the wild type (Fig. 4a, b). The mutation is involved in the redistribution of adjacent amino acids near the β-β’ interfaces (e.g. Glu330, Ala331, Pro332 in wild-type and Glu331, Ala332, Pro333 in the mutant). These changes have an important role in alteration of local hydrogen bonds (i.e. elimination of hydrogen bonds between Glu201 and Trp277 in the wild-type and formation of new hydrogen bonds between Thr279 and Glu331in the mutant structure).
As shown in Figs. 5a, b, mutation of Glutamate to Lysine at the position of 330 induced rearrangements of some local hydrogen bonds including the elimination of Glu330-Cys188. This mutation is located near the subunit interface of β-β’.

The structural illustration of the mutated region related to the Glu330Lys mutation a Glutamate 330 situation with exclusive hydrogen bonds in the wild-type conformation. b Lysine 330 position with exclusive hydrogen bonds in the mutant conformation. The mutated residue is colored yellow. E1α, E1β, E1α′, and E1β′ are shown as white, blue, green and pink, respectively. Oxygen, nitrogen, sulfur and carbon atoms are in red, blue, yellow and cyan, respectively. Hydrogen bonds are indicated by black dash-lines. For a detailed description see “Results and Discussion”
Failure to amplify genomic DNA covering the third exon of the BCKDHB gene indicated the possibility of the presence of a deletion or at least a SNP (single nucleotide polymorphism) in one of the primer annealing sites. Changing the primer sequences or its location did not solve the problem, therefore we designed a duplex PCR to simultaneously amplify the first and third exons of the gene for the patient. Since in a reaction of duplex PCR the internal control primers gave a bond and the third exon did not give its relative bond, it could be concluded that there was a deletion that covers at least one of the primer annealing sites. In addition, we tried to amplify the genomic DNA using the forward primer of exon 2 and the reverse primer for exon 3. The result was no amplification. We also tried the forward primer of exon 3 and the reverse primer of exon 4, yet no band was produced, indicating complete deletion of exon 3.
Further analysis of the exon-3 deletion was completed using I-TASSER to investigate the impact on protein structure. The model with C-score 1.91 and estimated TM-score 0.99 ± 0.04 was selected. The RMSD value between experimentally determined structure and predicted model was estimated as 2.5 ± 1.9 Å. As shown in Fig. 6, the predicted model and crystal structure were structurally compared after the superimposition. These results revealed that exon-3 deletion removes 25 residues within the protein sequence, ranging from Lys92 to Gly116 (Fig. 6b). Therefore, the mutation led to the elimination of the two β-sheets at the interface of the β /β’ with α/ α’ near the ligand binding sites (Fig. 6a).

The 3D structural representation of exon-3 deletion in the BCKDHB gene. a The wild-type (cyan) and predicted mutant E1β’ (pink) are shown within the E1b complex; the mutated part, deleted residues within the sequence of Lys92 to Gly116, is indicated in yellow; removed regions in the wild-type structure are presented in red. b The secondary structure of the wild-type and mutant conformations. Missing regions including two β-parallel sheets are indicated by the red arrow. T: Turn, E: Extended conformation (β-sheets), B: Isolated bridge, H: α-helix, G: 3–10 helix, I: Pi-pihelix, C: Coil. For a detailed description see “Results and Discussion”
Discussion
Homozygosity mapping is an efficient tool for identifying genetic defects underlying genetically heterogeneous disorders such as inborn errors of metabolism. This tool is especially useful in populations with a high rate of consanguineous marriages.
Haplotype analysis of the studied families showed ten families with homozygous haplotype block for the BCKDHB gene, six for the BCKDHA, two for the DBT and no family was homozygous for the markers flanking the DLD gene. Subsequently, we sequenced the corresponding gene in each family.
Iran is a country with a consanguineous marriage rate of approximately 38 % (Saadat et al. 2004), so naturally the rate of autosomal recessive disorders will be higher in comparison to the Western countries. The rarer a disease, the higher the probability it will occur in a consanguineous marriage. MSUD is a rather rare disease even in Iran with a high rate of consanguineous marriage. Very few studies have been published regarding mutation frequencies in Iran (Miryounesi et al. 2015). With this in mind, we conducted homozygosity mapping for twenty-one families with MSUD.
A very important finding was an observation of two common haplotypes among investigated families (Fig. 1a, b). Haplotype A was seen in four families. To determine whether these families are related or from the same ancestral origin, we interviewed the families and asked about their ethnicity. Our analysis showed the families are of Lak origin which lives in Lorestan province. Such findings in a country with high genetic heterogeneity and, on the other hand, disease rarity of MSUD brought us to the conclusion that founder effect may play a strong role in this situation. The four patients with this mutation either died or suffer from severe developmental delay. The three patients with the B haplotype carrying same mutation died before the age of one month. Of these three patients, two patients belong to the Lurs and one is of Kurdish origin.
Mutations discovered in this study are categorized into two groups based on their structural-functional impacts on the E1b complex.
The first category includes mutations that might directly or indirectly affect subunit interfaces i.e. the p. Arg170Cys which is directly in contact with α, β and β’ interfaces, and p. Glu330Lys and p. Gly278_Thr279insThr which might affect the local residues adjacent to the interface of the β–β’ subunits. The second category consists of mutations that might have an influence on the metal ion interaction and also ThDP cofactor binding (p. Arg170Cys) (Figs. 2 and 3).
Ævarsson et al. demonstrated that the specific conserved loop located between residues 206 and 211 of the α subunit is stabilized by the potassium ion interactions. This interaction is also critical for the enzyme’s activity due to the contribution of this loop to cofactor binding (ThDP) (Ævarsson et al. 2000). Our analysis indicated that the p. Arg170Cys mutation resulted in the breakage of salt bridges between Arg170-β and Glu163-β which subsequently affect the interactions between Ser206-α-Glu163-β and Ser206-α-Gln212-α. Due to the critical role of Ser206-α in the aforementioned loop, this mutation has an influence on both the structure and function of the E1b complex (Fig. 3a, b). Moreover, the proposed assembly pathway of E1b indicates that the tetrameric assembly process is composed of two steps: 1) The heterodimerization of α/β and α’/β’, and 2) Subsequent heterotetramerization of α/β-α’/β’(Ævarsson et al. 2000). Since the p. Arg170Cys mutation is placed at the α-β subunit interfaces, this might destabilize multimerization of the whole structure during complex assembly (Figs. 1 and 2).
As earlier studies showed the essential role of the residues which are located at the interface of the β-β’ subunits of E1b including His206 from the β’ and Trp277, Pro332, Tyr334, Glu201 and Phe203 from the β subunit (Ævarsson et al. 2000), the insertion of Threonine between Gly278 and Thr279 not only led to conformational changes in the following two extended sheets (sheets B and C) near this position, but also opened the loop structure between sheet A and the subsequent helix (Fig. 4). These alterations caused loss of hydrogen bonds between Glu201-β and Trp277-β and the formation of new hydrogen bonds between Thr279-β and Glu331-β (Fig. 4a, b). Due to the redisposition of the local residues of sheet A, followed by the helix, sheet B, and C, which are adjacent to the His206 of β’ subunit, this mutation might effect on the association of two β subunits.
Regarding the substitution of lysine for glutamic acid (E330K) (Fig. 2), substitution of negatively charged glutamate with positively charged lysine not only affected the local electrostatic interactions, but also the hydrogen bonds (Fig. 5). This mutation is close to the two β subunits which led to the rearrangement of inter-chain hydrogen bonds in the β subunit including the elimination of the Glu330-β-Cys188-β hydrogen bonds (Fig. 5a). Due to the close distance of this mutation to the β-β’ subunits, this might have an influence on E1b stability.
The structural effect of exon-3 deletion of the BCKDHB gene on protein structure was evaluated after homology modeling (Fig. 6). The deletion resulted in the elimination of residues from Lys92 to Gly116. Elimination of the mentioned residues resulted in the removal of two parallel sheets within the interface of α/β subunits. This might have strong negative influence not only in the assembly of the E1b complex, but also on the binding of ThDP nearby. Although the diagnosis of the disease was at 20 days, diet and medication were not effective on her disease recovery.
To further evaluate the pathogenicity of the identified variants, we checked for the presence of the identified changes in 50 healthy unrelated individuals from the same ethnic backgrounds. None of them showed the mentioned changes. Also, none of the identified variants are present in the single nucleotide polymorphism database (Exome Variant Server, http://evs.gs.washington.edu/EVS/). These predictions are also supported by bioinformatics predictions by Polyphen-2 (Adzhubei et al. 2010) which indicates that this is “probably damaging” and SIFT (Kumar et al. 2009) indicated that these mutations are “damaging” (Table 2).
To understand the effect of the mutation on the one patient with the insertion of three nucleotides (c.833_834ins3), which added a threonine to the protein structure, Human Splice Finder (HSF) (Desmet et al. 2009) was utilized. This program predicted that the insertion will alter the wild-type donor splice site and probably affect splicing. HSF prediction algorithms predict that it decreases the consensus value of wild type from 82.65 to 11.1 for the mutant. Also, the predicted variation score is −86.57 % (this prediction is much lower than the significance level of −10 % in HSF).
To establish genotype-phenotype correlations, more cases need to be studied. These findings will add to our molecular understanding of MSUD in Iran and other countries.
References
Adzhubei IA et al. (2010) A method and server for predicting damaging missense mutations. Nat Methods 7:248–249
Ævarsson A, Chuang JL, Wynn RM, Turley S, Chuang DT, Hol WG (2000) Crystal structure of human branched-chain α-ketoacid dehydrogenase and the molecular basis of multienzyme complex deficiency in maple syrup urine disease. Structure 8:277–291
Benson G (1999) Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res 27:573
Chuang DT SV (2001) Maple syrup urine disease (branchedchain ketoaciduria). In: Scriver CR Beaudet AL SW, Valle D, (eds) CB, Kinzler KW, Vogelstein B (assoc eds). (eds) The Metabolic and Molecular Basis of Inerited Disease. Mc Graw-Hil, New York, pp 1971–2006
Den Dunnen JT, Antonarakis SE (2000) Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat 15:7–12
Desmet F-O, Hamroun D, Lalande M, Collod-Béroud G, Claustres M, Béroud C (2009) Human Splicing Finder: an online bioinformatics tool to predict splicing signals. Nucleic Acids Res 37:e67–e67
Eswar N et al. (2006) Comparative protein structure modeling using Modeller. Current Protocols in Bioinformatics: 5.6. 1–5.6. 30
Foroozani H et al. (2015) Molecular Characterization of QDPR Gene in Iranian Families with BH4 Deficiency: Reporting Novel and Recurrent Mutations
Harris RA, Joshi M, Jeoung NH (2004) Mechanisms responsible for regulation of branched-chain amino acid catabolism. Biochem Biophys Res Commun 313:391–396
Humphrey W, Dalke A, Schulten K (1996) VMD: visual molecular dynamics. J Mol Graph 14:33–38
Korein J, Sansaricq C, Kalmijn M, Honig J, Lange B (1994) Maple syrup urine disease: clinical, EEG, And plasma amino acid correlations with a theoretical mechanism of acute neurotoxicity. Int J Neurosci 79:21–45
Kumar P, Henikoff S, Ng PC (2009) Predicting the effects of coding non-synonymous variants on protein function using the SIFT algorithm. Nat Protoc 4:1073–1081
Legendre M, Pochet N, Pak T, Verstrepen KJ (2007) Sequence-based estimation of minisatellite and microsatellite repeat variability. Genome Res 17:1787–1796
Lu G et al. (2009) Protein phosphatase 2Cm is a critical regulator of branched-chain amino acid catabolism in mice and cultured cells. J Clin Invest 119:1678–1687 doi:10.1172/jci38151
Miller SA, Dykes DD, Polesky HF (1988) A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 16:1215
Miryounesi M, Ghafouri-Fard S, Goodarzi H, Fardaei M (2015) A new missense mutation in the BCKDHB gene causes the classic form of maple syrup urine disease (MSUD). Journal of pediatric endocrinology & Metabolism: JPEM 28:673–675 doi:10.1515/jpem-2014-0341
Phillips JC et al. (2005) Scalable molecular dynamics with NAMD. J Comput Chem 26:1781–1802
Reed LJ, Damuni Z, Merryfield ML (1985) Regulation of mammalian pyruvate and branched-chain alpha-keto acid dehydrogenase complexes by phosphorylation-dephosphorylation. Curr Top Cell Regul 27:41–49
Roy A, Kucukural A, Zhang Y (2010) I-TASSER: a unified platform for automated protein structure and function prediction. Nat Protoc 5:725–738
Saadat M, Ansari-Lari M, Farhud DD (2004) Consanguineous marriage in Iran. Ann Hum Biol 31:263–269 doi:10.1080/03014460310001652211
Shaw V (2014) Clinical paediatric dietetics. John Wiley & Sons, Blackwell Science, London
Yang J, Zhang Y (2015) I-TASSER server: new development for protein structure and function predictions. Nucleic Acids Res 43:W174–W181
Yang J, Yan R, Roy A, Xu D, Poisson J, Zhang Y (2015) The I-TASSER Suite: protein structure and function prediction. Nat Methods 12:7–8
Yoshino M et al. (1999) Management of acute metabolic decompensation in maple syrup urine disease: A multi-center study. Pediatr Int 41:132–137
Zhang Y (2008) I-TASSER server for protein 3D structure prediction. BMC Bioinform 9:40
Acknowledgments
We thank the patients and their families for their cooperation in this study.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Abiri, M., Karamzadeh, R., Mojbafan, M. et al. In silico analysis of novel mutations in maple syrup urine disease patients from Iran. Metab Brain Dis 32, 105–113 (2017). https://doi.org/10.1007/s11011-016-9867-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11011-016-9867-1