
Abstract
Insulin resistance is common in type 2 diabetes mellitus (T2DM), neurodegenerative diseases, cardiovascular diseases, kidney diseases, and polycystic ovary syndrome. Impairment in insulin signaling pathways, such as the PI3K/Akt/mTOR pathway, would lead to insulin resistance. It might induce the synthesis and deposition of advanced glycation end products (AGEs), reactive oxygen species, and reactive nitrogen species, resulting in stress, protein misfolding, protein accumulation, mitochondrial dysfunction, reticulum function, and metabolic syndrome dysregulation, inflammation, and apoptosis. It plays a huge role in various neurodegenerative diseases like Parkinson’s disease, Alzheimer’s disease, Huntington’s disease, and Amyloid lateral sclerosis. In this review, we intend to focus on the possible effect of insulin resistance in the progression of neurodegeneration via the impaired P13K/Akt/mTOR signaling pathway, AGEs, and receptors for AGEs.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Insulin resistance is a condition where cells cannot respond to insulin, thus resulting in hyperglycemia and hyperinsulinemia [1]. Insulin resistance and hyperglycemia are associated with mitochondrial dysfunction, oxidative stress, and cell damage [2]. It is one of the common phenomena seen in metabolic diseases, obesity, Diabetes mellitus (DM), and neurodegenerative diseases (ND) [3]. It leads to impaired glucose metabolism, lipid metabolism, protein accumulation, inflammation, etc. In advanced Parkinson's diseases (PD), Alzheimer's diseases (AD), Huntington's disease HD), DM, cardiovascular diseases (CVD), and other age-related diseases, insulin resistance effects are observed [4, 5]. Hyperglycaemia and insulin resistance induce complications such as protein deposition and mitochondrial dysfunction [6]. Amyloid β, Amyloid β precursor protein (APP), Tau, α-synuclein, and Huntington (HTT) depositions in neurodegenerative diseases. Initially, these proteins are involved in neural cell growth, repair, microtubule stabilization, etc. Later, when they go mutated or under physiological stress, they lose their function that which is neural autophagy [7, 8].
High circulatory glucose in insulin resistance tends to bind with proteins and lipids, thus forming the Advanced glycation end products (AGEs) and advanced lipoxidation end products [9]. It also depends on reactive oxygen species, reactive nitrogen species, blood glucose level, and intracellular and extracellular pressure [10]. AGEs such as methylglyoxal, pentosidine, pyrimidine, and carboxymethyl lysine progress apoptosis via binding with AGE (RAGE) receptors, AGE-R1/R2/R3, SR A/B, and triggering oxidative stress [11]. AGEs such as methylglyoxal, pentosidine, pyrimidine, and carboxymethyl lysine progress apoptosis via binding with AGE (RAGE) receptors, AGE-R1/R2/R3, SR A/B, and triggering oxidative stress [12]. RAGE activates PI3K (Phosphatidylinositol 3 phosphate kinase)/Akt (AK strain transforming)/mTOR (mechanistic target of rapamycin), JAK/STAT, and ERK signaling pathways. Age-related diseases, DM, renal disorder, cardiovascular diseases, and neurodegenerative diseases have overexpression RAGE and AGEs. Studies revealed that AGEs induce inflammation, oxidative stress, and apoptosis [13].
PI3K/Akt /mTOR pathway plays a significant role in cell growth, cellular aging, apoptosis, glucose, protein, and lipid metabolisms [14, 15]. This pathway interrupts reactive oxygen species (ROS), mitochondrial stress, endoplasmic reticulum stress (ER stress), oxidative stress, mutations, and inadequate ligand–receptor interaction [16]. Dysregulation of PI3/Akt/mTOR causes various diseases such as metabolic diseases [17], aging, diabetes mellitus, neuron degradation, cancer, delayed wound healing [18], and psychological disorders. Insulin and insulin-like growth factors (IGF) regulate this pathway [19]. Increased activation of Akt (AK strain transforming) reduces the degradation of the neural cells as well as prevents diabetes mellitus [20]. Deterioration of PI3K/Akt/mTOR pathway induces the pro-apoptosis, apoptosis, autophagy [21], protein misfolding, formation of advanced glycation end products, and insulin resistance in cells like neurons, pancreatic cells, hepatocytes, and nephrons [15]. Glucose and its related metabolism are vital for normal and healthy body functions; defects in this pathway cause various diseases. This review intended to summarize how insulin resistance is associated with the disoriented PI3K/Akt/mTOR signaling pathway, its close relationship with AGEs, and the effect of RAGE–AGE interaction in the PI3K/Akt/mTOR pathway. Furthermore, it discusses these aspects with diabetes mellitus and common neurodegenerative diseases.
Mechanism of action of insulin via PI3/AKT/mTOR pathway
Insulin, insulin-like growth factors (IGF), and insulin-like hormones are primary activators for the induction of the PI3K/AKT/mTOR pathway and mitogen-activated protein kinase pathway (MAPK) [1, 22]. Insulin signaling starts with the binding of insulin with insulin receptor (IR) (IR-A and IR-B), insulin-like growth factor receptor (IGF-IR), and insulin receptor-related receptor (IRR). It simultaneously activates Insulin receptor substrate (IRS), extracellular signal-related kinase (ERK), mitogen-activated protein kinase (MAPK), and glycogen synthase kinase 3 (GSK 3) [23]. Brain, adipocytes, and hepatocytes express IR highly [24]. Depending on the binding of the IR receptor, different pathways get activated; if insulin bind with IR-A, it activates the c-jun N-terminal kinase (JNK) pathway via SHcA. In case it is IR-B, it would initiate IRS [22]. Deactivation of IR might incite insulin resistance and obesity, but it does not affect development, while overexpression might cause hypoglycemia [25]. Deactivation of IR might instigate insulin resistance and obesity, but it does not affect growth, while overexpression might cause hypoglycemia [17]. This family of insulin receptors contains a heterodimeric structure of two α subunits in the extracellular matrix and two β subunits in the transmembrane region. Ligand molecule binds with the α subunit of the receptor and induces the changes in the β subunit, which leads to autophosphorylation of Tyr1162, Tyr1158, Tyr1163, and Tyr972 [26]. Subsequently, this initiates the phosphorylation of the IRS family proteins, APS, GRB 10/14, SH2B1, and SH2B2. Protein-tyrosine phosphatase (PTP1B) regulates IR by dephosphorylation. Ironically, IR inhibits PTP1B by activating NAD(P)H oxidase through a feedforward mechanism [27, 28].
So far, studies have revealed six members of the IRS family proteins (IRS1 to IRS 6), but IRS-1 and IRS-2 are the most explored substrates [29, 30]. IRS acts as a regulator for insulin signaling; it mediates the phosphorylation of other kinases involved in insulin signaling [31]. Irs-1−/− knockout mice did not show diabetic symptoms, while Irs-2−/−-deleted mice presented with the symptoms of T1DM, reduced neural proliferation, and infertility. But interestingly, Irs-2−/− mice lived longer than the Irs-1−/− knockout mice. This outcome exposed the relation between the IRS and liver metabolism, glucose utilization, and glycogen biosynthesis [25] [24]. PI3K, Grab-2, SHP-2, Fyn, c-Crk, CrkII, and Nck are major regulators for Akt/mTOR pathway. It mainly begins with the IR receptor activation in mammals [32]. Drosophila and C.elegans have receptor proteins similar to the IR, namely, INR and Daf 2 [33]. IRS is a primary mediator of glucose metabolism and expresses in various types of cancer [23]. Phosphorylation of IRS1 in the Ser/Thr amino acid sequence may suppress the activation of PI3K [32].
PI3K is a secondary messenger and lipid kinase with class I, II, and III subtypes. But PI3K class I was the most analyzed subtype; it is composed of regulatory and catalytic subunits, respectively, P85 (α, β, and γ) and P110 (α, β, γ, and δ) [34]. P110 α and P110 β expression are largely seen in most of the tissues, while leucocytes were highly expressed P110 γ and P110 δ. P110 α and P110 β subunits play an important role in embryonic development [35]. Platelets-like growth factors and insulin receptor signaling activate PI3K. When it is activated, it converts phosphatidylinositol 4,5 bisphosphate (PIP2) to phosphatidylinositol 3,4,5 triphosphate (PIP3) [36]. PTEN dephosphorylates PIP3 to PIP2 [37]. It regulates many downstream pathways, including the Akt signaling pathway. It is one of the major targets for PI3K. PTEN inhibits Erk and Frk kinase proteins activation, which are involved in energy impairment and neuron degeneration [38]. Defect in this gene induces insulin resistance and hyperlipidemia [39]. Receptor tyrosine kinase (RTK) and G protein-coupled complex (GPCR) also upregulates PI3K [40].
The protein kinase B (PKB) or Akt belongs to the serine-threonine kinase family. It has three subtypes Akt 1—expressed in all cells, Akt 2—majorly in insulin-sensitive cells, and Akt 3—elevated in brain and reproductive organs [41]. Akt 2 knockout mouse express diabetic-like characteristics. Akt participates inactivation of mTOR complexes, phosphatidylinositol 3 phosphate, caspase 9, p21, p27 NFκ B, and Bad. It inhibits FOX O, GSK 3, and caspases [42]. Akt is an upregulator of mTOR 1, while it is activated by mTOR 2 and PDK 1 [27, 43]; PP2A, PHLPP, inhibits it. Akt stimulates the GLUT 2 and 4 receptor translocation, which plays a significant role in glucose uptake and metabolism [37, 44]. Akt inhibits TSC 2 by phosphorylating and activates Ras homologous enriched in the brain (RHEB) to stimulate mTOR 1 activity [45, 46]. Akt plays a crucial role in neurodevelopment and survival through the P13K/Akt pathway and the Akt/ERK pathway [47].
There are two types of mTOR identified so far: mTOR 1 (sensitive to rapamycin) and mTOR 2 (insensitive to rapamycin). Both mTOR 1 (6 subunits) and mTOR 2 (7 subunits) present as dimers but perform different functions in the presence of subunit proteins [48]. mTOR 1 complex consists of regulatory associated protein of mTOR (RAPTOR), DEP domain-containing interacting protein (DEPTOR), MLST8, PRAS 8, and mTOR 1. While mTOR 2 complex contains rapamycin-insensitive companion of mTOR (RICTOR), DEPTOR, mSIN 1, mLST 1, and mTOR 2 [14, 39]. It belongs to Ser/Thr kinase protein family. Both mTOR proteins have different downstream proteins, upstream regulators, and functions [39]. PTEN, AMPK, ATP deficiency, and TSC 2 repress the mTOR 1 activity. TSC 2 is phosphorylated by AMPK when the ATP concentration is low. Moreover, TSC1/TSC2 knockout mice showed better β cell proliferation, increased size, and hyperinsulinemia [49]. Growth factor-induced PIP3 activates mTOR 2. It is also positively regulated by Akt [50]. mTOR 1 initiates translation, cell growth, lipogenesis, and regulates autophagy, while mTOR 2 regulates cytoskeleton organization, anabolism, cell survival, and cell proliferation by regulating Akt, PPAR, SREBP1, Sgk, 4E BP1, and Pkc [49, 51]. Both complexes activate transcription factors, for example, c-Myc and HIF1a—these genes overexpression in hypoxia and diabetes (Fig. 1). In cancer and dementia, it induces tumorigenesis by inhibiting autophagy; even though rapamycin therapy might help due to S6K intervention, Akt is getting reactivated [52, 53]. Later, Torin 1 and PP- 242 were developed to tackle this issue [54]. mTOR has a huge part in immune response (multiplication and maturation of dendritic cells and T cells), sensing growth factors, and nutrition availability [55].

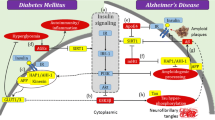
PI3K/Akt/mTOR signaling pathway. Insulin transmits growth, survival, and metabolic regulatory signals through IR-A and IR-B receptors. Then it activates JAK-STAT, Ras-Raf, ERK, PI3K/Akt/mTOR signaling pathways
Insulin resistance
Numerous hormones work to maintain metabolism, growth, and other cellular processes. Insulin, glucagon, thyroid hormones, and catecholamines are some of the best-known examples. Insulin was familiar for diminishing glucose concentration in blood by increasing glucose uptake in cells which is a direct antagonist to glucagon, cortisol, adrenalin, and growth hormones. Later hormones can prompt hyperglycemic conditions [28]. All these hormones, including leptin, ghrelin, amylin, and thyroid hormones, regulate homeostasis, energy metabolism, and appetite [56]. Insulin is a peptide hormone it has A (21 amino acid residues) and B (30 amino acid residues) subunits [57]. Insulin performs various functions such as glycogen synthesis, protein synthesis, lipogenesis, reduced lipolysis, increased glucose transport, lowering gluconeogenesis, and so on [58]. It can pass through the blood–brain barrier; in the case of IGF, it can be synthesized in all most all cells [59]. Even though insulin is sufficient or has a higher concentration, cells would not be sensitive enough to carry out different signaling mechanisms in specific circumstances called insulin resistance [57]. Insulin resistance decreases the ability of the cell to uptake glucose. It progresses into hyperglycemia and also causes hyperinsulinemia. Because of that, glucose metabolism would negatively impact [60]. Insulin resistance leads to T2DM, obesity, gestational DM, pregestational maternal obesity, metabolic syndrome, and diabetic complications [61]. In the absence of insulin activity due to insulin resistance, insulin antagonist hormones glucagon, corticoids, and catecholamines treat the cell as if it were fasting. It promotes gluconeogenesis, glycogen lysis, keto lysis, lipolysis, etc. [62]. Further, this would induce cell stress, apoptosis, oxidative stress, endoplasmic reticulum stress, lipid accumulation, AGEs formation, inflammation, and protein misfolding [63]. It also promotes the destruction of the Blood–Brain Barrier (BBB) via Endothelial Adora 2a activation, increasing vascular inflammation, synaptic plasticity, and cognitive impairment [64].
Insulin resistance cells would be unable to import glucose, while vascular endothelial cells would observe glucose by passive diffusion. Thus, the entered intracellular glucose would be converted into secondary metabolic products like sorbitol. Metabolism of secondary metabolites leads to AGEs [65]. AGEs are an oxidant it promotes oxidative stress, inflammation, and endothelial dysfunction [66]. ROS plays a considerable part in inducing insulin resistance through inflammation and influencing the electron transport chain. ROS and oxidative stress impact cellular signaling pathways, for instance, AMP-activated protein kinase (AMPK), ERK, JNK, cb1/CAP, MAPK, etc. (Fig. 2) [67, 68]. Mutations or inactivation of proteins in the insulin signaling pathway and downstream proteins induces insulin resistance [69]. Since IRS is a secondary messenger, and with the phosphorylation of IRS insulin signaling pathway begins. Impairment in irs1 and irs2 genes induces insulin resistance; the higher the mutations that occur in the gene increases the possibility of getting DM [70, 71]. Cellular molecules like free fatty acids, acetyl CoA, glucose, carbohydrate metabolic intermediates like diacylglycerol, and inflammatory molecules inhibit the activation of IRS [72]. Brain insulin resistance in AD and PD is closely related to PI3K/Akt/mTOR signaling pathway [73]. Besides, it obstructs the NO pathway, elevates inflammation, and causes insulin resistance [29]. Inhibition of these pathways increases autophagy markers (Atg5, Atg7, and Beclin-1), downregulation of cell survival markers, and mitophagy marker proteins like PINK1, BNIP 3, and HIF 1α [74]. PTEN is a regulatory protein for the insulin signaling pathway.

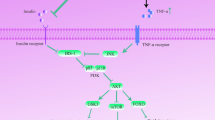
AGE–RAGE complex interaction induces apoptosis. AGE–RAGE complex blocks the insulin signaling pathways. It induces apoptosis by causing mitophagy, ER stress, protein misfolding, aggregation, and blocking of cell survival pathways
AGE–RAGE Interaction with signaling pathways
Synthesis and accumulation of AGEs increased with aging, hyperglycemia, and hyperlipidemia. There are more than 20 products synthesized out of a reaction between sugar moiety, lipids, proteins, and nucleic acids. Moreover, they tend to bind with the proteins like elastin and collagen [75, 76]. Synthesis of AGEs occurs through various pathways, including non-enzymatic (Maillard reaction) and enzymatic reactions (polyol pathway and lipid peroxidation). Besides these pathways, carbohydrates undergo a series of modifications and rearrangements through the Namiki pathway, Wolff pathway, Hodge pathway, and carbonyl stress pathway to produce AGE derivatives [9, 77]. Additionally, the glucose metabolic pathway and its intermediates also play a part in AGEs synthesis, for instance, methylglyoxal derived from glyceraldehyde 3 phosphate or its isomer dihydroxyacetone phosphate [76]. Reversible Schiff base (Amadori product) synthesized by these pathways forms a bond with them as lysine, arginine, alkylamine, and ribose. Pentosidine, Pyrraline, glyoxal/methylglyoxal lysine dimer, and carboxymethyl lysine (CML) are some typical AGEs in in-vivo. These compounds must undergo a series of glycation, oxidation, dehydration, condensation, and transition before finally becoming AGE products [78]. This structural modification assists in the glycation and oxidation of biomolecules in the cell [77, 79]. High-fat diet processed high protein diet, and cigarette smoking was known to improve the deposition of AGEs in tissue. A high concentration of AGE and its Interaction with RAGE encourage elevating the ROS and intracellular calcium level that incites endoplasmic reticulum stress, inflammatory pathways, protein misfolding, and apoptosis [13, 76]. RAGE is a multiligand transmembrane receptor member of the immunoglobin superfamily with 344 amino acids. Interaction of RAGE with its ligand (AGE) linked with ERK, PI3K, JAK/STAT, MAPK, Wnt signaling, and inflammatory pathways [10, 12]. Other than AGEs, it binds with cytokine S100 family, amphoterin, and amyloid β. Endothelial, kidney, glial, and astrocytes indicate the RAGE [80]. In contrast to the AGE–RAGE signaling interaction between AGE-R1 and AGE-R3, the latter seems responsible for the proinflammatory signaling pathway. Moreover, these interactions reduce AGE-mediated ROS, RAGE signaling, oxidative stress, and calcification, yet with age and higher AGEs concentration, these functions are declined [9, 81]. A study by Hu et al. indicated that AGEs-mediated autophagy by inactivating Akt and activating ERK that cells survived when treated with Akt activator and ERK inhibitor [82].
Insulin resistance, AGE-mediated diseases, and PI3K/Akt/mTOR signaling as comorbidities
Insulin resistance impacts numerous cellular, metabolic, inflammatory, etc. These conditions promote the diseases like diabetes mellitus, neurodegenerative diseases, atherosclerosis, obesity, renal diseases, liver diseases, and infection [63]. Evidence indicates the relationship between insulin resistance and AGEs (Fig. 3). Neurons are more sensitive to insulin; it aids neurons in synaptic development, neural remodeling, and memory deterioration and damages the blood–brain barrier [75, 83]. The emergence of insulin resistance in the brain cell loses a significant energy source and induces apoptosis. More precisely burden for the mitochondria to compensate for the absence of the energy requirement increases. Insulin resistance and AGEs raise the probability of triggering oxidative stress: brain magnetic resonance image PD and AD patients pointed out these changes [4, 84]. Impaired IRS restrain the synaptic plasticity and impose synaptic loss and dysfunction [75]. Insulin signaling impairment induced neurodegeneration in AD, PD, and Huntington's diseases [85]. Decreased insulin signaling in the brain altered glucose homeostasis, which might have led to cognitive dysfunction [41]. Increased amyloid β deposition due to hyperglycemia can potentially increase insulin resistance [86]. While AGEs form the cross-linking with the proteins by glycation and oxidation. It triggers β sheet formation in amyloid β, α synuclein, hemoglobin albumin, and prion proteins [87]. Apart from the natural tendency of AGEs to elevate ROS, it damages and oxidizes the protein in the typical scenario that the ubiquitin-proteasome system would degrade. But the presence of AGEs inhibits it and further promotes the aggregation of proteins, thus also contributing to the ROS and reactive nitrogen species (RNS) formed by nitric oxide synthase and NADPH oxidases. Insulin resistance mediated by PI3/Akt/mTOR pathway facilitates the AGEs synthesis and accumulation [9, 79]. It further raises the ER stress and therefore develops protein misfolding.

RAGE–AGEs interaction inhibits PI3K/Akt/mTOR and NF-κB signaling pathways. It affects cellular metabolism and protein folding, damages organelles, and causes diabetes mellitus, neurodegenerative diseases, and endothelial diseases
Various disease conditions like DM, DM-associated comorbidities like neuropathy, nephropathy, retinopathy, cardiovascular diseases, cataract, PD, AD, and ALS were associated with AGEs deposition. Since AGEs share a fair role in age-related diseases, metabolic diseases, cardiovascular diseases, and neurodegenerative diseases, it has the excellent ability to act as a biomarker [76, 88]. Mitochondrial mutations also cause insulin resistance; in that case, it can affect the expression factors, transcription factors, protein synthesis, etc., that may induce diseases like AD, PD, mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) [4]. A study in England disclosed that T2DM patients are more likely to get AD, PD, and CVD [89]. In contradicting a piece of recent evidence suggested that PD and insulin resistance does not have any relationship based on HOMO-IR, motor symptoms, non-motor symptoms, glucose, and insulin concentration in a fasting and fed state. They also concluded that Tau and blood amylin in the pancreas of DM patients causes neurodegeneration [90]. Apoptosis of neurons and Pancreatic β-cell destruction were observed in the streptozotocin-treated rat when PI3K/Akt/mTOR pathway was suppressed [41]. A study concluded that comorbid DM in severe PD patients worsens the cognitive impairment independently [91]. Depending on the treatment type, those patients who received DPP4 inhibitors and GLP1 agonists reduced the probability [92]. Some studies suggested that DM and neurodegenerative diseases may be comorbid conditions due to the common pathway they share, but managing neurodegeneration with insulin is not presented [93].
Diabetes mellitus
Diabetes mellitus (DM) is the most prevalent metabolic disease associated with insulin secretion and its mechanism of action. Insulin is the sole endocrine hormone responsible for decreasing glucose concentration in blood and glucose metabolism. Categories of DM include type 1 DM (T1DM), type 2 DM (T2DM), gestational diabetes, and pre-diabetic conditions (insulin resistance) based on age, obesity, and insulin association [25, 94]. In contrast, T1DM arises as pancreatic Langerhans' beta cells degrade (autoimmunity); T2DM rise due to insufficient synthesis of insulin, insulin resistance in the cells, and decreasing β cell mass with age [15, 17]. In most cases, mortality in T1DM depends on acute complications (children and young adults), while T2DM is related to chronic complications and relies on comorbidities [95]. Around 90% of the patients with DM suffer from T2DM, particularly youngsters [32, 96]. Around half (45.8%) of adult DM cases worldwide are not reported and are left untreated, leading to severe comorbidity conditions [97]. Insulin maintains glucose homeostasis via the insulin signaling pathway defect in this pathway is one of the major causes of insulin resistance which further leads to hyperglycemia and DM [1, 36]. Insulin activates IR, and then it phosphorylates IRS-2, which leads to the activation of PDK1, PI3K, and Akt simultaneously [1]. In most diabetic cases, being unable to phosphorylate IRS results in the deregulation of the PI3K/Akt pathway [49].
Regulating this pathway will induce hepatocyte proliferation, decrease blood glucose concentration, and regulate gluconeogenesis [25, 98]. Uncontrolled glucose concentration in DM patients leads to complications like neuropathy [99], nephropathy, stroke, cardiomyopathy, retinopathy, PD, vascular dementia, and AD, especially in chronic T1DM and T2DM [86]. There are two types of neuropathy: peripheral and autonomic neuropathy [100]. Diabetic neuropathy-related foot ulcers lead to amputations and complications like paraesthesia [101]. Approx. 50% of patients with DM develop neuropathy over their lifetime. Error in the Insulin signaling pathway and autophagy induces neuropathological changes and neuropathic conditions [102]. 30% of T2DM and 20% of T1DM patients eventually develop renal complications. Obesity, hypertension, memory impairment, anxiety, hepatopathy, organ damage microvascular, and macrovascular diseases are considered the comorbidity of DM [66]. It also gives rise to glucotoxicity, lipotoxicity, and proteotoxicity in β cells of the pancreas [2]. These all were closely associated with AGEs, ROS, RNS, oxidative stress, and hyperglycemia (Fig. 4) [25]. In diabetic nephropathy, AGEs stimulate TNFα and IL-6 by binding with RAGE; this instigates apoptosis of podocyte and glomerular heteropathy. Biosa et al. mentioned in their review that glycosylation products such as methylglyoxal interfere with the dopamine, syncline, and other proteins involved in AD and PD [103]. PI3/Akt/mTOR signaling pathway involves pain mechanism hyperexpression of it which stimulates hyperalgesia [102].

Pancreatic β cells apoptosis. Insulin resistance, Advanced glycation end products, inflammation, obesity, hyperlipidemia, and hypertension stimulates ROS, oxidative stress, apoptosis, and dysfunction of pancreatic β cells. These conditions were also associated with Hypersecretion of insulin
Some studies indicated that PI3K/Akt/mTOR maintains the β cell volume, prevents apoptosis, induces β cell proliferation, increases insulin secretion, and reduces endoplasmic reticulum (ER) stress autophagy and AGEs formation in β cells [44]. Irregulated PI3/Akt/mTOR pathway leads to hypoxic and reactive oxygen species (ROS)-induced apoptosis in myocardiopathy, nephropathy, and VEGF-induced atherosclerosis [104]. Suppression of this pathway negatively affects glucose uptake, which reduces cell proliferation and regulation of this phenomenon observed in hepatocellular carcinoma [105]. It also reduces lipotoxicity in a diabetic animal model by inhibiting Fox O1 [106]. PI3K/Akt/mTOR stimulates antidiabetic activity by inducing the immune system against inflammation. It helps immune cell activation (natural killer cells, dendritic cells, macrophages, etc.) and increases the secretion of cytokines (interleukins, interferons, etc.) [49, 107]. Fluctuations in phosphorylation and dephosphorylation of the proteins in the PI3K-mediated insulin signaling pathway and tumor necrosis factor (TNF) cause insulin resistance, and proteins like Nrf 2, Nf-κb, PPAR, and apoptotic proteins (caspases) indirectly control this pathway [85, 108]. Impairment of Akt 2 downregulates ATP7A in T2DM [42]. Braun et al. stated that decreasing insulin resistance is associated with increasing AKT/pAKT concentration when they treated T2DM patients with resveratrol, an antioxidant drug [109]. Liuwei Dihuang decoction and tangganjian (traditional Chinese medicines) improve glucose uptake in the liver by regulating PI3K/AKT pathway [1, 36]. Some experiments affirmed that IRS-2, AKT, PI3K, PPAR-γ, GLUT-4, Bax, Caspases, p62, mTOR, and INS-R protein downregulation increased haptic proliferation, reduced oxidative stress, and leads to depletion of glucose levels in the blood [21, 25]. GLUT-4 is a glucose undertaking transmembrane protein. It was synthesis, activation, and transport induced by the downstream process of the Akt/mTOR pathway [25, 67]. Akt 1 inhibits GSK 3 by phosphorylating, increasing glycogen synthesis, elevating glucose uptake, and improving wound healing properties [18].
Neurodegenerative diseases
Neurodegenerative diseases (NDs) affect the nervous system primarily. Neuronal degradation occurs due to aging, genetic factors, neurotransmitter depletion, mutations, toxins, insulin resistance, protein aggregation, and external factors like injury and pollution [110]. Regulation of neural signaling pathways, neurotransmitter synthesis, transport, cellular stress, and so on will aid in treating NDs Protein aggregation induces proteotoxicity in neural cells and promotes apoptosis in neural cells in many ways, for instance, via stimulating the RAGE-mediated cytotoxicity [79]. Fundamentally, RAGE involves in neural differentiation and proliferation via NF-κB, ERK, and JAK2/STAT3 pathways. RAGE plays a vital role in neuronal axon outgrowth and enhances neural survival [111]. When it activates AGEs, it damages neurons, thus causing apoptosis [112]. Some of the common neurodegenerative diseases are PD, AD, HD, Amyotrophic lateral sclerosis (ALS), ataxia, spinal muscular atrophy, supranuclear palsy, and motor neuron diseases are observed to have insulin resistance, high AGEs as well as disoriented PI3K/Akt/mTOR pathway [20]. PI3K/Akt/mTOR pathway boosts the elongation and growth of the axons and dendrites. Brain-derived neurotrophic factor protein (BDNF), insulin and growth factors activate it in the brain [113]. Error in any proteins of the PI3K/Akt/mTOR pathway and its regulatory pathway reduces the survival rate and growth of the neurons [114]. AGE–RAGE Interaction and RAGE intervention in the PI3K/Akt/mTOR pathway involved mitochondrial dysfunction, autophagy, and apoptosis. It promotes neurodegeneration in AD, PD, HD, and ALS [115].
Alzheimer’s diseases
AD is identified as T3DM because the neuron's resistance to insulin influences AD [116]. Insulin encourages the formation of synapses and dendrites, stem cell activation, neuro production, neural repair, and growth [24, 103]. D is mainly associated with aging and age-related conditions like reduced neural & synaptic plasticity, neural inflammation, depletion of neurotransmission, mitochondrial dysfunction, oxidative stress, and errors in DNA repair [33, 86]. AD symptoms comprise dementia, cognitive impairment, behavior changes, and psychological and mood disorders [6]. In AD, neuron degradation mainly occurs because of protein plaque formation (APP, Amyloid, Tau, and α synuclein), synaptic loss, and entanglement of neurofibrils [8]. Deposition of amyloid and tau proteins is known as amyloidoma, and tauopathy, respectively. Even though synuclein is a characteristic feature of the PD, detectable amount of deposition was noticed in AD [117]. Processing errors in APP seem to worsen AD progression and cause synaptic changes in AD-like mice [118, 119]. Deposition of these proteins in neurons induces neurotoxicity and further promotes inflammation and apoptosis [120]. Moreover, misfolded amyloid β binds with RAGE receptors and promotes neural degradation [121]. APP was a transmembrane protein; furthermore, it is a precursor molecule for amyloid β [8, 122]. Post-translational modifications of APP produce the isoforms of Amyloid β 1-40, 1-42, 1- 17, 1-15, and 1-14. Generally, it contributes to tumor suppression, is anti-microbial, prevents the leakage of Blood-borne solutes, and enhances brain injury recovery [8, 14]. Different cell types like platelets, immune cells, and fibroblast cells also expressed Amyloid β [123]. Tau (352 to 441 amino acid residues) is a microtubule-associated protein (MAP) present in the axonal compartment of neurons, and it works together with MAP 2 protein [7].
Investigations revealed the correlation between insulin, IGF, and its signaling pathway interference with AD-associated comorbidities such as DM, Amyloid accumulation, vascular inflammation the cognitive dysfunction for years. Insulin, IGF 1, and IGF 2 concentration seems to be low in AD patients even before they show their symptoms [124, 125]. Other related genes of the insulin signaling pathway, such as IRS, PI3K, Akt, GSK3, and PTEN, are low in activity [75]. Mutations in PTEN and SHIP2 dysregulate the insulin signaling pathway because it interferes with Akt and simultaneously causes insulin resistance. It affects glucose uptake and elevates tau phosphorylation in neural cells [38]. Akt mediates the activation of GSK3β, which is involved in the phosphorylation of APP, Tau, and amyloid β. Increased amyloid β (deposited) stimulates the activity of the GSK3β that interferes with the PI3K/Akt/mTOR pathway [14, 126]. Hypo-expression of mTORC1 and mTORC2 were observed with amyloid accumulated AD model [127]. JAK, SOCS 3, and PTPN1 promote APP's phosphorylation via Bcl-2 and Bax [128]. It also indicated that reduced insulin activity decreases amyloid protein degradation by inhibiting the α amyloid degrading enzyme and increases the extracellular α and β amyloid secretion and depletion in amyloid clearance in CNS fluid Deposition of amyloid and APP induces cytotoxicity and inflammation response by activating caspases, cytochromes, and NFκB [129]. A study pointed out that amyloid and tau protein clearance occurred via PI3/Akt/ mTOR/GSK 3β pathway (Fig. 5). Injecting melatonin in mice reduces the accumulation of beta-Amyloid and Tau in neurons by regulating PI3/Akt/ GSK 3β pathway [130]. Akt 1 phosphorylates GSK3β, which further activates Tau. Inactivated mTOR mediates amyloid clearance by stimulating autophagy in neurons [45].

Amyloid β-RAGE and AGE–RAGE interaction. Extracellular amyloid β invades the neurons through vesicles and aggregates as the result of dysregulating the amyloid β clearance. Besides, the AGE–RAGE signaling inhibits the PI3K/ Akt/mTOR pathway
AGEs contribute to APP, amyloid β, and tau accumulation and phosphorylation by glycating the respective proteins. It results in misfolding and aggregation in cells, thus promoting neuroinflammation. It also causes damage to Blood–Brain Barrier (BBB) and therefore encourages neuroinflammation by permitting the inflammatory cytokines and other molecules into the CNS and brain [11, 75]. It also has an adverse impact on memory and intellectual and cognitive ability [131]. Besides, AGEs glycate tau protein at the N-terminal site where tubulin has to bind, and this causes destabilization of the microtubule [78]. A study proved that the presence of AGEs depends on gender. For example, carboxymethyl lysine is in higher concentration in AD female patients than in male patients [11]. Pentoside and Upregulation of RAGE promote the extracellular amyloid β invasion into the neural cell. RAGE inhibitors inhibit by reserving amyloid β influx, reducing inflammation by regulating NF-κB signaling [123]. Elevated RAGE and its ligands are closely associated with cognitive impairment, Tau, and amyloid β accumulation which showed a closed relationship with the Akt/mTOR signaling. Increasing RAGE expression elevates the Akt phosphorylation and S100B expression; it promotes inflammatory cytokines and apoptosis of neurons [132].
Further analysis confirmed that cytoplasmic amyloid β-RAGE binding promotes the autophagosome, which leads to autophagy and neural apoptosis via dysregulating MAPK and inactivating mTORC 1 [12]. Synthetic RAGE targeted therapy against amyloid β interaction indicated that it can indeed prevent the caspase 3-mediated cell death. But they also stated that this need to be evaluated further since their mechanism seems a bit completed [121].
Parkinson’s disease
Parkinson's disease is the most prevalent neurodegenerative disease among older adults worldwide after Alzheimer's [133]. It occurs due to the degradation of the dopaminergic neuron in the brain's substantia nigra pars compacta because of the Lewy body formation (α synuclein deposition) [3, 134]. Dopamine is a dual neurotransmitter that has both exhibitory and inhibitory functions. As a consequence of the neural degradation, it instigates hyperactivity and a lack of inhibitory activity [135]. The Synuclein protein family consists of α synuclein, β synuclein, and γ synuclein. α and β synuclein located in the presynaptic nerve boutons and its deposition in dopaminergic neurons are typical characteristics of the PD brain.
The breast, colon, and pancreas also express γ synuclein [87, 117]. Synuclein has more lysine residue AGEs tend to bind with it. It goes through several post-translational modifications and unstable structural folding, thus revealing that it has more lysine residues [78, 87]. A study showed that lysine 58, 60, 80, 96, 97, and 102 positions of fructosamine have a high affinity towards it when treated with D-ribose. It shows that α synuclein tends to bind with AGEs [87, 136]. Similar synuclein dopamine derivative quinolones are also most likely to form a bond with AGEs like CML. Furthermore, it affects the homeostasis of the neurons and leads to apoptosis. Dopamine derivatives aggregation is associated with synuclein aggregation (Fig. 6) [136, 137]. Yang et al. proposed that mitochondrial stress or mitochondrial protein impairment also plays a significant role in neural degradation, specifically in energy-producing pathways [5]. Motor dysfunction, cognitive impairment, hallucination, mood disorders, and dementia are the most common symptoms of PD [3, 138]. In some cases, misdiagnosis of PD and movement disorder happens. They added because of the shared pathways, genetic mutations, and similar symptoms [89]. The executive dysfunction rate is higher in patients with DM comorbidity [138].

AGEs glycation of α synuclein and interfere with proteasomal degradation of misfolded proteins. α Synuclein glycated with carbohydrate moiety during post-translational modifications leads to misfolding and elevates the ER stress. Thereupon misfolded α Synuclein will be degraded by proteasomal degradation
A study in China revealed that DM patients with a long history have a 23% chance of developing PD and risk with age and gender (female), financial status, and occupation [5]. Some other experiments established that up to 8–30% of Parkinson's patients develop diabetes over time, and 50–80% of patients seem to have higher glucose concentrations during glucose tolerance tests. Magnetic resonance spectroscopy and Positron emission tomography results indicated glucose depletion and increased lactate concentration in substantia nigra [4]. The reduction of dopamine synthesis is associated with depleted insulin sensitivity [139]. For the patients who were under treatment for idiopathic PD with levodopa causes hyperglycemia and hyperinsulinemia, some studies suggest it may increase sensitivity towards insulin [140]. Magnetic resonance of the Parkinson's brain showed a decrease in glucose metabolism and ATP synthesis (Complex 1 of electron transport chain) [141]. The close bond between diabetes and PD indicates that these diseases might have a common phenomenon. To bring up the fact that insulin resistance and AGEs have a considerable role in PD progression [142], AGE–RAGE also contributes to the degradation of dopaminergic neurons by dysregulating the NF-κB signaling pathway, elevating inflammatory cytokine [143].
There seem to be an exciting interaction between PRRK7 and AGEs, specifically CML and methylglyoxal glycation of synuclein proteins. Increasing the expression of PARK 7 lessens the glycation and aggregation of α synuclein [144]. PARK family proteins are commonly involved in PD pathology. PTEN is inhibited by PTEN-induced kinase 1 (PINK 1) or PARK 6, which influences the phosphorylation of Akt via PIP3 to PIP2 conversion for normal cell survival and growth regulation in the absence of growth factors. In the presence of IGF 1, PINK 1 increases the PIP3 concentration.
Further, PINK 1 increases the Akt expression via mTOR 2 phosphorylating RICTOR [43, 145]. PINK 1 and Akt regulate each other reciprocally. Research disclosed the role of Akt, PINK 1, and parkin (PARK 2) in mitophagy. Erk and Akt/mTOR pathways modulate autophagy [146]. When mitochondria get damaged, PINK 1 and Akt will accumulate in the mitochondrial outer membrane and cytoplasm, respectively. It attracts the binding of the parkin to induce autophagy [115] (Fig. 6 PI3K/Akt pathway interferes with PD pathogenesis, including neurotransmitter synthesis and secretion via Cpd38 regulation, calcium channels, and NMDA glutamate receptor. Mediate neural survival, protein aggregation, and mitochondrial function through modulating the activity of FOX O, GSK 3β, and PARK [14, 134]. GLP 1 receptors are used in neuroprotective treatments in diseases like AD and progressed PD [133]. Interlink between PI3K/Akt/mTOR pathway and AGEs role in PD is not widely studied. Various scenarios explored the correlation between AGEs, insulin resistance, and its signaling pathway. Additionally, to add to this point, insulin resistance has an inevitable role in PD progression.
Huntington’s diseases
Huntington's disease (HD) is a chronic and progressive autosomal dominant disorder. Apoptosis of neurons in medium spiny neurons, mainly GABA, dopamine, and acetylcholine-producing neurons, due to the aggregation of Huntington protein (HTT) and APP reduction progress to HD [59, 147, 148]. Degeneration of the neurons occurs in the brain's striatum, hypothalamus, and cortex parts. HTT participates in various development-related pathways, such as hemopoiesis and embryonic development, and prevents apoptosis [147, 149]. Since it is associated with mitochondria, endoplasmic reticulum, and the Golgi complex, due to this HTT aggregation HD, patients show cognitive impairment, dementia, motor abnormalities, behavioral irregularity, psychiatric symptoms, and involuntary muscle movements (Chorea) [150]. Mutation in the CAG triplet repeats (more than 35 repeats) of the htt gene results in the polyglutamine (poly Q) synthesis in the N-terminal of HTT. Spinocerebellar ataxias, HD, dentatorubral–pallidoluysian atrophy, and spinal and bulbar muscular atrophy are some known poly Q diseases [151, 152]. HTT will act as a scaffold to interact with motor proteins to regulate protein transport, such as brain-derived neurotrophic factor (BDNF). Instead, the mutation in HTT reduces the protein transport [150]. HTT mutation may lead to transcriptional dysregulation, misfolding, aggregation, and neural dysfunction, which leads to apoptosis. Studies conducted on Drosophila manifested the same results [148]. At the same time, AGEs promote the glycation of HTT protein in HD patients [152]. Ehinger et al. confirmed that phosphorylation of HTT reduced motor coordination and did not cause any changes in the transportation of the brain-derived neurotropic hormones [150].
In a study with HD model mice, insulin, IGF, and magnesium participate neural survival, proliferation, and productive properties [147]. Akt phosphorylates the HTT protein, which regulates amyloid precursor protein (APP). APP maintains the density and quantity of the synapsis [118]. Moreover, they proposed that controlling Akt-HTT via PI3K, ERK, and JNK in HD and AD might reduce APP depletion. Reduced Akt-HTT has been seen in animal models and the brain of HD patients [59]. Few studies demonstrated that IGF 1 phosphorylates the Akt, caspases, BAD, and FOX O proteins to prevent autophagy and provide neuro productivity in the HD model. GSK 3β may induce apoptosis by inhibiting proteins like Camp-response element-binding protein and heat shock [153]. Reversing or inhibiting the Akt/mTOR-influenced autophagy by targeting metabotropic glutamate receptor subtype 5 (mGLUR5) in HD model animals reduced autophagy [154]. A study stated that manganese has a considerable part in regulating Akt by phosphorylating it. Akt and mTOR would phosphorylate HTT at Ser 421, which maintains mitochondrial autophagy and reduces the accumulation of proteins and axonal transport [147].
Amyloid lateral sclerosis (ALS)
ALS is a progressive degradation of motor neurons in the spinal cortex and anterior with sclerosis. It occurs through familial and sporadic history. The standard clinical features of ALS are muscular atrophy, spasticity, and respiratory failure due to paralysis of respiratory muscles and diaphragm [111, 155]. It is one of the fast progressive diseases; within five years after the diagnosis, most ALS patients lose their life [48]. Insulin resistance worsens muscular atrophy simultaneously with the gradual reduction in GLUT receptors; patients also showed abnormal glucose homeostasis [88, 156]. Superoxide dismutase 1 or Cu/Zn superoxide dismutase (SOD1), TAR DNA-binding protein (TDP-43), deposition, positive ubiquitin protease inclusion and mutations in C9orf72, and FUS/TLS were observed in motor neurons when/as ALS progression [155]. Most of the sporadic ALS seemed to have elevated SOD1. On the other hand, TDP mutations and deposition were seen in familial ALS [157]. Mutation in SOD 1 increases neural toxicity and neural loss by increasing ROS, nitrous oxide, glutamate toxicity, enzymes like nitric oxide synthase, cyclooxygenase, and inflammatory cytokines such as TNFα and IL1 stir up damage in neural cells. Mutation in SOD1 is more common in sporadic ALS (15–20%), whereas familial ALS patients showed 5–10% [112, 158]. Disease advanced with oxidative stress, mitochondrial dysfunction, inflammation of neurons, neurofilament disorientation, and loss of axonal terminals [158].
Oxidative stress induced by ROS and RNS give rise to AGEs further in binds with RAGE that activates inflammatory pathway like NF-κB. Amador products, CML, imidazole, pentosidine, and pyrraline, are present in the anterior horn of ALS patients' brains and cerebrospinal fluid and are considered a marker [159, 160]. AGEs are more tend to bind with the mutated SOD1. A study explained that blocking the activity of RAGE might be a helpful strategy to lessen the progression of ALS [158]. Even though ALS patients appear to have insulin resistance, some studies indicated no direct contact between the PI3K/Akt/mTOR pathway. Yet, gene analysis uncovered the dysregulation of Astrocyte elevated gene 1 (AGE-1) ALS patients. The PI3K/Akt/CREB pathway regulates this protein. It is CREB that mediates the activation of AGE-1 [161]. While Tolosa et al. reported that VEGF reduces the SOD1-induced glutamate toxicity through PI3K signaling pathway. Notably, PI3K signaling pathway prevented the bcl-2-mediated apoptosis in the ALS model [162]. Although numerous studies explored protein deposition, insulin resistance, and AGEs, yet did not explore the interconnecting link between these conditions in ALS. Since there is a common pathway, we must investigate these parameters for possible correlation. Focusing on these aspects might help to develop a new treatment strategy.
Conclusion
This review discussed that insulin resistance induces the formation of AGEs. It interferes with PI3K/Akt/mTOR pathway that regulates cell growth, cell survival, metabolism, and proliferation. Insufficient insulin secretion and mutations in the PI3K/Akt/mTOR pathway decrease the metabolism, development, and transcription, which induces apoptosis and inflammation. It is also affected by the binding of AGEs with receptors since they impact many signaling pathways. AGEs through PI3K/Akt/mTOR boost amyloid β fibrils, α synuclein aggregation, tau hyperphosphorylation, and HTT deposition. Maintaining the proper glucose level and improving insulin sensitivity would aid in the reduction of the formation of AGEs. So, it is necessary to focus on the treatment for reducing the AGEs synthesis or a way to facilitate its interaction with its receptor. It might be helpful in the diseases like AD, PD, ALS, and HD, where AGEs play a significant role.
Data Availability
Data sharing is not applicable for this study as no datasets are genarated or analyzed in the current study.
Abbreviations
- 4E BP1:
-
Eukaryotic translation initiation factor 4E (elF4E)-binding protein 1
- AD:
-
Alzheimer’s disease
- AGEs:
-
Advances glycation end products
- ALS:
-
Amyloid lateral sclerosis
- AKT:
-
AK strain transforming
- AMPK:
-
AMP-activated protein kinase
- APP:
-
Amyloid precursor protein
- APS:
-
Adaptor protein with Pleckstrin homology and Src homology 2 domains
- ATP:
-
Adenosine triphosphate
- ATG:
-
Autophagy-related gene
- Bax:
-
Bcl-2 associated X protein
- BBB:
-
Blood–Brain Barrier
- BNIP 3:
-
BCL2/adenovirus E1B 19 kDa protein-interacting protein 3
- CML:
-
Nε-carboxy-methyl-lysine
- c-Myc:
-
Master Regulator of Cell Cycle Entry and Proliferative Metabolism
- Cpd38:
-
6-(2,4-difluorophenoxy)-5-((ethylmethyl)pyridine-3-yl)-8-methylpyrrolo[1,2-a] pyrazin-1(2H)-one
- CREB:
-
cAMP-responsive element-binding protein
- CVD:
-
Cardiovascular diseases
- DEPTOR:
-
DEP domain-containing interacting protein
- DPP4:
-
Dipeptidyl-peptidase 4
- DM:
-
Diabetes mellitus
- ERK:
-
Extracellular signal-regulated kinase
- FOX O:
-
Forehead box O transcription factor
- FRK:
-
Fyn-related kinase
- FUS/TLS:
-
Fused in sarcoma/translocated in liposarcoma
- GLP 1:
-
Glucagon-like polypeptide 1
- GABA:
-
Gama-aminobutyric acid
- GLUT:
-
Glucose transporter
- GPCR:
-
G protein-coupled receptor
- GRB:
-
Growth factor receptor-bound protein
- GSK:
-
Glycogen synthase kinase
- HD:
-
Huntington’s disease
- HIF1a:
-
Hypoxia-inducible Factor-1a
- HTT:
-
Huntington protein
- IGF:
-
Insulin growth factor
- IL:
-
Interleukin
- IR:
-
Insulin receptor
- IRS:
-
Insulin receptor substrate
- JNK:
-
Jun kinase
- MAPK:
-
Mitogen-activated protein kinase
- MELAS:
-
Mitochondrial Encephalopathy, Lactic Acidosis, and Stroke-like episodes
- MLST8:
-
Mammalian lethal with SEC18 protein 8
- mSIN- mLST 1- mTOR:
-
Mechanistic/mammalian target of rapamycin
- NADPH:
-
Mitogen-activated protein kinase
- NF κb:
-
Nuclear factor kappa-light chain-enhancer of activated B cells
- Nrf:
-
Nuclear factor κ-light-chain-enhancer of activated B cell
- NMDA:
-
N-methyl-D-aspartate
- PD:
-
Parkinson’s disease
- PDK:
-
Phosphoinositide-dependent Protein Kinase
- Pkc:
-
Protein Kinase C
- PI3K:
-
Phosphatidyl inositol 3 phosphate kinase
- PIP3:
-
Phosphatidyl inositol 3,4,5 triphosphate
- PINK:
-
PTEN putative kinase 1
- PKB:
-
Protein kinase B
- PTEN:
-
Phosphatase and tensin homolog
- PTP1B:
-
Protein-tyrosine Phosphatase 1B
- PP-242:
-
mTOR inhibitor
- PPAR:
-
Peroxisome proliferator-activated receptor
- PRAS 8:
-
Protease-activated receptors 8
- RTK:
-
Receptor tyrosine kinase
- RAGE:
-
Receptor for AGEs
- RAPTOR:
-
Regulatory associated protein of mTOR
- RHEB:
-
Ras homologous enriched in the brain
- RICTOR:
-
Rapamycin-insensitive companion of mTOR
- ROS:
-
Reactive oxygen species
- RNS:
-
Reactive nitrogen species
- RXR:
-
Retinoid transcription factor
- SREBP1:
-
Sterol regulatory element-binding transcription factor 1
- SGK:
-
Serine/threonine-protein kinase
- SR:
-
Scavenger receptor
- S6K:
-
Ribosomal s6 kinase
- SOXS3:
-
Suppressor of cytokine signaling 3
- SOD1:
-
Superoxide dismutase 1
- STAT:
-
Signal transducer and activator of transcription
- SHcA:
-
SHC transforming protein 1
- TDP-43:
-
TAR DNA-binding protein 43
- TSC:
-
Tuberous sclerosis proteins
- TNF:
-
Tumor necrosis factor
- VEGF:
-
Vascular endothelial growth factor
- Wnt:
-
Wingless-related integration site
References
Dai B, Wu Q, Zeng C, Zhang J, Cao L, Xiao Z, Yang M (2016) The effect of Liuwei Dihuang Decoction on PI3K/Akt signaling pathway in liver of type 2 Diabetes Mellitus (T2DM) rats with insulin resistance. J Ethnopharmacol 192:382–389. https://doi.org/10.1016/j.jep.2016.07.024
He CJ, Ma LQ, Iqbal MS, Huang XJ, Li J, Yang GZ, Ihsan A (2020) Veratrilla Baillonii Franch exerts anti-diabetic activity and improves liver injury through IRS/PI3K/AKT signaling pathways in Type 2 Diabetic Db/Db mice. J Funct Foods 75:104204. https://doi.org/10.1016/j.jff.2020.104204
Yang L, Wang H, Liu L, Xie A (2018) The role of insulin/IGF-1/PI3K/Akt/GSK3β signaling in Parkinson’s disease dementia. Front Neurosci 12:1–8. https://doi.org/10.3389/fnins.2018.00073
Aviles-Olmos I, Limousin P, Lees A, Foltynie T (2013) Parkinson’s Disease, insulin resistance and novel agents of neuroprotection. Brain 136:374–384. https://doi.org/10.1093/brain/aws009
Yang YW, Hsieh TF, Li CI, Liu CS, Lin WY, Chiang JH, Li TC, Lin CC (2017) Increased risk of Parkinson disease with diabetes mellitus in a population-based study. Medicine (United States). https://doi.org/10.1097/MD.0000000000005921
Chu F, Li K, Li X, Xu L, Huang J, Yang Z (2021) Graphene oxide ameliorates the cognitive impairment through inhibiting PI3K/Akt/MTOR pathway to induce autophagy in AD mouse model. Neurochem Res 46:309–325. https://doi.org/10.1007/s11064-020-03167-z
Barbier P, Zejneli O, Martinho M, Lasorsa A, Belle V, Smet-Nocca C, Tsvetkov PO, Devred F, Landrieu I (2019) Role of Tau as a microtubule-associated protein: structural and functional aspects. Front Aging Neurosci 10:1–14. https://doi.org/10.3389/fnagi.2019.00204
Brothers HM, Gosztyla ML, Robinson SR (2018) The physiological roles of amyloid-β peptide hint at new ways to treat Alzheimer’s Disease. Front Aging Neurosci 10:1–16. https://doi.org/10.3389/fnagi.2018.00118
Ott C, Jacobs K, Haucke E, Navarrete Santos A, Grune T, Simm A (2014) Role of advanced glycation end products in cellular signaling. Redox Biol 2:411–429. https://doi.org/10.1016/j.redox.2013.12.016
Zhao G, Zhang X, Wang H, Chen Z (2020) Beta carotene protects H9c2 cardiomyocytes from advanced glycation end product-induced endoplasmic reticulum stress, apoptosis, and autophagy via the PI3K / Akt / MTOR signaling pathway. Ann Transl Med 8:1–13. https://doi.org/10.21037/atm-20-3768
Sharma A, Weber D, Raupbach J, Dakal TC, Fließbach K, Ramirez A, Grune T, Wüllner U (2020) Advanced glycation end products and protein carbonyl levels in plasma reveal sex-specific differences in Parkinson’s and Alzheimer’s disease. Redox Biol 34:101546. https://doi.org/10.1016/j.redox.2020.101546
Kim Y, Kim C, Son SM, Song H, Hong HS, Han SH, Mook-Jung I (2016) The novel RAGE Interactor PRAK is associated with autophagy signaling in Alzheimer’s disease pathogenesis. Mol Neurodegener 11:1–11. https://doi.org/10.1186/s13024-016-0068-5
Adamopoulos C, Farmaki E, Spilioti E, Kiaris H, Piperi C (2013) Advanced glycation end-products induce endoplasmic reticulum stress in human aortic endothelial cells. Clin Chem Lab Med. https://doi.org/10.1515/cclm-2012-0826
Xu F, Na L, Li Y, Chen L (2020) Roles of the PI3K/AKT/MTOR signalling pathways in neurodegenerative diseases and tumours. Cell Biosci 10:1–12. https://doi.org/10.1186/s13578-020-00416-0
Yan J, Wang C, Jin Y, Meng Q, Liu Q, Liu Z, Liu K, Sun H (2018) Catalpol ameliorates hepatic insulin resistance in type 2 diabetes through acting on AMPK/NOX4/PI3K/AKT pathway. Pharmacol Res 130:466–480. https://doi.org/10.1016/j.phrs.2017.12.026
Dos Santos JM, Tewari S, Mendes RH (2019) The role of oxidative stress in the development of diabetes mellitus and its complications. J Diabetes Res 2019:10–12. https://doi.org/10.1155/2019/4189813
Wang J, Yang X, Zhang J (2016) Bridges between mitochondrial oxidative stress, ER stress and MTOR signaling in pancreatic β cells. Cell Signal 28:1099–1104. https://doi.org/10.1016/j.cellsig.2016.05.007
Jere SW, Houreld NN, Abrahamse H (2019) Role of the PI3K/AKT (MTOR and GSK3β) signalling pathway and photobiomodulation in diabetic wound healing. Cytokine Growth Factor Rev 50:52–59. https://doi.org/10.1016/j.cytogfr.2019.03.001
Hassanpour M, Rezabakhsh A, Rahbarghazi R, Nourazarian A, Nouri M, Avci ÇB, Ghaderi S, Alidadyani N, Bagca BG, Bagheri HS (2017) Functional convergence of Akt protein with VEGFR-1 in human endothelial progenitor cells exposed to sera from patient with type 2 diabetes mellitus. Microvasc Res 114:101–113. https://doi.org/10.1016/j.mvr.2017.07.002
Bathina S, Gundala NKV, Rhenghachar P, Polavarapu S, Hari AD, Sadananda M, Das UN (2020) Resolvin D1 ameliorates nicotinamide-streptozotocin-induced type 2 diabetes mellitus by its anti-inflammatory action and modulating PI3K/Akt/MTOR pathway in the brain. Arch Med Res 51:492–503. https://doi.org/10.1016/j.arcmed.2020.05.002
Di Tu Q, Jin J, Hu X, Ren Y, Zhao L, He Q (2020) Curcumin improves the renal autophagy in rat experimental membranous nephropathy via regulating the PI3K/AKT/MTOR and Nrf2/HO-1 signaling pathways. BioMed Res Int. https://doi.org/10.1155/2020/7069052
Villalobos-Labra R, Silva L, Subiabre M, Araos J, Salsoso R, Fuenzalida B, Sáez T, Toledo F, González M, Quezada C, Pardo F, Chiarello DI, Leiva A, Sobrevia L (2017) Akt/MTOR role in human foetoplacental vascular insulin resistance in diseases of pregnancy. J Diabetes Res. https://doi.org/10.1155/2017/5947859
Mardilovich K, Pankratz SL, Shaw LM (2009) Expression and function of the insulin receptor substrate proteins in cancer. Cell Commun Signal 7:1–15. https://doi.org/10.1186/1478-811X-7-14
Tumminia A, Vinciguerra F, Parisi M, Frittitta L (2018) Type 2 diabetes mellitus and Alzheimer’s disease: role of insulin signalling and therapeutic implications. Int J Mol Sci. https://doi.org/10.3390/ijms19113306
Bao S, Wu YL, Wang X, Han S, Cho SB, Ao W, Nan JX (2020) Agriophyllum oligosaccharides ameliorate hepatic injury in type 2 diabetic Db/Db mice targeting INS-R/IRS-2/PI3K/AKT/PPAR-γ/Glut4 signal pathway. J Ethnopharmacol 257:112863. https://doi.org/10.1016/j.jep.2020.112863
Rabiee A, Krüger M, Ardenkjær-Larsen J, Kahn CR, Emanuelli B (2018) Distinct signalling properties of insulin receptor substrate (IRS)-1 and IRS-2 in mediating insulin/IGF-1 action. Cell Signal 47:1–15. https://doi.org/10.1016/j.cellsig.2018.03.003
Zoncu R, Efeyan A, Sabatini D (2012) MTOR: from growth signal integration to cancer. Diabetes Ageing. 12:21–35. https://doi.org/10.1038/nrm3025.mTOR
Kubota T, Kubota N, Kadowaki T (2017) Imbalanced insulin actions in obesity and type 2 diabetes : key mouse models of insulin signaling pathway. Cell Metab 25:797–810. https://doi.org/10.1016/j.cmet.2017.03.004
Wang Q, Cheng XL, Zhang DY, Gao XJ, Zhou L, Qin XY, Xie GY, Liu K, Qin Y, Liu BL, Qin MJ (2013) Tectorigenin attenuates palmitate-induced endothelial insulin resistance via targeting ROS-associated inflammation and IRS-1 pathway. PLoS One. https://doi.org/10.1371/journal.pone.0066417
Deyev IE, Sohet F, Vassilenko KP, Serova OV, Nadezhda V, Zozulya SA, Burova EB, Houillier P, Rzhevsky DI, Berchatova AA, Murashev AN, Chugunov AO, Roman G, Nikol NN, Bertelli E, Eladari D, Alexander G (2012) Insulin receptor-related receptor as an extracellular alkali sensor. Cell Metab. 13:679–689. https://doi.org/10.1016/j.cmet.2011.03.022.Insulin
Boura-Halfon S, Zick Y (2009) Phosphorylation of IRS proteins, insulin action, and insulin resistance. Am J Physiol Endocrinol Metab. https://doi.org/10.1152/ajpendo.90437.2008
Gao S, Guo Q, Qin C, Shang R, Zhang Z (2017) Sea Buckthorn fruit oil extract alleviates insulin resistance through the PI3K/Akt signaling pathway in type 2 diabetes mellitus cells and rats. J Agric Food Chem 65:1328–1336. https://doi.org/10.1021/acs.jafc.6b04682
O’Neill C (2013) PI3-Kinase/Akt/MTOR signaling: impaired on/off switches in aging, cognitive decline and Alzheimer’s disease. Exp Gerontol 48:647–653. https://doi.org/10.1016/j.exger.2013.02.025
Geering B, Cutillas PR, Nock G, Gharbi SI, Vanhaesebroeck B (2007) Class IA phosphoinositide 3-kinases are obligate P85–P110 heterodimers. Proc Natl Acad Sci USA 104:7809–7814. https://doi.org/10.1073/pnas.0700373104
Xie Y, Shi X, Sheng K, Han G, Li W, Zhao Q, Jiang B, Feng J, Li J, Gu Y (2019) PI3K/Akt signaling transduction pathway, erythropoiesis and glycolysis in hypoxia (review). Mol Med Reports 19:783–791. https://doi.org/10.3892/mmr.2018.9713
Fan Y, He Z, Wang W, Li J, Hu A, Li L, Yan L, Li Z, Yin Q (2018) Tangganjian decoction ameliorates type 2 diabetes mellitus and nonalcoholic fatty liver disease in rats by activating the IRS/PI3K/AKT signaling pathway. Biomed Pharmacother 106:733–737. https://doi.org/10.1016/j.biopha.2018.06.089
Brachmann SM, Ueki K, Engelman JA, Kahn RC, Cantley LC (2005) Phosphoinositide 3-kinase catalytic subunit deletion and regulatory subunit deletion have opposite effects on insulin sensitivity in mice. Exp Gerontol 25:1596–1607. https://doi.org/10.1128/MCB.25.5.1596
Gupta A, Dey CS, Chernoff J (2009) PTEN, a widely known negative regulator of insulin / PI3K signaling Positively Regulates Neuronal Insulin Resistance. Mol Biol Cell. https://doi.org/10.1091/mbc.E12-05-0337
Dibble CC, Cantely LC (2015) Regulation of MTORC1 by PI3K signaling. Trends Cell Biol 25:545–555. https://doi.org/10.1016/j.tcb.2015.06.002.Regulation
Ersahin T, Tuncbag N, Cetin-Atalay R (2015) The PI3K/AKT/MTOR interactive pathway. Mol BioSyst 11:1946–1954. https://doi.org/10.1039/c5mb00101c
Bathina S, Das UN (2018) Dysregulation of PI3K-Akt-MTOR pathway in brain of streptozotocin-induced type 2 diabetes mellitus in Wistar rats. Lipids Health Dis 17:1–11. https://doi.org/10.1186/s12944-018-0809-2
Sudhahar V, Okur MN, Bagi Z, Bryan JPO, Hay N, Makino A, Patel VS, Phillips SA, Stepp D, Fukai T, Genetics M, Veterans B, Medical A, Veterans N, Medical A (2019) Akt2 stabilizes ATP7A, a Cu transporter for SOD3, in vascular smooth muscles: novel mechanism to limit endothelial dysfunction in Type2 diabetes. Arterioscler Thromb Vasc Biol. 38:529–541. https://doi.org/10.1161/ATVBAHA.117.309819.Akt2
Furlong RM (2019) The Parkinson’s gene PINK1 activates Akt via PINK1 kinase-dependent regulation of the phospholipid PI(3,4,5) P3. J Cell Sci 3:132. https://doi.org/10.1242/jcs.233221
Bian C, Bai B, Gao Q, Li S, Zhao Y (2019) 17β-estradiol regulates glucose metabolism and insulin secretion in rat islet β cells through GPER and Akt/MTOR/GLUT2 pathway. Front Endocrinol 10:1–12. https://doi.org/10.3389/fendo.2019.00531
Karki R, Tom A, Hofmann-Apitius M (2017) Comorbidity analysis between Alzheimer’s disease and type 2 diabetes mellitus ( T2DM ) based on shared pathways and the role of T2DM drugs. J Alzheimers Dis 60:721–731. https://doi.org/10.3233/JAD-170440
Tuohetaerbaike B, Zhang Y, Tian Y, nan Zhang N, Kang J, Mao X, Zhang Y, Li X (2020) Pancreas protective effects of urolithin a on type 2 diabetic mice induced by high fat and streptozotocin via regulating autophagy and AKT/MTOR signaling pathway. J Ethnopharmacol. https://doi.org/10.1016/j.jep.2019.112479
Rai SN, Dilnashin H, Birla H, Singh S. Sen, Zahra W, Rathore AS, Singh BK, Singh SP (2019) The role of PI3K/Akt and ERK in neurodegenerative disorders. Neurotox Res 35:775–795. https://doi.org/10.1007/s12640-019-0003-y
Wang Y, Duan W, Wang W, Wen D, Liu Y, Liu Y, Li Z, Hu H, Lin H, Cui C, Li D, Dong H (2016) ScAAV9-VEGF prolongs the survival of transgenic ALS mice by promoting activation of M2 microglia and the PI3K/Akt pathway. Brain Res. https://doi.org/10.1016/j.brainres.2016.06.043
Tuo Y, Xiang M (2019) MTOR: a double-edged sword for diabetes. J Leukocyte Biol 106:385–395. https://doi.org/10.1002/JLB.3MR0317-095RR
Katta A, Kakarla S, Wu M, Paturi S, Gadde MK, Arvapalli R, Kolli M, Rice KM, Blough ER (2009) Altered regulation of contraction-induced Akt/MTOR/P70S6k pathway signaling in skeletal muscle of the Obese Zucker Rat. Exp Diabetes Res 2009:384683. https://doi.org/10.1155/2009/384683
Muthukumaran P, Thiyagarajan G, Arun Babu R, Lakshmi BS (2018) Raffinose from Costus speciosus attenuates lipid synthesis through modulation of PPARs/SREBP1c and improves insulin sensitivity through PI3K/AKT. Chemico-Biol Interact 284:80–89. https://doi.org/10.1016/j.cbi.2018.02.011
Lu X, Paliogiannis P, Calvisi DF, Chen X (2021) Role of the mammalian target of rapamycin pathway in liver cancer: from molecular genetics to targeted therapies. Hepatology 73:49–61. https://doi.org/10.1002/hep.31310
Zheng G, Wang L, Li X, Niu X, Xu G, Lv P (2021) Rapamycin alleviates cognitive impairment in murine vascular dementia: the enhancement of mitophagy by PI3K/AKT/MTOR axis. Tissue Cell 69:101481. https://doi.org/10.1016/j.tice.2020.101481
Yang J, Nie J, Ma X, Wei Y, Peng Y, Wei X (2019) Targeting PI3K in cancer: mechanisms and advances in clinical trials 06 biological sciences 0601 biochemistry and cell biology. Mol Cancer 18:1–28
Kezic A, Popovic L, Lalic K (2018) MTOR inhibitor therapy and metabolic consequences: where do we stand? Oxid Med Cell Longev. https://doi.org/10.1155/2018/2640342
Koliaki C, Liatis S, Dalamaga M, Kokkinos A (2020) the implication of gut hormones in the regulation of energy homeostasis and their role in the pathophysiology of obesity. Curr Obesity Reports 9:255–271. https://doi.org/10.1007/s13679-020-00396-9
Wilcox G (2005) Insulin and insulin resistance. Aliment Pharmacol Ther Suppl 22:61–63. https://doi.org/10.1111/j.1365-2036.2005.02599.x
Peterson CT, Vaughn AR, Sharma V, Chopra D, Mills PJ, Peterson SN, Sivamani RK (2018) Effects of turmeric and curcumin dietary supplementation on human gut microbiota: a double-blind, randomized, placebo-controlled pilot study. J Evid Based Integr Med 23:1–8. https://doi.org/10.1177/2515690X18790725
Bryan MR, Nordham KD, Rose DIR, Joshi P, Foshage AM, Nitin R, Michael A, Aschner M, Bowman AB, Lafayette W (2021) Manganese acts upon insulin/IGF receptors to phosphorylate AKT and increase glucose uptake in Huntington’s disease cells. Mol Neurobiol 57:1570–1593. https://doi.org/10.1007/s12035-019-01824-1.Manganese
Aguirre V, White MF (2000) Dysregulation of IRS-proteins causes insulin resistance and diabetes. Current Opin Endocrinol Diabetes 7:1–7. https://doi.org/10.1097/00060793-200002000-00001
Gao L, Yuan P, Zhang Q, Fu Y, Hou Y, Wei Y, Zheng X, Feng W (2020) Taxifolin improves disorders of glucose metabolism and water-salt metabolism in kidney via PI3K/AKT signaling pathway in metabolic syndrome rats. Life Sci 263:118713. https://doi.org/10.1016/j.lfs.2020.118713
Petersen MC, Shulman GI (2018) Mechanisms of insulin action and insulin resistance. Physiol Rev 98:2133–2223. https://doi.org/10.1152/physrev.00063.2017
Samuel VT, Shulman GI (2012) Review mechanisms for insulin resistance: common threads and missing links. Cell 148:852–871. https://doi.org/10.1016/j.cell.2012.02.017
Miyake Y, Tanaka K, Fukushima W, Sasaki S, Kiyohara C, Tsuboi Y, Yamada T, Oeda T, Miki T, Kawamura N, Sakae N, Fukuyama H, Hirota Y, Nagai M (2010) Case-control study of risk of Parkinson’s disease in relation to hypertension, hypercholesterolemia, and diabetes in Japan. J Neurol Sci 293:82–86. https://doi.org/10.1016/j.jns.2010.03.002
Song Q, Liu J, Dong L, Wang X, Zhang X (2021) Novel advances in inhibiting advanced glycation end product formation using natural compounds. Biomed Pharmacother 140:111750. https://doi.org/10.1016/j.biopha.2021.111750
Domingueti CP, Dusse LMSA, Carvalho MDG, De Sousa LP, Gomes KB, Fernandes AP (2016) Diabetes mellitus: the linkage between oxidative stress, inflammation, hypercoagulability and vascular complications. J Diabetes Complicat 30:738–745. https://doi.org/10.1016/j.jdiacomp.2015.12.018
Horii N, Hasegawa N, Fujie S, Uchida M, Iemitsu M (2020) Resistance exercise-induced increase in muscle 5α-dihydrotestosterone contributes to the activation of muscle Akt/MTOR/P70S6K- and Akt/AS160/GLUT4-signaling pathways in type 2 diabetic rats. FASEB J 34:11047–11057. https://doi.org/10.1096/fj.201903223RR
Choi J, Kim KJ, Koh EJ, Lee BY (2018) Gelidium elegans extract ameliorates type 2 diabetes via regulation of MAPK and PI3K/Akt signaling. Nutrients. https://doi.org/10.3390/nu10010051
Archuleta TL, Lemieux AM, Saengsirisuwan V, Teachey MK, Lindborg KA, Kim JS, Henriksen EJ (2009) Oxidant stress-induced loss of IRS-1 and IRS-2 proteins in rat skeletal muscle: role of P38 MAPK. Free Radic Biol Med 47:1486–1493. https://doi.org/10.1016/j.freeradbiomed.2009.08.014
Kido Y, Burks DJ, Withers D, Bruning JC, Kahn CR, White MF, Accili D (2000) Tissue-specific insulin resistance in mice with mutations in the insulin receptor, IRS-1, and IRS-2. Journal of Clinical Investigation 105:199–205. https://doi.org/10.1172/JCI7917
Yang Z, Zhang L, Liu J, Lu F, Wang L, Chen Y, Li D (2019) Hypoglycemic effects of Esculeoside A are mediated via activation of AMPK and upregulation of IRS-1. BMC Complement Altern Med 19:1–9. https://doi.org/10.1186/s12906-019-2543-3
White MF (2002) IRS proteins and the common path to diabetes. Am J Physiol Endocrinol Metab. https://doi.org/10.1152/ajpendo.00514.2001
Zheng M, Wang P (2021) Role of insulin receptor substance - 1 modulating PI3K / Akt insulin signaling pathway in Alzheimer’s Disease. 3 Biotech 11:1–17. https://doi.org/10.1007/s13205-021-02738-3
Maiti P, Scott J, Sengupta D, Al-Gharaibeh A, Dunbar GL (2019) Curcumin and solid lipid curcumin particles induce autophagy, but inhibit mitophagy and the PI3K-Akt/MTOR pathway in cultured glioblastoma cells. Int J Mol Sci. https://doi.org/10.3390/ijms20020399
Ferreira LSS, Fernandes CS, Vieira MNN, De Felice FG (2018) Insulin resistance in Alzheimer’s disease. Front Neurosci 12:1–11. https://doi.org/10.3389/fnins.2018.00830
Chaudhuri J, Bains Y, Guha S, Kahn A, Hall D, Bose N, Gugliucci A, Kapahi P (2018) The role of advanced glycation end products in aging and metabolic diseases: bridging association and causality. Cell Metab 28:337–352. https://doi.org/10.1016/j.cmet.2018.08.014
Salahuddin P, Rabbani G, Khan RH (2014) The role of advanced glycation end products in various types of neurodegenerative disease: a therapeutic approach. Cell Mol Biol Lett 19:407–437. https://doi.org/10.2478/s11658-014-0205-5
Li J, Liu D, Sun L, Lu Y, Zhang Z (2012) Advanced glycation end products and neurodegenerative diseases: mechanisms and perspective. J Neurol Sci 317:1–5. https://doi.org/10.1016/j.jns.2012.02.018
Abedini A, Derk J, Schmidt AM (2018) The Receptor for advanced glycation endproducts is a mediator of toxicity by IAPP and other proteotoxic aggregates: establishing and exploiting common ground for novel amyloidosis therapies. Protein Sci 27:1166–1180. https://doi.org/10.1002/pro.3425
Giridharan VV, Generoso JS, Collodel A, Dominguini D, Faller CJ, Tardin F, Bhatti GS, Petronilho F, Dal-Pizzol F, Barichello T (2021) Receptor for advanced glycation end products (RAGE) mediates cognitive impairment triggered by pneumococcal meningitis. Neurotherapeutics 18:640–653. https://doi.org/10.1007/s13311-020-00917-3
Cai W, He JC, Zhu L, Chen X, Striker GE, Vlassara H (2008) AGE-receptor-1 counteracts cellular oxidant stress induced by AGEs via negative regulation of P66shc-dependent FKHRL1 phosphorylation. Am J Physiol Cell Physiol. https://doi.org/10.1152/ajpcell.00350.2007
Hu P, Lai D, Lu P, Gao J, He H (2012) ERK and Akt signaling pathways are involved in advanced glycation end product-induced autophagy in rat vascular smooth muscle cells. Int J Mol Med 29:613–618. https://doi.org/10.3892/ijmm.2012.891
Yamamoto M, Guo DH, Hernandez CM, Stranahan AM (2019) Endothelial Adora2a activation promotes blood-brain barrier breakdown and cognitive impairment in mice with diet-induced insulin resistance. J Neurosci 39:4179–4192. https://doi.org/10.1523/JNEUROSCI.2506-18.2019
Sun Y, Ma C, Sun H, Wang H, Peng W, Zhou Z, Wang H, Pi C, Shi Y, He X (2020) Metabolism: a novel shared link between diabetes mellitus and Alzheimer’s disease. J Diabetes Res. https://doi.org/10.1155/2020/4981814
Akhtar A, Sah SP (2020) Insulin signaling pathway and related molecules: role in neurodegeneration and Alzheimer’s disease. Neurochemistry International 135:104707. https://doi.org/10.1016/j.neuint.2020.104707
Takeda S, Sato N, Uchio-Yamada K, Sawada K, Kunieda T, Takeuchi D, Kurinami H, Shinohara M, Rakugi H, Morishita R (2010) Diabetes-accelerated memory dysfunction via cerebrovascular inflammation and Aβ deposition in an Alzheimer mouse model with diabetes. Proc Natl Acad Sci USA 107:7036–7041. https://doi.org/10.1073/pnas.1000645107
Chen L, Wei Y, Wang X, He R (2010) Ribosylation rapidly induces α -synuclein to form highly cytotoxic molten globules of advanced glycation end products. PLoS One. https://doi.org/10.1371/journal.pone.0009052
Reyes ET, Perurena OH, Festoff BW, Jorgensen R, Moore WV (1984) Insulin resistance in amyotrophic lateral sclerosis. J Neurol Sci 63:317–324. https://doi.org/10.1016/0022-510X(84)90154-0
De Pablo-Fernandez E, Goldacre R, Pakpoor J, Noyce AJ, Warner TT (2018) Association between diabetes and subsequent Parkinson disease: a record-linkage cohort study. Neurology 91:e139–e142. https://doi.org/10.1212/WNL.0000000000005771
Delamarre A, Rigalleau V, Meissner WG (2020) Insulin resistance, diabetes and Parkinson’s disease: the match continues. Parkinsonism Relat Disord 80:199–200. https://doi.org/10.1016/j.parkreldis.2020.10.013
Kotagal V, Albin RL, Müller MLTM, Koeppe RA, Frey KA, Bohnen NI (2013) Diabetes is associated with postural instability and gait difficulty in Parkinson disease. Parkinsonism Relat Disord 19:522–526. https://doi.org/10.1016/j.parkreldis.2013.01.016
Brauer R, Wei L, Ma T, Athauda D, Girges C, Vijiaratnam N, Auld G, Whittlesea C, Wong I, Foltynie T (2020) Diabetes medications and risk of Parkinson’s disease: a cohort study of patients with diabetes. Brain 143:3067–3076. https://doi.org/10.1093/brain/awaa262
Schernhammer E, Hansen J, Rugbjerg K, Wermuth L, Ritz B (2011) Diabetes and the risk of developing Parkinson’s disease in Denmark. Diabetes Care 34:1102–1108. https://doi.org/10.2337/dc10-1333
Chen K, Xie K, Liu Z, Nakasone Y, Sakao K, Hossain A, Hou DX (2019) Preventive effects and mechanisms of garlic on dyslipidemia and gut microbiome dysbiosis. Nutrients. https://doi.org/10.3390/nu11061225
Gagnum V, Stene LC, Jenssen TG, Berteussen LM, Sandvik L, Joner G, Njølstad PR, Skrivarhaug T (2017) Causes of death in childhood-onset type 1 diabetes: long-term follow-up. Diabetic Med 34:56–63. https://doi.org/10.1111/dme.13114
Yin X, Xu Z, Zhang Z, Li L, Pan Q, Zheng F, Li H (2017) Association of PI3K/AKT/MTOR pathway genetic variants with type 2 diabetes mellitus in Chinese. Diabetes Res Clin Pract 128:127–135. https://doi.org/10.1016/j.diabres.2017.04.002
Zheng Y, Ley SH, Hu FB (2018) Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat Rev Endocrinol 14:88–98. https://doi.org/10.1038/nrendo.2017.151
Li Y, Liu Y, Liang J, Wang T, Sun M, Zhang Z (2019) Gymnemic acid ameliorates hyperglycemia through PI3K/AKT- and AMPK-Mediated signaling pathways in type 2 diabetes mellitus rats. J Agric Food Chem 67:13051–13060. https://doi.org/10.1021/acs.jafc.9b04931
Walter-höliner AI, Barbarini DS, Lütschg J, Zanier U, Saely CH, Simma B (2017) High prevalence and incidence of diabetic peripheral neuropathy in children and adolescents with type 1 diabetes mellitus: results from a 5-year prospective cohort study. Pediatric Neurol. https://doi.org/10.1016/j.pediatrneurol.2017.11.017
Kallinikou D, Soldatou A, Tsentidis C, Louraki M, Kanaka-Gantenbein C, Kanavakis E, Karavanaki K (2019) Diabetic neuropathy in children and adolescents with type 1 diabetes mellitus: diagnosis, pathogenesis, and associated genetic markers. Diabetes/Metab Res Rev 35:1–14. https://doi.org/10.1002/dmrr.3178
Dong J, Li H, Bai Y, Wu C (2019) Muscone ameliorates diabetic peripheral neuropathy through activating AKT/MTOR signalling pathway. J Pharm Pharmacol 71:1706–1713. https://doi.org/10.1111/jphp.13157
Liu K, Yang Y, Zhou F, Xiao Y, Shi L (2020) Inhibition of PI3K/AKT/MTOR signaling pathway promotes autophagy and relieves hyperalgesia in diabetic rats. NeuroReport. https://doi.org/10.1097/WNR.0000000000001461
Biosa A, Outeiro TF, Bubacco L, Bisaglia M (2018) Diabetes mellitus as a risk factor for Parkinson’s disease: a molecular point of view. Mol Neurobiol 55:8754–8763. https://doi.org/10.1007/s12035-018-1025-9
Gao JR, Qin XJ, Fang ZH, Li-Shan, Han LP, Hui-Jian Guo MF, Jiang NN (2019) To explore the pathogenesis of vascular lesion of type 2 diabetes mellitus based on the PI3K/Akt signaling pathway. J Diabetes Res. https://doi.org/10.1155/2019/4650906
Hou Y, Wang K, Wan W, Cheng Y, Pu X, Ye X (2018) Resveratrol provides neuroprotection by regulating the JAK2/STAT3/PI3K/AKT/MTOR pathway after stroke in rats. Genes Dis 5:245–255. https://doi.org/10.1016/j.gendis.2018.06.001
Du C, Wu M, Liu H, Ren Y, Du Y, Wu H, Wei J, Liu C, Yao F, Wang H, Zhu Y, Duan H, Shi Y (2016) Thioredoxin-interacting protein regulates lipid metabolism via Akt/MTOR pathway in diabetic kidney disease. Int J Biochem Cell Biol 79:1–13. https://doi.org/10.1016/j.biocel.2016.08.006
Ran Z, Zhang Y, Wen X, Ma J (2019) Curcumin inhibits high glucose-induced inflammatory injury in human retinal pigment epithelial cells through the ROS-PI3K/AKT/MTOR signaling pathway. Mol Med Reports 19:1024–1031. https://doi.org/10.3892/mmr.2018.9749
Akhtar A, Sah SP (2020) Neurochemistry international insulin signaling pathway and related molecules: role in neurodegeneration and Alzheimer’s disease. Neurochem Int 135:104707. https://doi.org/10.1016/j.neuint.2020.104707
Brasnyó P, Molnár GA, Mohás M, Markó L, Laczy B, Cseh J, Mikolás E, Szijártó IA, Mérei Á, Halmai R, Mészáros LG, Sümegi B, Wittmann I (2011) Resveratrol improves insulin sensitivity, reduces oxidative stress and activates the Akt pathway in Type 2 diabetic patients. Br J Nutr 106:383–389. https://doi.org/10.1017/S0007114511000316
Chi H, Chang H, Sang T (2018) Neuronal cell death mechanisms in major neurodegenerative diseases. Int J Mol Sci. https://doi.org/10.3390/ijms19103082
Juranek JK, Daffu GK, Wojtkiewicz J, Lacomis D, Kofler J, Schmidt AM (2015) Receptor for advanced glycation end products and its inflammatory ligands are upregulated in amyotrophic lateral sclerosis. Front Cell Neurosci 9:1–12. https://doi.org/10.3389/fncel.2015.00485
Ray R, Juranek JK, Rai V (2016) RAGE axis in neuroinflammation, neurodegeneration and its emerging role in the pathogenesis of amyotrophic lateral sclerosis. Neurosci Biobehav Rev 62:48–55. https://doi.org/10.1016/j.neubiorev.2015.12.006
Heras-Sandoval D, Pérez-Rojas JM, Hernández-Damián J, Pedraza-Chaverri J (2014) The role of PI3K/AKT/MTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell Signal 26:2694–2701. https://doi.org/10.1016/j.cellsig.2014.08.019
Fakhri S, Iranpanah A, Mehdi M, Zachariah S (2021) Phytomedicine natural products attenuate PI3K / Akt / MTOR signaling pathway: a promising strategy in regulating neurodegeneration. Phytomedicine 91:153664. https://doi.org/10.1016/j.phymed.2021.153664
Soutar MPM, Kempthorne L, Miyakawa S, Annuario E, Melandri D, Harley J, O’Sullivan GA, Wray S, Hancock DC, Cookson MR, Downward J, Carlton M, Plun-Favreau H (2018) AKT signalling selectively regulates PINK1 mitophagy in SHSY5Y cells and human IPSC-derived neurons. Sci Reports 8:1–11. https://doi.org/10.1038/s41598-018-26949-6
Nguyen TT, Ta QTH, Nguyen TKO, Nguyen TTD, Van Giau V (2020) Type 3 diabetes and its role implications in Alzheimer’s disease. Int J Mol Sci 21:1–16. https://doi.org/10.3390/ijms21093165
Bendor J, Logan T, Edwards RH (2014) The function of α -synuclein. Neuron 79:1044–1066. https://doi.org/10.1016/j.neuron.2013.09.004
Bruyère J, Abada YS, Vitet H, Fontaine G, Deloulme JC, Cès A, Denarier E, Pernet-Gallay K, Andrieux A, Humbert S, Potier MC, Delatour B, Saudou F (2020) Presynaptic APP levels and synaptic homeostasis are regulated by Akt phosphorylation of huntingtin. elife 9:1–30. https://doi.org/10.7554/eLife.56371
Singh SS, Jois SD (2018) Homo- and heterodimerization of proteins in cell signaling: inhibition and drug design. Adv Protein Chem Struct Biol 111:1–59
Ghasemi R, Moosavi M, Zarifkar A, RastegarMaghsoudi KN (2015) The interplay of Akt and ERK in Aβ toxicity and insulin-mediated protection in primary hippocampal cell culture. J Mol Neurosci 57:325–334. https://doi.org/10.1007/s12031-015-0622-6
Kamynina AV, Esteras N, Koroev DO, Bobkova NV, Balasanyants SM, Simonyan RA, Avetisyan AV, Abramov AY, Volpina OM, Walker DG (2018) Synthetic fragments of receptor for advanced glycation end products bind beta-amyloid 1–40 and protect primary brain cells from beta-amyloid toxicity. Front Neurosci 12:1–9. https://doi.org/10.3389/fnins.2018.00681
Angeloni C, Zambonin L, Hrelia S (2014) Role of methylglyoxal in Alzheimer’s disease. Biomed Res Int 2014:238485
Zeng F, Liu Y, Huang W, Qing H, Kadowaki T, Kashiwazaki H, Ni J, Wu Z (2021) Receptor for advanced glycation end products up-regulation in cerebral endothelial cells mediates cerebrovascular-related amyloid β accumulation after porphyromonas gingivalis infection. J Neurochem 158:724–736. https://doi.org/10.1111/jnc.15096
Westwoo W, Beiser A, DeCarli C, Harris TB, Chen TC, He XM, Roubenoff R, Pikula A, Au R, Braverman LE, Wolf PA, Vasan RS, Seshadri S (2014) Insulin-like growth factor-1 and risk of Alzheimer dementia and brain atrophy. Neurology 82:1613–1619. https://doi.org/10.1212/WNL.0000000000000382
Sedzikowska A, Szablewski L (2021) Insulin and insulin resistance in Alzheimer’s disease. Int J Mol Sci 22:9987
Su M, Naderi K, Samson N, Youssef I, Fülöp L, Bozso Z, Laroche S, Delatour B, Davis S (2019) Mechanisms associated with type 2 diabetes as a risk factor for Alzheimer-related pathology. Mol Neurobiol 56:5815–5834. https://doi.org/10.1007/s12035-019-1475-8
Lee H-K, Kwon B, Lemere CA, de la Monte S, Itamura K, Ha AY, Querfurth HW (2016) MTORC2 (Rictor) in Alzheimer’s disease and reversal of amyloid- β expression-induced insulin resistance and toxicity in rat primary cortical neurons. J Alzheimers Dis 56:1015–1036
Chong ZZ, Shang YC, Wang S, Maiese K (2012) A critical kinase cascade in neurological disorders: PI3K, Akt and MTOR. Future Neurol 7:733–748. https://doi.org/10.2217/fnl.12.72
Sun E, Motolani A, Campos L, Lu T (2022) The pivotal role of NF-KB in the pathogenesis and therapeutics of Alzheimer’s disease. Int J Mol Sci 23:8972. https://doi.org/10.3390/ijms23168972
Ali T, Kim MO (2015) Melatonin ameliorates amyloid beta-induced memory deficits, tau hyperphosphorylation and neurodegeneration via PI3/Akt/GSk3β pathway in the mouse hippocampus. J Pineal Res 59:47–59. https://doi.org/10.1111/jpi.12238
Wang A, Xiao C, Zheng J, Ye C, Dai Z, Wu Q, Liu J, Strappe P, Zhou Z (2020) Terpenoids of ganoderma lucidum reverse cognitive impairment through attenuating neurodegeneration via suppression of PI3K/AKT/MTOR expression in vivo model. J Funct Foods 73:104142. https://doi.org/10.1016/j.jff.2020.104142
Gasparotto J, Girardi CS, Somensi N, Ribeiro CT, Moreira JCF, Michels M, Sonai B, Rocha M, Steckert AV, Barichello T, Quevedo J, Dal-pizzol F, Gelain DP (2018) Cro receptor for advanced glycation end products mediates sepsis-triggered amyloid-  accumulation, tau phosphoryla- tion, and cognitive impairment. J Biol Chem 293:226–244. https://doi.org/10.1074/jbc.M117.786756
Athauda D, Foltynie T (2016) The glucagon-like peptide 1 (GLP) receptor as a therapeutic target in Parkinson’s disease: mechanisms of action. Drug Discov Today 21:802–818. https://doi.org/10.1016/j.drudis.2016.01.013
Cheong JLY, de Pablo-Fernandez E, Foltynie T, Noyce AJ (2020) The association between type 2 diabetes mellitus and Parkinson’s disease. J Parkinson’s Dis 10:775–789. https://doi.org/10.3233/jpd-191900
Nakano N, Matsuda S, Ichimura M, Minami A, Ogino M, Murai T, Kitagishi Y (2017) PI3K/AKT signaling mediated by g protein-coupled receptors is involved in neurodegenerative Parkinson’s disease (review). Int J Mol Med 39:253–260. https://doi.org/10.3892/ijmm.2016.2833
Annekatrin K (2018) Alpha-synuclein glycation and the action of anti-diabetic agents in Parkinson’s disease. J Parkinsons Dis 8:33–43. https://doi.org/10.3233/JPD-171285
Natale G, Pompili E, Biagioni F, Paparelli S, Lenzi P, Fornai F (2013) Histochemical approaches to assess cell-to-cell transmission of misfolded proteins in neurodegenerative diseases. Eur J Histochem 57:31–35. https://doi.org/10.4081/ejh.2013.e5
Ashraghi MR, Pagano G, Polychronis S, Niccolini F, Politis M (2016) Parkinson’s disease, diabetes and cognitive impairment. Recent Pat Endocr Metab Immune Drug Discov 10:11–21. https://doi.org/10.2174/1872214810999160628105549
Wang SY, Wu SL, Chen TC, Chuang C. Sen (2020) Antidiabetic agents for treatment of Parkinson’s disease: a meta-analysis. Int J Environ Res Public Health 17:1–11. https://doi.org/10.3390/ijerph17134805
Becker C, Brobert GP, Johansson S, Jick SS, Meier CR (2008) Diabetes in patients with idiopathic Parkinson’s disease. Diabetes Care 31:1808–1812. https://doi.org/10.2337/dc08-0479
Hu MTM, Taylor-Robinson SD, Chaudhuri KR, Bell JD, Labbé C, Cunningham VJ, Koepp MJ, Hammers A, Morris RG, Turjanski N, Brooks DJ (2000) Cortical dysfunction in non-demented Parkinson’s disease patients. A combined 31P-MRS and 18FDG-PET study. Brain 123:340–352. https://doi.org/10.1093/brain/123.2.340
Shaikh S, Nicholson LFB (2008) Advanced glycation end products induce in vitro cross-linking of a -synuclein and accelerate the process of intracellular inclusion body formation. J Neurosci Res 2082:2071–2082. https://doi.org/10.1002/jnr.21644
Teismann P, Sathe K, Bierhaus A, Leng L, Martin HL, Bucala R, Weigle B, Nawroth PP, Schulz JB (2012) Receptor for advanced glycation endproducts (RAGE) deficiency protects against MPTP toxicity. NBA 33:2478–2490. https://doi.org/10.1016/j.neurobiolaging.2011.12.006
Sharma N, Pavani S, Kalivendi SV (2019) Free radical biology and medicine the deglycase activity of DJ-1 mitigates α -synuclein glycation and aggregation in dopaminergic cells : role of oxidative stress mediated downregulation of DJ-1 in Parkinson’s disease. Free Radic Biol Med 135:28–37. https://doi.org/10.1016/j.freeradbiomed.2019.02.014
Murata H, Sakaguchi M, Jin Y, Sakaguchi Y, Futami JI, Yamada H, Kataoka K, Huh NH (2011) A new cytosolic pathway from a Parkinson disease-associated kinase, BRPK/PINK1: activation of AKT via MTORC2. J Biol Chem 286:7182–7189. https://doi.org/10.1074/jbc.M110.179390
Gugliandolo A, Pollastro F, Bramanti P, Mazzon E (2020) Cannabidiol exerts protective effects in an in vitro model of Parkinson’s disease activating AKT/MTOR pathway. Fitoterapia 143:104553. https://doi.org/10.1016/j.fitote.2020.104553
Bryan MR, Bowman AB (2017) Manganese and the insulin-IGF signaling network in huntington’s disease and other neurodegenerative disorders. Adv Neurobiol 18:113–142. https://doi.org/10.1007/978-3-319-60189-2_6
Ersoy Tunalı N (2021) Molecular mechanisms of polyglutamine pathology and lessons learned from Huntington’s disease. Neurodegener Dis. https://doi.org/10.5772/intechopen.93508
Colin E, Régulier E, Perrin V, Dürr A, Brice A, Aebischer P, Déglon N, Humbert S, Saudou F (2005) Akt is altered in an animal model of Huntington’s disease and in patients. Eur J Neurosci 21:1478–1488. https://doi.org/10.1111/j.1460-9568.2005.03985.x
Ehinger Y, Bruyère J, Panayotis N, Abada Y, Borloz E, Matagne V, Scaramuzzino C, Vitet H, Delatour B, Saidi L, Villard L, Saudou F, Roux J (2020) Huntingtin phosphorylation governs BDNF homeostasis and improves the phenotype of Mecp 2 knockout mice. EMBO Mol Med. https://doi.org/10.15252/emmm.201910889
Liévens JC, Iché M, Laval M, Faivre-Sarrailh C, Birman S (2008) AKT-sensitive or insensitive pathways of toxicity in glial cells and neurons in drosophila models of Huntington’s disease. Hum Mol Genet 17:882–894. https://doi.org/10.1093/hmg/ddm360
Bras IC, Konig A (2019) Glycation in Huntington’s disease : a possible modifier and target for intervention. J Huntingtons Dis 8:245–256. https://doi.org/10.3233/JHD-190366
Timothy Hansen CT (2019) Excess active P13K rescues Huntingtin-mediated neuronal cell death but has no effect on axonal transport defects. Apoptosis 176:341–358. https://doi.org/10.1007/s10495-019-01520-4.Excess
Abd-Elrahman KS, Ferguson SSG (2019) Modulation of MTOR and CREB pathways following MGluR5 blockade contribute to improved Huntington’s pathology in ZQ175 mice. Mol Brain 12:1–9. https://doi.org/10.1186/s13041-019-0456-1
Saberi S, Stauffer JE, Schulte DJ, Ravits J (2015) Neuropathology of amyotrophic lateral sclerosis and its variants. Neurol Clin 33:855–876. https://doi.org/10.1016/j.ncl.2015.07.012
Pradat P-F, Bruneteau G, Gordon PH, Dupuis L, Bonnefont-Rousselot D, Simon D, Salachas F, Corcia P, Frochot V, Lacorte J-M, Jardel C, Coussieu C, Le Forestier N, Lacomblez L, Loeffler J-P, Vincent VM (2010) Impaired glucose tolerance in patients with amyotrophic lateral sclerosis. Amyotroph Lateral Scler 11(1–2):166–171
Kato S, Nakashima K, Horiuchi S, Nagai R, Cleveland DW, Liu J, Hirano A, Takikawa M, Kato M, Nakano I, Sakoda S, Asayama K, Ohama E (2001) Formation of advanced glycation end-product-modified superoxide dismutase-1 (SOD1) is one of the mechanisms responsible for inclusions common to familial amyotrophic lateral sclerosis patients with SOD1 gene mutation, and transgenic mice expressing human. Neuropathology. https://doi.org/10.1046/j.1440-1789.2001.00359.x
Juranek JK, Daffu GK, Geddis MS, Li H, Rosario R, Kaplan BJ, Kelly L, Schmidt AM (2016) Soluble RAGE treatment delays progression of amyotrophic lateral sclerosis in SOD1 mice. Front Cell Neurosci 10:1–9. https://doi.org/10.3389/fncel.2016.00117
Kikuchi S, Shinpo K, Ogata A, Tsuji S, Takeuchi M, Makita Z, Tashiro K (2002) Detection of Nε-(Carboxymethyl)Lysine (CML) and non-CML advanced glycation end-products in the anterior horn of amyotrophic lateral sclerosis spinal cord. Amyotroph Lateral Scler Other Motor Neuron Disord 3:63–68. https://doi.org/10.1080/146608202760196020
Kaufmann E, Boehm BO, Süssmuth SD, Kientsch-Engel R, Sperfeld A, Ludolph AC, Tumani H (2004) The advanced glycation end-product N ε-(Carboxymethyl)Lysine level is elevated in cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Neurosci Lett 371:226–229. https://doi.org/10.1016/j.neulet.2004.08.071
Yin X, Ren M, Jiang H, Cui S, Wang S, Jiang H, Qi Y, Wang J, Wang X, Dong G, Leeds P, Chuang DM, Feng H (2015) Downregulated AEG-1 together with inhibited PI3K/Akt pathway is associated with reduced viability of motor neurons in an ALS model. Mol Cell Neurosci 68:303–313. https://doi.org/10.1016/j.mcn.2015.08.009
Tolosa L, Mir M, Asensio J, Olmos G (2008) Vascular endothelial growth factor protects spinal cord motoneurons against glutamate-induced excitotoxicity via phosphatidylinositol 3-kinase. J Neurochem 105:1080–1090. https://doi.org/10.1111/j.1471-4159.2007.05206.x
Funding
The authors declare that no funding and grants were accepted for the preparation of the manuscript.
Author information
Authors and Affiliations
Contributions
All authors contributed in manuscript design and study conception. Kanagavalli Ramasubbu collected the data and wrote the manuscript. Devi Rajeswari V contributed to the drafting of the manuscript. All the authors approved the final version of the manuscript.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ramasubbu, K., Devi Rajeswari, V. Impairment of insulin signaling pathway PI3K/Akt/mTOR and insulin resistance induced AGEs on diabetes mellitus and neurodegenerative diseases: a perspective review. Mol Cell Biochem 478, 1307–1324 (2023). https://doi.org/10.1007/s11010-022-04587-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-022-04587-x