
Abstract
Intermittent maple syrup urine disease (MSUD) is a potentially life-threatening metabolic disorder caused by a deficiency of branched chain α-ketoacid dehydrogenase (BCKD) complex. In contrast to classic MSUD, children with the intermittent form usually have an atypical clinical manifestation. Here, we describe the presenting symptoms and clinical course of a Chinese boy with intermittent MSUD. Mutation analysis identified two previously unreported mutations in exon 7 of the BCKDHB gene: c.767A > G (p.Y256C) and c.768C > G (p.Y256X); the parents were each heterozygous for one of these mutations. In silico analysis predicted Y256C probably affects protein structure; Y256X leads to a premature stop codon. This case demonstrates intermittent MSUD should be suspected in cases with symptoms of recurrent encephalopathy, especially ataxia or marked drowsiness, which usually present after the neonatal period and in conjunction with infection. symmetrical basal ganglia damage but normal myelination in the posterior limb will assist differential diagnosis; alloisoleucine is a useful diagnostic marker and mutation analysis may be of prognostic value. These novel mutations Y256C and Y256X result in the clinical manifestation of a variant form of MSUD, expanding the mutation spectrum of this disease.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
Maple syrup urine disease (MSUD, OMIM248600) is an autosomal recessive metabolic disease caused by mutations in the BCKDHA, BCKDHB, DBT and DLD genes, which encode the E1α, E1β, E2 and E3 subunits of the BCKD complex. These mutations lead to defective decarboxylation of branched-chain amino acids (BCAAs), of which the main constituents are leucine, isoleucine and valine, which causes an accumulation of BCAAs and their branched-chain alpha-ketoacids (BCKAs) in the body. Accumulation of BCAAs and BCKAs induces nervous system damage by interfering with the synthesis of important neurotransmitters and process of myelination in the brain (Strauss et al. 1993; Strauss and Morton 2003; Zinnanti et al. 2009). Based on the age of onset, clinical manifestation and residual BCKD activity, patients with MSUD are classified as having the classic form or variant form. The variant form is further divided into the intermediate, intermittent, thiamine responsive and dihydrolipoyl dehydrogenase (E3)-deficient forms. In classic MSUD, BCKD complex activity is low (<2 %) or undetectable. In patients with higher residual BCKD activity, milder variants of the disease are observed, inducing the intermittent form in which BCKD activity ranges from 5 to 20 %. Classic MSUD accounts for 75 % of cases and is characterized by neonatal-onset encephalopathy, maple syrup odor in the urine, significantly elevated branched chain amino acids in the blood and alpha-ketoacids in the urine are prerequisite for the diagnosis. If untreated, this condition is often lethal. In contrast to classic MSUD, children with the variant form can present at any age, and the clinical manifestation is often less severe or atypical, especially the intermittent form as the patients can undergo normal development and have normal intelligence and, when asymptomatic, normal levels of BCAAs and BCKAs. The symptoms of intermittent MSUD are usually trigged by infection (Axler and Holmquist 2014). As a result, intermittent MSUD is easy to misdiagnose or often remains undiagnosed. In this study, we reviewed the clinical data and laboratory results of a case of variant MSUD to improve the awareness of this disease among physicians.
Case report
Clinical data
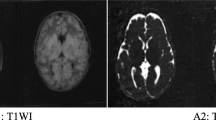
At the age of 18 months, during an episode of fever, a boy was admitted to hospital due to drowsiness, unsteadiness and low food intake, with no symptoms of convulsion or vomiting. The psychomotor development of the boy before admission was normal and there was no abnormal family history. A clinical systematic examination was normal and neurological examination showed gait instability with the body tilted to the right and slightly reduced muscle tension. Viral encephalitis was considered; electroencephalography, lumbar puncture and head computed tomography examinations were normal. After receiving acyclovir, the clinical condition of the patient improved rapidly and the boy was discharged from hospital 1 week after admission. 2 months later, the boy was readmitted to hospital with similar but more severe symptoms while suffering from fever; at the same time, a peculiar, fruity odor was noted from the urine. Twenty-four hours after admission, plasma amino acid analysis was performed and revealed 431.67 μmol/L leucine (normal range 4.2–367.0 μmol/L), 167.13 μmol/L isoleucine (normal range 26.9–146.00 μmol/L), 471.81 μmol/L valine (normal range 117.6–495.7 μmol/L) and 44.88 μmol/L alloisoleucine (normal range 0–7.7 μmol/L). Urine organic acid analysis revealed elevated ketone bodies, α-ketoisovaleric acid, α-keto-β-methylvaleric acid and α-ketoisocaproic acid. Axial T2-weighted and inversion-recovery MRI sequences showed bilateral symmetrical hyperintensities in the globus pallidus, thalami, dorsal of pons, medulla oblongata, denate body of the cerebellum, cerebellar cortices of the frontal lobe and the insular lobe (Fig. 1a–b). The posterior limb of the internal capsule was normal (Fig. 1c). Diffusion weighted imaging (DWI) and the corresponding apparent diffusion coefficient (ADC) maps revealed restricted diffusion in the areas with bilateral symmetrical hyperintensities (Fig. 2). Multivoxel proton MR spectroscopy, arterial blood gas analysis, blood lactic acid, blood ammonia, blood fat, renal parameters, liver function tests and a myocardial zymogram were normal. After treatment by dietary protein restriction, L-carnitine and glucose, lipids and non–branched-chain amino acids, the clinical condition of the patient gradually recovered and the boy was released from the hospital 2 weeks later. Before discharge, plasma amino acid analysis showed normal levels of leucine, isoleucine and valine, though alloisoleucine was above normal (7.95 μmol/L).

Axial T2-weighted MR images showing bilateral symmetrical hyperintensities in the globus pallidus, thalami, dorsal of pons, medulla oblongata, dentate body of the cerebellum, cerebellar cortices of the frontal lobe and insular lobe a–b. Note the sparing of the posterior limb of the internal capsule c, arrows)

DWI with corresponding ADC maps demonstrating restricted diffusion in the globus pallidus, thalami, dorsal of pons, medulla oblongata, dentate body of the cerebellum, cerebellar cortices of the frontal lobe and the insular lobe
Genetic testing
The BCKDHA, BCKDHB, DBT and DLD genes were screened for the presence of mutations using second generation sequencing technology. Two mutations were identified in exon 7 of the BCKDHB gene: c.767A > G (adenine > guanine) and c.768C > G (cytosine > guanine), which lead to an amino acid substitution (p.Y256C, tyrosine > cysteine) and premature stop codon (p.Y256X, tyrosine > termination), respectively. The patient was heterozygous for both the Y256C missense mutation and Y256X nonsense mutation (Fig. 3). Neither of these mutations have previously been reported. Direct sequencing analysis of the parents revealed the mother was heterozygous for the c.767A > G mutation and the father was heterozygous for the c.768C > G mutation (Fig. 4).

Identification of the novel c.767A > G and c.768C > G BCKDHB mutations in the patient by second generation sequencing

Identification of the heterozygous BCKDHB c.767A > G mutation in the boy’s mother a and heterozygous BCKDHB c.768C > G mutation in the boy’s father b by second generation sequencing
In silico analysis of the novel substitution mutation
The BCKDHB gene encodes branched-chain alpha-keto acid dehydrogenase E1 component β-chain (BCKDH E1-beta, Uniprot ID: P21953). Polyphen2 software (http://genetics.bwh.harvard.edu/cgi-bin/ggi/ggi2.cgi) was used to assess the significance of the novel missense mutation on protein function. In silico analysis predicted Y256C to be potentially damaging to protein function (Fig. 5). The program predicted that this mutation adversely affects enzymatic activity as a result of a change in hydrophobicity at a buried site. These results suggest that Tyr256 and other amino acids besides coiled between residues 249–254 and residues 264–267 of the β strand. Tyrosine is a hydrophilic amino acid, which often appears on protein surfaces. When mutations lead to cysteine replacing tyrosine, the -SH group of cysteine has a low hydrogen bond force and never appears on the protein surface; this can markedly affect the structure of the protein (Fig. 6). Crystal structure analysis revealed that the Tyr256Cys mutation belongs to the transketolase pyrimidine (ThDP)-binding domain site, from Gln67 to Ala263 (CATHID:3.40.50.970), and is likely to deactivate the enzyme (Fig. 7a–b).

The PolyPhen program predicts that the Y256C mutation may adversely affect enzymatic activity with a score of 0.998

The second structure of BCKDH E1-beta

BCKDH 1-beta 3D Visualization: a transketolase pyrimidine binding domain; b the Tyr256Cys mutation affects the transketolase pyrimidine binding domain sites at Gln67 to Ala263, which may deactivate the enzyme
Y256X is a truncating mutation, which means that not all of the transketolase C-terminal domain can be coded. His291 of this transketolase C-terminal domain has been shown to be important for ThDP binding to BCKDH E1-beta (Wynn et al. 2003); it interacts with the ThDP phosphate groups via water-mediated hydrogen bonds. This residue and the following Ser292-α belong to the phosphorylation loop (residues 285-313-α). Phosphorylation of this serine residue is sufficient to deactivate the enzymatic activity (Zhao et al. 1994). Taken together, these results suggest that a Y256X mutation will damage BCKD activity.
Discussion
Clinically, this case is typical of intermittent MSUD. In patients with the intermittent form of this disease, symptoms usually first appear between 12 and 18 months of age, as the requirement for the amino acids necessary for normal growth decrease during this stage of development. Clinical or biochemical abnormalities are usually undetectable unless the patient is under stress, such as infection, or has an excessive dietary intake of protein. At such times, the symptoms progress from ataxia, anorexia, vomiting, hypertonicity and lethargy to coma, and the condition can become irreversible and fatal within a few days if inadequately diagnosed or untreated, or the patient may recover spontaneously or in response to treatment. Neurological and intellectual function generally appear to return to normal in patients who recover.
Diffuse bilateral symmetrical hyperintensity of the white matter, globus pallidus and thalamus on magnetic resonance imaging, as observed in this case, are considered characteristic of MSUD (Kilicarslan et al. 2012; Bindu et al. 2010), Additionally, the residual BCKD activity in patients with the intermediate form of MSUD may be high enough to allow some myelination to occur, so note that the absence of hyperintensity in the posterior limb of the internal capsule on MRI can be used to differentiate between the classic and intermediate forms of MSUD (Bindu et al. 2007 Jul).
Compared to the classic form, the elevations in serum leucine, isoleucine and valine are not as severe in patients with the intermittent form, especially during asymptomatic periods. The levels of these amino acids can even be normal, as observed in this case, leading to a high risk of misdiagnosis. Additionally, slightly elevated levels of branched-chain amino acids can also be observed secondary to ketotic hypoglycemia, diabetes and hunger; therefore, the intermittent form of MSUD is highly likely to be misdiagnosed. Alloisoleucine is probably formed from isoleucine via a tautomeric reaction involving transaminated ketoacids. Alloisoleucine is elevated in the classical and variant forms of MSUD, even in asymptomatic cases or cases with mild clinical symptoms, but not elevated in response to secondary disturbances to branched-chain amino acid metabolism; therefore, elevated alloisoleucine is recognized as the most sensitive and specific diagnostic marker for MSUD (Schadewaldt et al. 1999).
MSUD can be divided into the IA, IB, II and III types based on involvement of the BCKDHA, BCKHB, DBT and DLD genes; the frequency of the mutations in these genes varies in different populations (Jaafar et al. 2013). In Chinese patients with MSUD, BCKDHA and BCKDHB point mutations are most common (Yang et al. 2012; Hou and Hwang 2014 Jun). Most studies consider that the IA and IB types are prone to the classic form of MSUD, whereas the II and III types are often associated with the variant form (Wang et al. 2012 Apr 25). In this case, the boy possessed the IB genotype but demonstrated the clinical manifestation of the intermediate form. This may be related to the effect of the nonsense mutation in BCKDHB, however, it should be noted that strict genotype-phenotype correlations are not easily defined for MSUD. In addition to genotype, the clinical manifestation of MSUD is influenced by a large number of variables, including the rate of growth (and net protein synthesis), calorie intake, the quality and quantity of dietary protein, the frequency and severity of precipitating illnesses, and the developmental timing of metabolic disturbances.
References
Axler O, Holmquist P (2014) Intermittent maple syrup urine disease: two case reports. Pediatrics 133(2):e458–60
Bindu PS, Shehanaz KE, Christopher R, Pal PK, Ravishankar S (2007) Intermediate maple syrup urine disease: neuroimaging observations in 3 patients from South India. J Child Neurol 22(7):911–3
Bindu PS, Kovoor JM, Christopher R (2010) Teaching NeuroImages: MRI in maple syrup urine disease. Neurology 74(3):516–524
Hou JW, Hwang TL (2014) Different gene preferences of maple syrup urine disease in the aboriginal tribes of Taiwan. Pediatr Neonatol 55(3):213–7
Jaafar N, Moleirinho A, Kerkeni E et al (2013) Molecular characterization of maple syrup urine disease patients from Tunisia. Gene 517(1):116–119
Kilicarslan R, Alkan A, Demirkol D et al (2012) Maple syrup urine disease: diffusion-weighted MRI findings during acute metabolic encephalopathic crisis. Jpn J Radiol 30(6):522–525
Schadewaldt P, Bodner-Leidecker A, Hammen HW et al (1999) Significance of L-alloisoleucine in plasma for diagnosis of maple syrup urine disease. Clin Chem 45(10):1734–1740
Strauss KA, Morton DH (2003) Branched-chain ketoacyl dehydrogenase deficiency: maple syrup disease. Curr Treat Options Neurol 5:329–341
Strauss KA, Puffenberger EG, Morton DH (1993) Maple syrup urine disease. In: Pagon RA, Bird TD, Dolan CR, Stephens K, Adam MP (eds) eneReviews™. University of Washington, Seattle
Wang YP, Qi ML, Li TT, Zhao YJ (2012) Two novel mutations in the BCKDHB gene (R170H, Q346R) cause the classic form of maple syrup urine disease (MSUD). Gene 498(1):112–5
Wynn RM, Machius M, Chuang JL, Li J, Tomchick DR, Chuang DT (2003) Roles of His291-αand His146-βin the reductive acylation reaction catalyzed by human ranched-chain α-ketoacid dehydrogenase: refined phosphorylation loop structure in the active site. J Biol Chem 278:43402–43410
Yang N, Han L, Gu X et al (2012) Analysis of gene mutations in Chinese patients with maple syrup urine disease. Mol Genet Metab 106(4):412–418
Zhao Y, Hawes J, Popov KM, Jaskiewicz J, Shimomura Y, Crabb DW, Harris RA (1994) Site-directed mutagenesis of phosphorylation sites of the branched chainα-ketoacid ehydrogenase complex. J Biol Chem 269:18583–18587
Zinnanti WJ, Lazovic J, Griffin K et al (2009) Dual mechanism of brain injury and novel treatment strategy in maple syrup urine disease. Brain 132(4):903–918
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Guo, Y., Liming, L. & Jiang, L. Two novel compound heterozygous mutations in the BCKDHB gene that cause the intermittent form of maple syrup urine disease. Metab Brain Dis 30, 1395–1400 (2015). https://doi.org/10.1007/s11011-015-9711-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11011-015-9711-z