
Abstract
Disruption of Akt and Erk-mediated signal transduction significantly contributes in the pathogenesis of various neurodegenerative diseases (NDs), such as Parkinson’s disease, Alzheimer’s diseases, Huntington’s disease, and many others. These regulatory proteins serve as the regulator of cell survival, motility, transcription, metabolism, and progression of the cell cycle. Therefore, targeting Akt and Erk pathway has been proposed as a reasonable approach to suppress ND progression. This review has emphasized on involvement of Akt/Erk cascade in the neurodegeneration. Akt has been reported to regulate neuronal toxicity through its various substrates like FOXos, GSK3β, and caspase-9 etc. Akt is also involved with PI3K in signaling pathway to mediate neuronal survival. ERK is another kinase which also regulates proliferation, differentiation, and survival of the neural cell. There has also been much progress in developing a therapeutic molecule targeting Akt and Erk signaling. Therefore, improved understanding of the molecular mechanism behind the regulatory aspect of Akt and Erk networks can make strong impact on exploration of the neurodegenerative disease pathogenesis.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Neurodegenerative disorders (NDs) consist of a large number of disorders affecting the nervous system. It includes the heterogeneous group of disorders such as Alzheimer’s disease (AD), Parkinson’s disease (PD), amyotrophic lateral sclerosis (ALS), Huntington’s disease (HD), and many more. The heterogeneous disorders are characterized by progressive and selective loss of neurons (Sheikh et al. 2013). In most cases, the loss of function of the affected neuronal type defines the major phenotype of the disease. The main cause of most NDs is still unknown and, therefore, there is no effective cure. NDs treatment depends on retardation or alleviation of symptoms (Jaiswal et al. 2012). The well-accepted hypotheses accounting for the loss of neurons in neurodegeneration include neurotrophic factor deprivation, the exposure to exogenous or endogenous toxic substances [e.g., MPTP, acetaldehyde, pesticides, 6-OHDA, proteins with expanded polyglutamine stretches], oxidative stress, and genetic factors. Later, it can either predispose to noxious stimuli or directly cause the disease. A number of genes associated to specific NDs have been uncovered (Colucci-D'Amato et al. 2003).
For instance, a well-studied neurodegeneration is in the case of PD. Mutations in three genes, involved in hereditary PD, have been so far identified: α-synuclein, parkin, and DJ-1 (Nuytemans et al. 2010). Phenotypically, the effect of mutations overlaps with the most common sporadic PD but still differs from each other. HD is the well-known dominantly inherited autosomal ND with high penetrance; it is caused by a polyglutamine (polyQ) expansion in the protein huntingtin, which is thus presumed to acquire a toxic gain of function. According to recent findings, the mutated huntingtin can cause the death of specific neuronal population by a mechanism that leads to insufficient neurotrophic support to target neurons. Mutant huntingtin activates Nrf2 responsive genes which in turn impair synthesis of dopamine and so cause dopaminergic neuronal death (van Roon-Mom et al. 2008). Specifically, huntingtin regulates transcription in cortical neurons of BDNF, which is transported to the striatum where it sustains striatal neuron survival. This property is lost with mutated huntingtin (Reichardt 2006). Thus HD pathogenesis represents an example of how neurotrophins, toxicity, and genetics can coordinate resulting in a neurodegenerative disease. Glial cells (microglia and astroglia) are the key regulators of various neurodegenerative diseases because they are the resident immune cells of the brain (Xu et al. 2018). Activation of these cells leads to the pro-inflammatory mediator’s production such as cytokines, chemokines, and NO. Akt and Erk pathway plays significant role in the progression of above discussed various NDs.
Therefore, ERK and Akt/PKB signaling pathways can be an effective therapeutic target to prevent the progression of neurodegeneration, regulation of pro-inflammatory mediators, oxidative damage, and neurotrophins such as nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF).
Akt, also known as PKB (protein kinase B), belongs to the cAMP-dependent AGC (PKA, PKG, and PKC) protein kinase super family as they share structural homology within their catalytic domain (Song et al. 2005). The activation of Akt/PKB, a serine/threonine-specific protein kinase, is affected by various factors like insulin, growth factors, cytokines, and cellular stress. The signal transduction pathways regulate diverse biological functions including nutrient metabolism, proliferation, cell growth, transcriptional regulation, and cell survival (Brazil et al. 2004). All AKT isoforms are highly expressed in the nervous system and play important roles in the neuroprotection. The measurement of the active activation is calculated by the ratio of phosphorylated Akt at Ser473 to the total membrane-associated phospho-independent Akt. Active phosphorylated Akt promotes neuronal cell survival by increasing the phosphorylation of downstream substrates including Bad, FOXOs, GSK3β, and caspase-9 via Akt/PKB signaling pathways.
Recently, the evidence that ERK, also known as p42/p44 or MAPK, plays vital role in neural function seeks the immense attention and experimental support. ERK signaling with its involvement in proliferation, differentiation, and survival can lead to the promotion of neural cell death and can be involved in the pathogenesis of neurodegeneration. Thus, ERK can be seen as a unifying motif in the pathogenetic mechanisms of different etiological agents (Colucci-D'Amato et al. 2003).
Neurotrophins
The survival, differentiation, growth, and apoptosis of neurons are mediated by the neurotrophins, by binding to two types of cell surface receptors, i.e., the Trk tyrosine kinases and the p75 neurotrophin receptor (p75NTR). These receptors, often present on the same cell, coordinate and modulate the responses of neurons to neurotrophins. The functions of the neurotrophin receptors vary identically from the sculpting of the developing nervous system to the regulation of the survival and regeneration of injured neurons. It is noticeable that while Trk receptors transmit positive signals such as enhanced survival and growth, p75NTR transmits both positive and negative signals. The signals generated by the two neurotrophin receptors can either augment or oppose each other. Trk and p75NTR thus exist in a contradictory relationship, each acting to suppress or enhance the other’s actions (Reichardt 2006).
The key question in neurotrophin signal transduction is “How the two neurotrophin receptors act together to regulate the responses of cells to neurotrophins, and the nature of the intracellular signals used by these receptors exert their effects” (Saba et al. 2018).
In the past years, the intracellular signaling proteins and signal transduction pathways used by these receptors to promote neurotrophin actions have been identified. The latest finding in neurotrophin signaling, emphasizing on the mechanisms, is used by the neurotrophin receptors to promote neuronal survival and the apoptosis in primary neurons and in vivo systems. A potent activity of neurotrophins, particularly in sympathetic and sensory neurons, is the neuronal survival. Both during development and in culture, the survival of these neurons is absolutely dependent upon a constant exposure to optimal amounts of neurotrophins (Kaplan and Miller 2000).
The first neurotrophin-activated signaling protein shown to mediate survival of these neurons was the small GTP-binding protein Ras. Inhibition of Ras activity decreased survival of most, but not all, populations of sympathetic neurons, whereas increasing Ras activity as a result of deletion of neurofibromatosis-1 (NF-1), a Ras regulatory inhibitor, allowed peripheral neurons to survive in culture in the absence of neurotrophins. Ras, which in most cases is responsible for 40–60% of neurotrophin-dependent survival, does not act directly to promote survival. Rather, it functions by translating and directing neurotrophin-initiated signals into multiple signaling pathways. Recent data indicate that two of these signaling pathways, PI-3K/Akt and MEK/MAPK, are the major effectors of neurotrophin and Ras-activated survival (Kaplan and Miller 2000; Luttrell et al. 1999).
Akt/PKB Pathway
Among the signaling proteins that respond to a large variety of signals, protein kinase B (PKB, also known as Akt) found to be a significant player in regulation of metabolism, cell survival, motility, transcription, and cell-cycle progression. PKB belongs to the AGC subfamily of the protein kinase super family, which consists of 518 members in humans (Manning et al. 2002) which is conserved from primitive metazoans to humans.
In mammals, there are three isoforms of PKB such as PKBα, PKBβ, and PKBγ (Akt1, Akt2, and Akt3, respectively); these are products of separate genes and share a conserved structure that consist of three important functional domains: an N-terminal pleckstrin homology (PH) domain, a central kinase domain, and a C-terminal regulatory domain containing the hydrophobic motif (HM) phosphorylation site [FxxF(S/T)Y] (Hanada et al. 2004). Among the AGC members, both the central kinase domain and the HM are highly conserved. Interestingly, the PH domain, which is essential for binding to lipids such as PIP3, is absent in the S. cerevisiae ortholog (Sch9).
Role of PKB in signaling became noticeable when it was shown to be a downstream component in PI3-K signaling pathway, which is activated upon (Alessi et al. 1996a) auto-phosphorylation of receptor tyrosine kinases induced by various ligands (such as insulin or other growth factors) (Alessi et al. 1997), stimulation of G protein-coupled receptors, or activation of integrin signaling (Fayard et al. 2005; Wymann et al. 2003).
PI 3-kinase is a significant component in the formation of the second messenger PIP3 from PIP2. With the help of its PH domain, it allows the translocation of PKB from the cytoplasm to the plasma membrane. Upon recruited to the plasma membrane, PKB is activated by a multistep process that requires phosphorylation of Thr308 in the activation loop of the kinase domain and Ser473 within the HM of the regulatory domain. The serine/threonine kinase phosphoinositide-dependent kinase 1 (PDK1), once recruited to the plasma membrane by PIP3 through its PH domain, is the kinase responsible for the phosphorylation of Thr308 (Bayascas and Alessi 2005; Stephens et al. 1998).
Phosphorylation of Ser473 is a very vital step in the activation of PKB since it stabilizes the active conformation state (Jacinto et al. 2006). When activated at the plasma membrane, phosphorylated PKB can translocate to the cytosol or the nucleus (Alessi et al. 1996a). Number of PKB-binding proteins has been shown to modulate PKB activity further, in either a positive or negative way. This clearly suggests transient regulation of the kinase by adaptor proteins (Brazil et al. 2002; Song et al. 2005). Furthermore, several phosphatases negatively regulate PKB activity also.
Moreover, PKB activity is negatively inhibited by several phosphatases. The tumor suppressor phosphatase along with tensin homology deleted on chromosome 10 (PTEN) (Abdulkareem and Blair 2013) and the SH2-domain-containing inositol polyphosphate 5-phosphatase (SHIP) (Choi et al. 2002) inhibit PKB activity indirectly by converting PIP3 to PIP2 and PtdIns(3,4)P2. Protein phosphatase 2A (PP2A) and PH domain leucine-rich repeat protein phosphatase (PHLPPα) do so directly by dephosphorylating Ser473 and/or Thr308 on PKB (Dyson et al. 2001). Although PKB is also proposed to be activated in a PI3K-independent manner (Vanhaesebroeck and Alessi 2000), the physiological significance of these findings requires further study (Fayard et al. 2005).
Substrates of Akt/PKB
Akt has only been shown to regulate the survival in neurons, and not in any other response such as neurite outgrowth or differentiation. Therefore, all of the proposed direct targets of Akt activity identified in the past year have been proteins that regulate cell survival in many cell systems: these include Bad, an inhibitor of the Bcl-2 anti-apoptotic protein; procaspase-9, which is cleaved into the pro-apoptotic caspase-9; and Forkhead, a transcription factor that induces apoptosis by increasing levels of Fas ligand (FasL). Akt suppresses apoptosis by phosphorylating the apoptotic protein in the Akt consensus phosphorylation site RXRXXS/T (single-letter amino acid code) in each case. How Akt regulates the activity of its many targets, as well as the mechanisms whereby PI-3K regulates Akt activity, has been extensively discussed (Read and Gorman 2009). Therefore, it is discussed here that whether, Akt phosphorylation of one or more of these targets is responsible for neurotrophin-induced neuronal survival. The first reported target of Akt-mediated survival activity was Bad (Bcl-2-associated death promoter). Phosphorylation by Akt at Ser136 in Bad induced its association with 14-3-3, and prevented it from associating with and inactivating the anti-apoptotic Bcl-2 and Bcl-XL proteins (Orike et al. 2001). Evidence for the importance of Akt-induced Bad phosphorylation is derived from overexpression the experiments in cerebellar neurons, whereby insulin-like growth factor 1 (IGF-1) or constitutively active Akt suppressed the apoptotic activity of wild-type Bad, but not of Bad mutated at Ser136 (van Golen et al. 2001).
These experiments provide compelling evidence for Akt regulation of ectopically expressed Bad, but three other lines of evidence indicate that endogenous Bad phosphorylation might not be important for growth factor-mediated neuronal survival. First, endogenous Bad has not been reported to be phosphorylated in neurotrophic factor-treated neurons, except for increases in brain-derived neurotrophic factor (BDNF)-treated cerebellar neurons (Bonni et al. 1999). Second, analysis of the Bax knockout mice indicates that Bax is the apoptotic Bcl-2 family member that is required for cerebellar neuron cell death (Harris and Johnson 2001). Third, neurons from the Bad knockout mouse do not show alterations in apoptosis (Jiao and Li 2011). The proteolytic cleavage and activation of procaspase-9 was also shown to be effectively inhibited by Akt in vitro and in overexpression experiments. However, the lack of conservation of the Ser196 Akt phosphorylation site in non-human procaspase-9 (Parrish et al. 2013), as well as Akt-induced phosphorylation of endogenous procaspase-9 in neurons, do not support the role for this phosphorylation event in neurotrophic factor-mediated neuronal survival. The third and best candidate for a direct Akt target in neurons is Forkhead 1 (FKHRL1). Genetic studies in C. elegans first indicated that the activity of a Forkhead family member, DAF16 (Dauer-formation-16), which contains an Akt consensus phosphorylation site, was suppressed by Akt (Paradis and Ruvkun 1998). Greenberg’s group then showed that ectopic expression of FKHRL1 mutated at the Thr32 and Ser315 Akt phosphorylation sites increased apoptosis of cerebellar neurons cultured in IGF-1 by 20% (Brunet et al. 1999). Apoptosis induced by FKHRL1 was reduced by inhibition of FasL binding to its receptor, indicating that FKHRL1 stimulates apoptosis, in part, by inducing the transcription of cell death ligands such as Fas. While endogenous Forkhead has not been shown to be phosphorylated by Akt in neurons, the strong genetic evidence for this protein as an Akt target, coupled with the compelling in vitro and non-neuronal cell data showing regulation of Forkhead by Akt phosphorylation (Brunet et al. 1999), makes Forkhead an attractive target for Akt in mammalian neurons.
These results suggest that a signaling pathway consisting of Ras/PI-3K/Akt is the major regulator of neuronal survival. Akt may suppress apoptosis directly by inhibiting the activities of Forkhead or Bad, indirectly by suppressing GSK-3 apoptotic activities (Pap and Cooper 1998), increasing IAP, Bcl-2, or Bcl-XL levels, or by blocking the function of the primary neuronal apoptotic pathway in neurons, JNK-p53-Bax. Akt probably mediates cell survival at a number of levels, depending upon the cell type, target availability, and the requirement for transcriptional or posttranscriptional events to suppress apoptosis (Kaplan and Miller 2000).
Activation of PI3K/Akt
PI3K–Akt pathway seems to be very significant component for mediating neuronal survival under a wide variety of circumstances. The trophic factors such as NGF, insulin-like growth factor I, or BDNF activate a variety of signaling cascades, including the phosphatidylinositol-3-OH kinase (PI3K)–Akt (Akt; protein kinase identified in the AKT virus [also known as protein kinase B]), the Ras–mitogen-activated protein kinase (MAPK), and the cAMP/protein kinase A (PKA) pathways (Schlessinger 2000). These above mentioned pathways contribute to cell survival under certain conditions that depend on the neuronal cell type and the survival factor (Pettmann and Henderson 1998). The survival factors, by binding to their cognate tyrosine kinase receptors, bring out the recruitment of PI3K to the vicinity of the plasma membrane. The catalytic subunit of PI3K generates the phosphoinositide phosphates PIP2 and PIP3 at the inner surface of the plasma membrane. PIP2 and PIP3 lead to the activation of several serine/threonine kinases, including Akt/PKB, serum glucocorticoid-inducible kinase (SGK), ribosomal S6 kinase (RSK), and atypical forms of protein kinase C (PKC) (Vanhaesebroeck and Alessi 2000). Akt is recruited to the inner surface of the plasma membrane via interaction of its pleckstrin homology domain with the phospholipid products of PI3K. The activation of Akt is dependent on phosphorylation, which is achieved at least in part by the protein kinase PDK1 (phosphoinositide-dependent protein kinase-1) at the plasma membrane (Dudek et al. 1997).
Previously, the PI3K–Akt pathway has been found to be adequate and, in some cases, needed for the trophic factor-induced cell survival of a number of neuronal cell types (Vaillant et al. 1999; Yao and Cooper 1995). Ginty and co-workers have suggested that the capacity of neurotrophins to promote neuronal survival requires a functional PI3K–Akt pathway both inside the cell body and in the distal axons that are in contact with the dendrites of target neurons (Kuruvilla et al. 2000). In neurons, the experimental approach used in most studies to show that Akt is necessary for cell survival relies on the expression of dominant-interfering alleles of this protein kinase. This approach has considerable limitations because expressing dominant-negative alleles of Akt most likely affects the activity of other closely related kinases, such as SGK, that are activated by the same upstream kinase, PDK1 (Vanhaesebroeck and Alessi 2000).
Certainly, SGK, a protein kinase related to Akt and activated by PI3K, also takes part in mediating the survival signals in cerebellar granule neurons along with other cell types (Liu et al. 2000). Because the selective disruption of Akt or SGK remains a noteworthy challenge due to the presence of three distinct genes for each kinase, the identification of chemical inhibitors of this family of kinases will be useful in future efforts to describe the specific functions of Akt, SGK, and other PDK1-regulated kinases, such as the RSKs and PKCs (Cohen 1999; Deshmukh and Johnson Jr 1998). The recognition of Akt substrates has been notably aided by the characterization of a consensus peptide motif (RXRXXpS/T) that is preferred by Akt (Alessi et al. 1996b). Database searches suggest that this motif is found in a large number of proteins. Several of these proteins have been shown to be Akt substrates in vitro and in vivo. The important substrate like transcription factors may regulate the expression of components of the cell death machinery, Bcl-2 family members, a regulator of translation (4E BP-1), endothelial nitric oxide synthase, the telomerase reverse transcriptase subunit (Fulton et al. 1999; Gingras et al. 1998; Kang et al. 1999), the tumor suppressor BRCA1 (Altiok et al. 1999), and protein kinases such as Raf (Zimmermann and Moelling 1999), IκB kinase, or glycogen synthase kinase-3 (GSK-3) (Ozes et al. 1999; Romashkova and Makarov 1999). As the PI3K–Akt pathway regulates cell proliferation and metabolism as well as cell survival, though, it will be vital to differentiate which particular Akt targets mediate the neuronal survival effects of Akt (Brunet et al. 2001).
ERK Pathway
The MAPKs are serine/threonine protein kinases that promote a large diversity of cellular functions in many cell types. Three major mammalian MAPK subfamilies have been described: the extracellular signal-regulated kinases 1 and 2 (ERK1/2), the c-Jun NH2-terminal kinases (JNK), and the p38 kinases. There is a widely accepted perception that JNK/SAPK (stress-activated protein kinase) and p38 MAPK promote cell death, whereas ERK1/2 opposes cell death (Subramaniam and Unsicker 2006). However, this view is overly simplistic. A growing number of studies have suggested a death-promoting role for ERK1/2 in both in vitro and in vivo models of neuronal death. A new member of MAPKs that is ERK5 (also called big mitogen-activated kinase 1; BMK1) has been identified recently and implicated in neuronal survival (Cavanaugh 2004). The instant activation of ERK5 was identified in the hippocampal cornuammonis (CA3) and dentate gyrus regions after cerebral ischemia (Wang et al. 2005). In medulloblastoma cell lines, overexpression of ERK5 was shown to promote apoptotic cell death (Sturla et al. 2005). Most of the pharmacological studies implicating ERK1/2 have been carried out using PD98059 or U0126 (which inhibits mitogen-activated protein kinase/ERK kinase (MEK), an upstream activator of ERK1/2). Both of these inhibitors also inhibit ERK5 activation (Nishimoto and Nishida 2006). Therefore, it remains to be seen whether ERK5 is also involved in ERK1/2-implicated cell death paradigms (Subramaniam and Unsicker 2010).
Components of ERK
The activation of the ERK1/2 cascade is typically initiated at membrane receptors, such as receptor Tyr kinases (RTKs), G protein-coupled receptors (GPCRs), ion channels, and others (Naor et al. 2000; Rane 1999). These receptors transmit the signal by recruiting adaptor proteins (e.g., Grb2) and exchange factors (e.g., SOS) that, in turn, induce the activation of Ras at the plasma membranes, or membranes of other organelles. The activated GTP bound Ras then transmits the signal by activating the protein kinases Raf-1, B-Raf, and A-Raf (Rafs) inside the MAP 3K level of this cascade (Kyriakis et al. 1993). This activation occurs by recruiting Rafs to the membranes, where they are then phosphorylated and activated by a mechanism that is not fully understood (Wellbrock et al. 2004). Under specific conditions, other MAP 3K components may participate in the activation of ERK1/2. Examples that depict this are c-Mos, which acts specifically in the reproductive system, the proto-oncogene TPL2, which seems to act in transformed cells, and MEKK1, which may act as a MAP 3K in the ERK1/2 cascade under stress conditions (Gotoh and Nishida 1995; Lange-Carter et al. 1993). The Rafs transmit their signal by phosphorylating the MAPKKs, MEK1/2 (Kyriakis et al. 1992). The MEK1/2 were first identified as ERK1/2 activators, and their study revealed two main proteins with molecular masses of 45 kDa (MEK1) and 46 kDa (MEK2), which share a high degree (more than 85%) of homology on activation (Ahn et al. 1991; Gómez and Cohen 1991; Zheng and Guan 1993).
The composition of these proteins comprises of a large regulatory N-terminal domain containing a nuclear export signal (NES), followed by a catalytic kinase domain and a shorter C-terminal region. MEK1/2 is activated through serine phosphorylation at the MAPKK-typical Ser-Xaa-Ala-Xaa-Ser-Thr motif in their activation loop (residues 218–222 in human MEK1) (Alessi et al. 1994). In turn, MEK1/2 activate their only known substrates, native ERK1/2, that functions as their sole downstream targets, suggesting that the MEK1/2 serve as the specificity-determining components of the ERK1/2 cascade. The MEK1/2 are the only kinases that can be phosphorylated by both regulatory Thr and Tyr residues of ERK1/2, and therefore, they belong to the small family of dual-specificity protein kinases (Dhanasekaran and Reddy 1998). The next components of the cascade, namely ERK1/2, belong to its MAPK level and are important executers of the upstream signals. They are evolutionary conserved kinases that are gene products of ERK1 (MAPK3) and ERK2 (MAPK1). Each gene encodes one main product, which are the primary 44-kDa (ERK1) and 42-kDa (ERK2) proteins. In addition, these genes encode several splice variants, including ERK1b (46 kDa), ERK1c (42 kDa), and others (Yung et al. 2017). The MEK1/2-mediated phosphorylation of ERK1/2 on the Thr and Tyr residues in their Thr-Xaa-Tyr signature motif (Thr202 and Tyr204 in human ERK1) is the mechanism that induces their full activation (Payne et al. 1991). Because of high similarity between ERK1 and ERK2, their function and regulation are similar under most circumstances (Lefloch et al. 2008), although some differences between their functions have been recently elucidated under distinct conditions (Shin et al. 2010). The ERK1/2 is “Pro-directed” protein kinases, meaning that they phosphorylate Ser or Thr residues, neighbors to Pro residues. Pro-Xaa-Ser/Thr-Pro is the most common consensus sequence for substrate recognition by ERK1/2, although Ser/Thr-Pro can serve as a substrate as well (Gonzalez et al. 1991).
There are 200 different substrates of ERK1/2 which have been identified to date; it seems that it is responsible for the induction and regulation of many ERK1/2-dependent processes (Von Kriegsheim et al. 2009; Yoon and Seger 2006). Among the first identified substrates/interactors of the ERK1/2 cascades are the cytoplasmic PLA2, cytoskeletal elements, and the intercellular domains of membranal receptors (Kyriakis et al. 1993; Northwood et al. 1991; Reszka et al. 1995). However, large number of nuclear ERK1/2 targets identified are not less important, including transcription factors such as Elk1, c-Fos, and c-Jun (Chuderland et al. 2008). These latter substrates require stimulation-dependent nuclear translocation of ERK1/2 for their phosphorylation, and this is run by the novel nuclear translocation signal (NTS) on ERK1/2, recently identified (Chuderland et al. 2008). Finally, among the large number of substrates, several MAPKAPKs may further extend the ERK1/2 cascade. The first identified MAPKAPK was RSK in which its phosphorylation by ERK1/2 was one of the main building blocks for the elucidation of the whole signaling cascade (Sturgill et al. 1988). MAPK-interacting protein kinase (MNK) and mitogen and stress-activated protein kinase (MSK) were also identified as MAPKAPKs of the cascade (Deak et al. 1998; Fukunaga and Hunter 1997; Waskiewicz et al. 1997). However, the activation of these kinases is not restricted to the ERK1/2 cascade, as they are also activated by the p38 cascade, mainly under stress conditions. Therefore, it is estimated that a few minutes after extracellular stimulation, the cascade is responsible for not less than 500 phosphorylations throughout the cells. The signals of the ERK1/2 cascade are well-disseminated (Wortzel and Seger 2011).
Activation of ERK
ERK activation has been involved in neural cell survival. The experiments using dominant-negative constructs or MEK inhibitors, carried out both in vivo and in neuronal primary cultures or neuroepithelial cell lines, show that ERK promotes survival by antagonizing cell death (Mazzoni et al. 1999; Xia et al. 1995). ERK shows that it has a protective effect on oxidative stress. In the transgenic mice which express the activated form of Ha-ras gene selectively in neurons, ERK is activated, but differs from the pro-survival kinase Akt-1. At the same time, the chronic activation of the Ras pathway and subsequent ERK phosphorylation turn out to be neuroprotective in these mutants (Guyton et al. 1996). The prevention of degeneration of motor neurons after facial nerve lesion and neurotoxin-induced degeneration of dopaminergic neurons of substantia nigra and their striatal projection is immensely attenuated in the heterozygous transgenic mice and other experimental results show that inhibition of ERK reduces or blocks cytotoxicity and thus brain injuries (such as seizures, cerebral ischemia, trauma) or neurotoxin exposure. Reactive oxygen species (ROS) activates the ERK and its presence reduces ROS-induced cell death (Murray et al. 1998; Sugino et al. 2000). This dual effect of ERK is similar to that described during oxidative stress (Alessandrini et al. 1999). As in the case of ischemia, the noxious stimulus leads to ERK activation may result in survival or death; it depends on different experimental models, such as the duration of ischemia and the animal species (Sugino et al. 2000).
These support that ERK activation alone may not be predictive of the subsequent cellular response, thus rebutting a simplistic parallelism between ERK activation and cell survival or cell death. The impact of glutamate or the neurotoxin 6-OHDA on cultured primary neurons and neural cell lines indicates that the chronic activation of ERK may contribute in the pathogenesis of neurodegeneration. 6-OHDA has long been used to induce dopaminergic cell death and therefore to generate animal models of Parkinson’s disease. Recently, studies using the dopaminergic cell line B65 have demonstrated that 6-OHDA-induced cell death requires chronic ERK activation and that the MEK blocker conferred protection against 6-OHDA cytotoxicity (Kulich and Chu 2001).
On the contrary, in the same cellular system, cell death induced by hydrogen peroxide, which is also capable of a transient ERK activation, is unaffected by MEK blockers, thus showing that the particular ERK-dependent mechanism of neuronal death may be specific to a given toxic agent (Kulich and Chu 2001). Also MPTP, the well-studied neurotoxin largely used in PD experimental paradigms, induces activation of ERK, while its inhibition rescues the affected cell (Gómez-Santos et al. 2002; Langston et al. 1984; Xia et al. 2001). Phospho-ERK was found in aggregates in the substantia nigra of PD patients (Zhu et al. 2002a). Therefore, it has been hypothesized that ERK activation may be critical to neuronal cell death mechanisms in PD. Numerous reports have shown a relationship between increased β-amyloid (Aβ) and ERK activity, suggesting a link between ERK activation and Alzheimer’s disease (AD). β-amyloid peptides are elevated in brain tissue of AD patients and are the principal component of amyloid plaques, a major criterion for postmortem of the disease. Chronic activation of ERK has been found in acute and organotypic hippocampal slides from the transgenic animal model of AD hyper-expressing Aβ. The latter stimulates the a-7 nicotinic acetylcholine receptors, which in turn activate the ERK cascade (Zhu et al. 2002b). In addition, increased phospho-ERK has also been found in brain extracts from AD patients (Russo et al. 2002). Most NDs are characterized by abnormal fibrillary deposition in the CNS, which can be due to accumulation of neurofilaments, tau protein, a-synuclein, or β-amyloid.
Some of these components are cytosolic targets of ERK, thus implicating MAPK pathway in formation/maintenance of such pathological hallmark, and therefore in the noxious events that lead to the specific neurodegeneration (Gerfen et al. 2002; Grazia Spillantini et al. 1998). It is worth noting that changes of neuronal and synaptic plasticity play a crucial role in neurodegenerative processes. For instance, in neurodegeneration involving the basal ganglia, such as HD, human PD or animal models and also during L-DOPA treatment of PD patients, there are important changes of signaling by dopamine D1 and D2 receptor types, with consequent alterations of gene expression and neuronal firing patterns (Calabresi et al. 2001; Simonian and Coyle 1996). As mentioned above, ERK cascade is centrally involved in dopaminergic signaling and, in turn, activates both CREB and AKT-1. The latter are key regulators of neuronal plasticity and survival. Thus, ERK signaling seems to play an important role in regulation of synaptic plasticity, which is oftenly observed disrupted in NDs. Compelling evidence linking chronic ERK activation to neurodegeneration comes from work on glutamate-induced neuronal cell death. It is well known that glutamate, a widespread excitatory neurotransmitter, can also exert noxious effects that lead to cell death by excitotoxicity or oxidative stress by increasing ROS (Simonian and Coyle 1996). DeFranco and co-workers suggested that kinetics and duration of ERK activation, as well as its subcellular localization, may determine whether downstream targets will trigger beneficial or detrimental effects on neuronal cells.
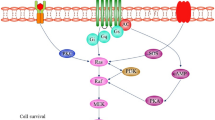
They showed that prolonged exposure of cultured cortical neuron or hippocampal cell lines to glutamate results in prolonged activation of ERK following ROS elevation. MEK inhibitors were capable of blocking the glutamate effect. The prolonged glutamate exposure results in ERK nuclear retention which requires protein synthesis and is also essential for the cell death effects: when nuclear retention is impaired by cycloheximide, glutamate-induced cell death is impaired, although ERK activation remains unaffected (Stanciu and DeFranco 2002; Stanciu et al. 2000). Thus, in both PC12/MEN2 cells and in the glutamate-treated neurons, chronic activation of ERK is accompanied by alteration of its nuclear import/export, although the biological functions assessed and the cellular models were different. Furthermore, the altered ERK compartmentalization appears crucial for the biological effects observed, such as NGF unresponsiveness in PC12 cells and cell death in CNS neurons. The mechanism(s) subserving the compartmentalization of ERK within the cells remains still unknown, although its importance in physiology and pathology appears crucial (Colucci-D'Amato et al. 2003). The following figure explains regarding Akt/Erk pathways in the area of neurodegenerative disease (Fig. 1).

Akt/PKB activation and regulation by receptor tyrosine kinase (RTK) via phosphoinositide-3-kinase (PI3K) pathway. Ligand binding cause dimerization and activate kinase activity of RTK by cross-phosphorylation of tyrosine residue in the cytosolic domain. SH2 domain of p85 (regulatory subunit of PI3K) binds to phosphorylated tyrosine in RTK and activates the kinase activity of SH3 domain. Activated p85 binds to the p110 (catalytic subunit of PI3K) and forms an active PI3K enzyme. Active PI3K causes phosphorylation of PIP2 by adding phosphate group to 3′-OH position of inositol ring and forms PIP3. PH domain of Akt/PKB and PDK1 has high binding affinity to PIP3. Akt binding to PIP3 via PH domain causes conformational change and results in the exposure of phosphorylation sites (Thr308 and Ser473). mTORC2 acts as PDK2 molecule and partially activates Akt/PKB by phosphorylating Ser473 as well as Ser450. Ser phosphorylation stimulates Thr 308 phosphorylation by PDK1 which leads to the full activation of Akt/PKB. Active Akt/PKB phosphorylates its downstream target substrates and regulates diverse biological functions. PI3K-Akt/PKB pathway is negatively regulated by PTEN (phosphatase and tensin homolog deleted on chromosome 10), TRB3 (tribble homolog 3), CTMB (carboxy-terminal modulator protein), and PP2A (protein phosphatase 2A). Active Akt/PKB causes inhibitory phosphorylation of Bad (S136), GSK3β (Y216), TSC1/2(S), and RAF (S259). It also inhibits the nuclear translocation of FKHRL1 and promotes NFκB translocation to the nucleus by phosphorylating IκB. Shc, Grb2, and SoS binding to RTK promote RAS to bind with GTP (active). Active Ras-GTP activate RAF which then activate MEK. MEK activate ERK by phosphorylation. ERK inhibit caspase-8 and active GSK3β. Direct inhibition of GSK3β by Akt and ERK prevents cell from apoptosis and promotes cell survival
Neurodegeneration
It is reported that in neurodegenerative disorders including AD and PD, the PI3K/AKT pathway is altered, leading to autophagy disruption. The activity of the PI3K/AKT/mTOR pathway suppresses the autophagic process and is commonly related to pro-survival effects, and the suppression of mTOR activity promotes autophagy and it has been shown to be protective for neuronal cells, thus indicating that a coordinate mechanism is involved in neuroprotection (Jacinto et al. 2006). Most importantly, the presence of AP in neuronal tissue is suggestive of disrupted autophagy. Autophagy suppression in animal models shows the similar histopathological characteristics, depending upon the disease and duration of the disorder; autophagy may or may not be associated to PI3K/AKT/mTOR pathway disruption. It is important to understand the interplay between pro-survival pathways and the autophagic process to understand the role of mTOR complexes as sensors of cellular environmental cues, to activate or to inhibit downstream and upstream molecular effectors. The negative feedback loop of the PI3K/AKT/mTOR pathway may be involved in the switching between activation of pro-survival effectors and induction of constitutive autophagy that may be involved in neuroprotective effects (Griffin et al. 2005).
Disruption of the molecular effectors of the negative feedback loop of the PI3K/AKT/mTOR pathway may have impact on cell death with several outcomes. For instance, blocking the PI3K/AKT pro-survival signal at the mTOR level would mainly compromise cellular growth and structure, and promote increased autophagy. On the other hand, blocking the S6K/IRS pathway would ultimately close the loop by stopping mTOR activity, which would increase mTOR activity and thus affect constitutive autophagy, allowing abnormal protein aggregation and creating a toxic environment. In both cases, the presence of amyloid proteins (APs) must be increased which would finally lead to cell death; however, the trigger mechanisms are different, making it difficult to select one approach to protect neuronal tissue (Elstner et al. 2011).
Importantly, for neurodegenerative diseases accompanied by cognitive deficits, it must be contemplated that PI3K/AKT/mTOR pathway activation is important for synaptic activity and normal neuronal functions. Thus, a desirable compound aiming to protect neuronal tissue must therefore act in two ways: stimulating or sustaining the normal PI3K/AKT/mTOR survival pathway and promoting constitutive autophagy. Most compounds that induce autophagy promote clearance of aggregated proteins and induce neuroprotection, downregulating the PI3K/AKT/mTOR pathway or inhibiting mTOR. Therefore, evaluating its role in synaptic functions and behavior is relevant to integrate the molecular events associated to neuroprotection and the functional parameters in vivo (Heras-Sandoval et al. 2014).
HDAC3 Neurotoxicity: Regulation by Akt and GSK3β
It is well established that pharmacological inhibitors of classical HDACs are protective in various cell culture and in vivo models of neurodegenerative disease; the identity of the neurotoxic HDAC(s) targeted by these inhibitors to exert their protective effects is less clear. It has been shown that HDAC3 has strong neurotoxic activity and therefore represents a potential target of HDAC inhibitors in ND-related animal models (Borsello and Forloni 2007). HDAC3 is a potent stimulator of neuronal death when overexpressed; CGNs and cortical neurons express HDAC3 normally. Moreover, its expression is not increased when these neurons are induced to die. Yet the suppression of endogenous HDAC3 expression protects neurons from death. This suggests that under normal conditions, the apoptotic activity of HDAC3 is kept in check by survival-promoting signaling molecules. Studies proposed that in neurons primed to die, the levels or activities of these survival-promoting molecules are reduced, permitting HDAC3 to induce neuronal death. The results suggest that Akt is such a survival-promoting signaling molecule and expression of an active form of Akt or treatment with IGF-1, a physiological activator of Akt, protects against the neurotoxic effect of HDAC3 (Morrison et al. 2006).
A well-established target of Akt is GSK3β. Inhibition of GSK3 β either pharmacologically or caused by expressing a dominant-negative form of the kinase protects against HDAC3-mediated neurotoxicity. Activation of GSK3 β has been implicated in the pathogenesis of different neurodegenerative diseases, and the inhibition of this kinase has been considered a therapeutic approach in the treatment of these disorders (Hernandez et al. 2009). Despite the long-standing acceptance that GSK3 β plays a key role in promoting neuronal death, little is known about what its substrates are in the context of neurodegeneration. It has been shown that HDAC3 is directly phosphorylated by GSK3 β, suggesting that the neurodegeneration-promoting effect of GSK3 β could be mediated through the phosphorylation of HDAC3. The specific residues within HDAC3 that are phosphorylated by GSK3 β remain to be delineated (sequence analysis of HDAC3 reveals three consensus sites for GSK3 β phosphorylation). More work is also needed to determine the downstream mechanism by which phosphorylation of HDAC3 promotes neurotoxicity. An obvious possibility is that GSK3 β-mediated phosphorylation stimulates the deacetylase activity of HDAC3, resulting in the deacetylation of specific proteins that regulate neuronal death.
Although GSK3 β is activated in neurons primed to die (Forlenza et al. 2011), and that active GSK3 β phosphorylates HDAC3, and that the inhibition of GSK3 β protects against HDAC3 toxicity, we have not found a strict correlation between the total levels of Akt or GSK3 β or their activity (evaluated indirectly by looking at phosphorylation status) and vulnerability to HDAC3 toxicity in the cell. This suggests that activation of GSK3 β is by itself not sufficient to explain HDAC3-induced toxicity, pointing to the involvement of other cell-specific components and mechanisms. For example, the accessibility of HDAC3 to GSK3 β could be regulated differently in different cell types. It is also possible that the toxic activity of HDAC3 is dependent on the participation of another molecule that is expressed selectively in neurons, or that a molecule capable of blocking the toxic activity HDAC3 could be missing in neurons. A study by Farah et al. has produced three important findings. First, we have identified HDAC3 as a protein with neurotoxic activity. Although protection by HDAC inhibitors in experimental models of neurodegeneration has pointed to the existence of neurotoxic proteins within the HDAC family, conclusive identification of such a protein has been elusive. Second, we provide evidence suggesting that IGF-1, a trophic factor for many neuronal populations in vivo, promotes neuronal survival at least in part by suppressing the neurotoxic activity of HDAC3. Finally, we show that HDAC3 is a substrate of GSK3 β. Although the involvement of GSK3 β in promoting neurodegeneration both in experimental models and human pathologies is well accepted, the downstream mechanism by which this kinase promotes neuronal death is poorly understood. Evidence suggested that HDAC3 represents a downstream effector of GSK3 β neurotoxicity (Bardai and D'Mello 2011).
Neuroprotection: ERK and PI3K/Akt Pathway
It is identified that acteoside, an anti-oxidative phenylethanoid glycoside is a new activator of the transcription factor Nrf2 and the inducer of HO-1 expression. One of the most salient features of this study is that acteoside activates Nrf2-mediated-HO-1 induction through ERK1/2 and PI3 K-Akt pathways, thereby protecting the PC12 cells from Aβ 25–35-induced oxidative neurotoxicity. Study revealed that the acteoside is an activator of Nrf2. Recently, it has been hypothesized that Nrf2-ARE activation is a novel neuroprotective pathway that confers resistance to a variety of oxidative stress-related, neurodegenerative insults (Vargas et al. 2008). Using many chemical activators, the activation of the Nrf2-ARE pathway has been studied in a variety of tissues and cell types. For example, tertbutyl hydroquinone (tBHQ) was shown to have displayed cytoprotective efficacy against toxins in neuroblastoma cells and primary neuronal culture systems (Jakel et al. 2007) in vitro and in vivo.
Kanninen et al. found that it provides a direct evidence that the Nrf2-ARE pathway may be involved in the neuropathology of AD, and activation of this endogenous antioxidant pathway provides protection against the toxicity of Aβ peptide (Kanninen et al. 2008). Itoh et al. showed that acteoside efficiently increases the nuclear levels of the Nrf2. The core sensor of stress is the cytosolic Keap1-Nrf2 complex. In response to oxidative stress, Nrf2 is released from Keap1 and transmits the stress signal to the nucleus for activation of distinct set of genes encoding phase II detoxifying enzymes as well as several stress responsive proteins including hemeoxygenase-1 (HO-1) (Itoh et al. 1999). Several evidences suggest that HO-1 has neuroprotective effects against oxidative stress-induced neuronal damage (Bae and Schlessinger 2010). HO-1 can be highly upregulated during the formation of neurofibrillary tangles in Alzheimer’s brain. Particularly interesting is the role played by HO-1 in AD, which is associated with both oxidative brain injury and Aβ-associated pathology (Calabrese et al. 2010).
The significant increases in the levels of HO-1 have been observed in AD brains in association with neurofibrillary tangles, and HO-1 mRNA was found to be increased in AD neocortex and cerebral vessels; the HO-1 increase also co-localized with senile plaques and glial fibrillary acidic protein-positive astrocytes in AD brains. The previous findings support the importance of HO-1 factor in the response to oxidative injury, in protecting neurons against Aβ-induced oxidative stress-dependent injury (Wang et al. 2010). The protective role played by HO-1 in AD raised new possibilities regarding the possible use of natural substances, which are able to increase HO-1 levels, as potential drugs for the prevention and treatment of AD. The present study has illustrated that acteoside induces HO-1 in vitro and in vivo. Treatments of the PC12 cells with acteoside resulted in an increase in HO-1 protein expression. The acteoside-induced HO-1 expression requires Nrf2 activation, because the acteoside-induced expression of HO-1 was markedly suppressed by siRNA knockdown of Nrf2 gene. Andreadi et al. suggested that ERK and PI3 K/Akt are part of a central pathway involved in Nrf2 activation and translocation for highly specialized protein synthesis including HO-1 (Andreadi et al. 2006). In order to identify the signaling pathways used by acteoside to activate Nrf2 and induce HO-1 expression, Andreadi et al. tested to determine whether acteoside-induced expression of HO-1 occurs through a specific MAPK and PI3 K/Akt pathway. It is found that acteoside activated the ERK MAPK cascade, and PI3 K/Akt, but did not activate JNK and p38MAPK. In addition to this, use of specific inhibitors for ERK1/2 and PI3 K/Akt pathways confirmed the involvement of these two pathways in acteoside-induced HO-1 expression. In accordance with the postulation that an elevation of HO-1 by various stimuli may be a protective cellular response to delay the cell death and HO-1 play a role in protecting neurons against Aβ-induced oxidative stress (Komatsu et al. 2010).
The protective effects against Aβ-induced cytotoxicity by acteoside were greatly reduced by ZnPP. In order to determine the potential role of HO-1 in the Aβ-induced PC12 cell damage and acteoside-mediated neuroprotection, these results suggest that acteoside-stimulated HO-1 may protect PC12 cells against the Aβ-induced cytotoxicity. To determine whether such activation of ERK and PI3 K/Akt could contribute to the acteoside-mediated protection against the cytotoxic effect of Aβ 25–35, pharmacological inhibitors of these two kinases were utilized. Acteoside-mediated cytoprotection against Aβ 25–35-induced cytotoxicity was attenuated by PD98059 and LY294002. Altogether, these results demonstrate that acteoside is an activator of Nrf2 and inducer of HO-1 expression. Acteoside attenuates Aβ 25–35-induced neurotoxicity by induction of HO-1 via ERK and PI3 K/Akt signaling. These results shed some light on the mechanisms whereby acteoside protects against β-amyloid-induced cytotoxicity (Wang et al. 2012).
Astrocytes Protection by BDNF
BDNF that has impact on neuronal survival, synaptic activity, learning, memory, and neurogenesis are well documented (Binder and Scharfman 2004). Very little is known about BDNF role in astrocytes. BDNF prevents apoptotic cell death of rat astrocytes induced by two strong apoptotic stimuli such as SD (succinate dehydrogenase) and 3-NP (3-nitropropionic acid). BDNF increases astrocyte viability after SD as a consequence of apoptosis reduction. The mechanisms of BDNF protection include the reduction in p53 and active caspase-3 expression induced by SD. Tallying with this, it was recently shown that BDNF increases cell survival and reduces caspase-3 levels induced by amyloid-β in cultured neurons and by ischemic injury in rat cortex and hippocampus. Although, neurotrophin cocktail (BDNF, NT-3, and NT-4) reduces phosphorylation of p53 in cerebral cortical neurons (Jerónimo-Santos et al. 2015).
BDNF exerts its effects mainly through TrkB receptors. Of the TrkB isoforms, TrkB-FL and TrkB-T2 are those mainly expressed in neurons. Expression of TrkB-T1 in astrocytes is high and some reports have shown TrkB-FL expression. Studies showed that TrkB-T1 is highly expressed in astrocytes and that although TrkB-FL mRNA is present, TrkB-FL protein is undetectable in these cells, in line with several other reports (Aroeira et al. 2015; Binder and Scharfman 2004; Lebrun et al. 2006). TrkB-FL can also be cleaved by calpain protease to elicit a truncated TrkB-FL isoform very similar to TrkB-T1 (Vidaurre et al. 2013). Therefore, it is possible that TrkB-FL is expressed but may be cleaved to a truncated TrkB-FL isoform that may not be functional. It remains to be determined whether this cleavage occurs in astrocytes. BDNF reduction in astrocyte apoptosis was blocked by the TrkB selective antagonist ANA-12 and by broad-spectrum Tyr kinase inhibitor K252a. In the adult rat central nervous system, p75NTR expression was described as being very low in; p75NTR mRNA expression was negligible in experimental conditions. The fact that TrkB inhibition with ANA-12 blocked BDNF effects, plus the very low expression of p75NTR in our culture system indicates that p75NTR is not likely to be involved in BDNF action in astrocytes. TrkB-T1 mediates BDNF-induced internalization of glycine transporters in astrocytes and modulates GABA transporters. A recent report describes that TrkB-T1 knockout mice-derived astrocytes exhibit decreased migration and proliferation in vitro (Morris et al. 1994). In line with these reports, Tejeda, G.S., et al. results suggest involvement of TrkB-T1 in BDNF protective effects, and show for the first time the protective role of astrocyte TrkB by mediating BDNF effects (Tejeda et al. 2016).
BDNF protective mechanism of action involves ERK, Akt, and, the non-receptor tyrosine kinase, Src activation in astrocytes. BDNF was reported to increase pERK and pAkt in primary cortical astrocytes, to modulate GABA transporters through ERK and Akt pathways, and to activate Src in cortical neurons through TrkB-FL (Huang et al. 2013). Huang et al. have shown for the first time that in astrocytes BDNF activates Src and also demonstrated that ERK activation is mediated by Src in astrocytes. Therefore, ERK and Akt activation by BDNF may depend on Src in astrocyte, which is likely to contribute to cell survival. K252a is a non-specific tyrosine kinase inhibitor that inhibits Trks and BDNF-induced ERK activation. Roback et al. reported that K252a inhibited BDNF-induced MAPK activation in glial cells. K252a completely inhibited BDNF-induced ERK and Src activation in astrocytes as well as the protective effect of BDNF on SD-induced cell death (Menzies et al. 2017).
This effect is not because of inhibition of TrkB-FL since Hoover et al. demonstrated that astrocytes do not express TrkB-FL protein. K252a was also described to inhibit MAPKs. The results indicate that, in the absence or at low levels of TrkB-FL, K252a can affect other cellular targets such as MEK or Src kinases, resulting in inhibition of BDNF-induced ERK activity. TrkB-T1 was shown to increase calcium in astrocytes, whereas TrkB-FL did not. Concordantly, BAPTA markedly inhibits both ERK and Src activation by BDNF, suggesting that TrkB-T1 can activate intracellular signaling pathways through calcium (Hoover et al. 2010).
Although, K252a partially inhibited Src activation induced by BDNF; BAPTA had a more robust effect, thus establishing calcium as a central signaling molecule in TrkB signaling in astrocytes. Therefore, activation of calcium-Src-ERK-Akt signaling pathways by TrkB-T1 mediates protection of astrocytes by BDNF. These results corroborate with Vaz et al. who showed that BDNF enhances GABA transport in astrocytes through TrkB-T1 coupled to ERK/MAPK pathway. In contrast, BDNF acting on TrkB-T1 inhibits glycine uptake in astrocytes not involving ERK or Akt pathways (Aroeira et al. 2015). For past many years, research has focused on neuronal death without taking the role of glial cells into account in the neurodegenerative diseases. In fact, dysregulation of astrocytes functions is a common component of many neurodegenerative diseases (Liddelow et al. 2017) supporting that astrocytes can be targeted for treating neurodegeneration. The expansion of CAG repeats in Htt protein caused HD is a neurodegenerative disorder. Mitochondrial dysfunction is observed in HD patients and systemic administration of 3-NP induces HD-like symptoms in animals and humans. Thus, 3-NP administration is a widely used model that resembles HD. HD patients and animal models showed astrogliosis (Glass et al. 2010) and reduced glutamate transporter GTL-1 expression (Ross and Akimov 2014). Nevertheless, little is known about effects of HD on astrocyte physiology. Mitochondrial dysfunction induces astrocyte death and that BDNF is able to reduce it. Moreover, our data show that TrkB-T1-mediated activation of calcium-Src-ERK pathway mediates BDNF protection, further highlighting the importance of TrkB in astrocytes.
Ruiz et al. in vitro studies and Perucho et al. in vivo studies showed that Glial CM is rich in antioxidants and neurotrophic factors. Studies showed that fetal glial CM protects a striatal cell model of HD from death induced by 3-NP. Casarejos et al. showed that astrocyte-conditioned medium (ACM) from postnatal control astrocytes, though protective, cannot fully rescue neurons from 3-NP-induced cell death. This difference may be because of due to the fact that fetal astrocytes and postnatal astrocytes are different (Casarejos et al. 2013). BDNF shows antioxidant effect in astrocytes whereas ERK, calcium, and Src inhibition prevent BDNF anti-apoptotic effect on astrocytes, inhibition of none of them could prevent BDNF-induced reduction in ROS levels. This says that other unidentified mechanisms may mediate BDNF antioxidant action. Moreover, Shaughnessy et al. reported that BDNF increases expression of xCT subunit of system Xc- that imports cystine, essential to produce GSH. xCT light chain is the specific subunit, while 4F2 heavy chain is the promiscuous subunit of system xCT. Decreased xCT expression was shown in the striatum of HD mice and in neurons expressing mHtt (Shaughnessy et al. 2014). BDNF increases GSH intracellular levels and xCT expression in astrocytes. Higher GSH levels most likely contribute to ROS neutralization by BDNF. Nrf2 regulates the cell’s antioxidant response inducing expression of antioxidant enzymes, and neuroprotection.
In neurons, BDNF preconditioning was shown to exert an antioxidant action not involving Nrf2 (Faden et al. 2016). However, neurons depend on astrocyte-derived antioxidant factors to survive and lower level of Nrf2 than astrocytes. Also, since the GSH system of peroxide detoxification in neurons is less efficient than that in astroglial cells. Astrocytes fulfill a protective function for neurons through ROS elimination and GSH from astrocytes was shown to be essential for neuroprotection. BDNF exerted a protective effect in astrocytes through TrkB-T1, involving calcium-Src-ERK and Akt pathways, and a potent antioxidant action, probably through induction of Nrf2. Moreover, BDNF-treated astrocytes release protective factors that prevent death and oxidative stress in neurons. We hope that this work will encourage further studies on glial cells as therapeutic targets for neurodegenerative disorders (Saba et al. 2018).
VEGF Activates PI3K/Akt and ERK
Currently, Slater et al. have investigated that VEGF prevents MPP+-induced apoptosis in primary CGNs and the key signaling pathways are involved. It shows that in response to MPP+ in CGNs, VEGF activates both the PI3-K/Akt and ERK pathways, and these two pathways play opposite roles in the prevention of neuronal apoptosis, i.e., activation of the PI3-K/Akt and ERK pathways implicated in neuroprotection and neurotoxicity by VEGF against MPP+, respectively (Slater et al. 1996). VEGF reversed the inhibition of VEGFR-2 and a VEGFR-2-specific inhibitor abolished the protection of VEGF, indicating that VEGF blocked neuronal loss via acting on VEGFR-2 in our model. VEGFR-2 is critical for VEGF-mediated angiogenesis, proliferation, migration, and survival of endothelial cells. VEGF also protects neurons against death induced by a wide variety of insults, including hypoxia, serum withdrawal, and excitotoxic stimuli, mainly via activating VEGFR-2. Evidences support the finding that VEGFR-2 stimulation is linked to neuronal protection (Cui et al. 2011b).
Endogenous VEGF binds to the extracellular domain and induces dimerization and auto-phosphorylation of VEGFR-2 at tyrosine sites. Therefore, the factors capable of regulating the translation and/or transcription of VEGF may affect the endogenous level of VEGF and subsequently change the phosphorylation level of VEGFR-2. For example, hypoxia-inducible factor 1a (HIF-1a), a key factor in neuronal survival, can increase the endogenous level of VEGF by promoting its mRNA expression. Recent studies have shown that MPTP downregulates the expression of HIF-1a in vivo and MPP+ reduces the protein level of HIF-1a by enhancing its degradation in SH-SY5Y cells, suggesting that MPP+ may decrease the phosphorylation of VEGFR-2 by reducing the expression of endogenous VEGF, possibly through an HIF-1a-dependent mechanism (Slater et al. 1996). The PI3-K/Akt and ERK pathways are two key signaling pathways involved in the protection led by the activation of growth factor receptors. Some neurotrophic factors, including nerve growth factor and brain-derived neurotrophic factor, prevent neuronal cells from MPP+-induced apoptosis via activating the PI3-K/Akt pathway (Wick et al. 2002).
VEGF attenuated MPP+-induced neurotoxicity from activating the PI3-K/Akt signaling pathway confirm the importance of the PI3-K/Akt pathway in the protection against MPP+. GSK3β, inhibited by the activated Akt, is reported to be an important mediator in MPP+-induced neurotoxicity. Activation of GSK3β facilitates mitochondrial failure, and inhibiting the activity of GSK3β prevents neuronal loss from suppressing pro-apoptosis proteins such as p53 and caspase-3. The activation of GSK3β is induced by MPP+ and is inhibited in response to VEGF stimulation, and the GSK3β-specific inhibitor can prevent the neurotoxicity induced by MPP+. Together, these findings suggest that GSK3β mediates the neurotoxicity of MPP+ and inactivation of GSK3β is involved in the protection of VEGF (Petit-Paitel et al. 2009). The ERK pathway is activated by stimuli that are either pro-survival such as growth factor stimulation or pro-apoptosis such as oxidative stress. In our study, MPP+ increased the activation of ERK as shown by the increased phosphorylation of ERK1/2. Moreover, PD98059 reduced neuronal death caused by MPP+, suggesting that ERK activation may play a pro-apoptosis rather than pro-survival role in parkinsonian model. PD98059 binds to the inactive form of MEK and prevents the activation of the ERK pathway induced by MPP+. It is reported that the activation of the ERK pathway is involved in oxidative stress-induced apoptosis and results in the induction of a variety of pro-apoptotic factors. As a result, the activation of the ERK pathway promotes ROS production, increases degradation of specific proteins, and enhances expression of inappropriate cell cycle-related. Therefore, PD98059 might increase cell survival via inhibiting the activity of various pro-apoptotic factors. The increased activity of ERK and decreased activity of Akt are observed not only in the in vitro and in vivo PD models, but also found in the brain of PD patients (González-Polo et al. 2003). These alterations occur relatively early in the disease progress. Several studies have revealed that reversion of either alteration may cause partial neuroprotection. For example, pharmacological MEK inhibitor lessens the neurotoxicity in neuronal cell lines and in primary midbrain DA neurons, lithium, the inhibitor of GSK3β, possesses limited therapeutic benefits for PD. These may be explained by the insufficiency of regulating only one pathway to stop neuronal loss (Cuny 2009). Zhu et al. have found that VEGF alone cannot fully protect, but co-application of VEGF and PD98059 synergistically prevent neuronal death induced by MPP+. Moreover, MPP+-induced neuronal apoptosis can be fully protected by simultaneously reversing the inhibition of the PI3-K/Akt pathway and the activation of the ERK pathway (Zhu et al. 2007). These findings not only indicate a therapeutic usage of co-application of VEGF and the agents that inhibit the ERK pathway, but also provide a therapeutic potential of regulating different signaling pathways in treating PD. It could be expected that co-application of agents capable of inhibiting the ERK pathway and activating PI3-K/Akt pathway or using multi-functional drugs capable of regulating these pathways simultaneously might have therapeutic significances in the treatment of PD (Cui et al. 2011a). The following table explains the research conducted in the area of neurodegeneration (Table 1).
Role of PI3K/Akt and ERK in Clearance of Inclusion Bodies Through Autophagy in Neurodegenerative Diseases
PD is characterized by the presence of specific inclusions called Lewy bodies (LBs) in neurons of several regions of brain. LBs mostly composed of misfolded proteins such as α-synuclein, tubulin, microtubule-associated proteins, ubiquitin, amyloid precursor protein, synaptic vesicle proteins, various enzymes, and chaperons/co-chaperons. Several mitogen-activated protein kinases like Akt/Erk found in cytosol as well as in intracellular compartment like endoplasmic reticulum and mitochondria have been involved in the removal of these inclusions through phosphorylation and dephosphorylation activities (Bohush et al. 2018).
The major cause of neuronal death in PD is oxidative stress. Various studies suggested that the neurotoxins like 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) or 6-hydroxydopamine (6-OHDA) elicit PD-like symptoms in related animal models. It has also shown that these neurotoxins result in the activation of microglial cells due to ROS generation consequently attacking neighboring dopaminergic neurons. α-Synuclein promotes inflammation via activating p38, ERK, and JNK pathways in human microglial cells, resulting in the production of IL-1β and TNF-α. α-Synuclein promotes chronic inflammation by inducing IL-6 and intercellular adhesion molecule-1 (ICAM-1) expression in human astrocytes. The activation of MAPK signaling pathways is associated with the enhanced expression of these two proteins. Furthermore, α-synuclein regulates the neuronal apoptosis by releasing cytochrome c from mitochondria and increasing mitochondrial oxidative stress (Kim and Choi 2010).
Studies reveal that oxidative stress in AD patients promotes hyperphosphorylation of tau mediated by several kinases including JNK, p38, and ERK in neurofibrillary tangles. Aβ42 mediate APP dimerization resulting in tau phosphorylation by activating ASK1-MKK6-p38 signaling pathway. As a result of a neural injury, ASK1 induces either neuronal apoptosis or neurite outgrowth by forming a signaling complex with APP, MKK6, JIP1, and JNK1. Production of ROS and pro-inflammatory cytokines (TNF-α and IL-1β) is the result of microglial activation induced by Aβ42 aggregation, all of which consequently stimulates MAPK signaling pathways. Oxidative stress activates JNK and p38 and induces β-secretase gene expression, whereas β-secretase gene expression is negatively regulated by ERK1/2 (Kim and Choi 2010).
In motor neurons and microglia, aberrant expression and activation of p38 MAPK are thought to be important for ALS progression. In transgenic mice expressing mutant SOD1, G93A, degeneration of motor neurons is caused by persistent activation of p38. Moreover, mutant SOD1 induces apoptosis of motor neurons which is prevented by SB203580, a p38-MAPK inhibitor. Through aberrant phosphorylation and consequent aggregation of neurofilaments, both p38 and JNK1 are also implicated in cytoskeletal abnormalities of spinal motor neurons, a feature of ALS (Kim and Choi 2010).
A major family of signaling proteins, mitogen-activated protein kinases (MAPK), regulates neuronal survival, differentiation, and plasticity. Particularly, the extracellular signal-regulated protein kinase (ERK) is involved in neuronal development, hippocampal learning, and survival. Recently, it has been studied that ERK signaling may respond in neuronal stress. Inhibitor of an upstream kinase that activates ERK, MEK, confers significant protection in animal models of cerebral ischemia-reperfusion. In addition, suppression of phosphorylated ERK protects neuronal cell lines and primary neuronal cultures subjected directly to oxidative stress. In conclusion, studies highlight a potentially beneficial role for ERK signaling in oxidative neurotoxicity (Zhu et al. 2002a).
Huntington’s disease is caused by huntingtin protein, an expanded polyglutamine repeat, which actuates a diverse set of pathogenic mechanisms. A mutation in huntingtin causes activation of ERK and directs a protective transcriptional response along with inhibiting caspase activation. In contradiction, Htt also interferes with several signaling events of the ERK pathway. Mutant huntingtin compromises the ERK-dependent transcriptional response to cortico-striatal BDNF signaling. It also hinders glutamate uptake from the synaptic cleft by downregulating ERK-dependent glutamate transporter expression and leaving cells susceptible to excitotoxicity. Some of this cellular complexity can be used to achieve selective ERK activation which can be neuroprotective (Bodai and Marsh 2012).
Previous studies conducted in human neuroblastoma SK-N-SH cellular model of PD suggested the role of p38 MAPKs in autophagy since autophagy disorders are more commonly associated with neurodegenerative diseases. A study revealed that microRNA (miR)-181a causes inhibition of p38 MAPK/JNK pathway resulting in regulation of apoptosis and autophagy (Bohush et al. 2018).
Autophagy, a lysosome-dependent intracellular degradation process, plays an important role in maintaining homeostasis as it helps in recycling the cytoplasmic constituents into bio-energetic and biosynthetic materials. As autophagy process is related to different stress conditions, aberrant function of the process can cause cellular dysfunction and diseases. Degradation of accumulated abnormally misfolded proteins, the common cause of neurodegenerative disorders such as AD, PD, HD, and ALS can be efficiently done through autophagy. Defects in autophagy have been observed in different cases of neurodegenerative disorders as reported by recent studies. Also, neurodegeneration is seen to be associated with deregulated excessive autophagy. Therefore, normal functioning of autophagy is necessary for its implication in the treatment of neurodegenerative diseases and various autophagy-regulating compounds are being developed for therapeutic purposes (Nah et al. 2015).
Evidences from several studies have suggested the importance of autophagy in the healthy development of neurons. Researchers have revealed several links that associate autophagy with neurodegenerative diseases. However, direct links and molecular mechanisms remain is required to be further investigated. Lysosomal function impairment and fusion of autophagosome/lysosome are observed in most neurodegenerative disorders. Hence, it is very important to overcome lysosomal dysfunction for possible therapeutic strategies in neurodegenerative diseases. Nevertheless, the treatments implicated for treating abnormally deregulated autophagy in neurodegeneration targets on its initial stages and the research based on this is at its preliminary stage. The clinical trials for some compounds for the treatment of AD have been done in humans and the compounds being used for the treatment of other neurodegenerative diseases are now in the preclinical phase. Despite research limitations, targeting autophagy for the treatment of neurodegenerative diseases are highly expected (Nah et al. 2015).
Conclusion
Neurotrophins regulate neuronal survival and apoptosis at several levels. Trk uses at least two mechanisms, Ras/PI 3K/Akt-induced suppression of apoptotic proteins and pathways, and MEK/MAPK activation of anti-apoptotic proteins, to stimulate survival. p75NTR can potentiate Trk activity through the activation of NF-kB. Trk, through Ras and probably PI-3K/Akt, can interfere with p75NTR-induced apoptosis by suppressing the JNK–p53–Bax pathway upstream of JNK, or by inhibiting the activities of cell death proteins such as Forkhead. p75NTR, in turn, can suppress Trk-induced cell survival and growth pathways, possibly through ceramide-mediated inhibition of Akt and Raf activities. This functional crosstalk between Trk and p75NTR signaling pathways appears to be a key process in determining how the nervous system develops and is repaired following injury (Kaplan and Miller 2000). Dysregulation of the ERK1/2 cascade is known to result in various pathologies, inducing neurodegenerative diseases. Therefore, interference with the localization of certain components, and in particular with the translocation of ERK1/2 to the nucleus, may serve as potential therapeutic targets for some diseases (Wortzel and Seger 2011).
Recently, the evidence for ERK involvement also known as p42/p44 or MAPK plays vital role in neural function seeks the immense attention and experimental support thereby ERK can be seen as a unifying motif in the pathogenetic mechanisms of different etiological agents (Colucci-D'Amato et al. 2003). PI3-K/Akt and ERK pathways play opposite roles in the prevention of neuronal apoptosis, i.e., activation of the PI3-K/Akt and ERK pathways implicated in neuroprotection and neurotoxicity by VEGF. These findings not only indicate a therapeutic usage of co-application of VEGF and the agents that inhibit the ERK pathway, but also provide a therapeutic potential of regulating different signaling pathways in treating NDs (Cui et al. 2011a).
Autophagy disruption is also involved as shown in the brain of patients and in animal models the PI3K/AKT pathway is also altered in NDs including AD and PD. The activity of the PI3K/AKT/mTOR pathway suppresses the autophagic process and is commonly related to pro-survival effects, and the suppression of mTOR activity promotes autophagy and it has been shown to be protective for neuronal tissue, thus indicating that a coordinate mechanism is involved in neuroprotection. So compounds aiming to protect neuronal tissue must act in two ways: stimulating or sustaining the normal PI3K/AKT/mTOR survival pathway and promoting constitutive autophagy. Most compounds that induce autophagy promote clearance of aggregated proteins and induce neuroprotection, downregulating the PI3K/AKT/mTOR pathway or inhibiting mTOR. Therefore evaluating its role in synaptic functions and behavior is relevant to integrate the molecular events related to neuroprotection and the functional parameters in vivo (Heras-Sandoval et al. 2014). Acteoside is an activator of the transcription factor Nrf2, and inducer of HO-1 expression. Acteoside attenuates Aβ-induced neurotoxicity by induction of HO-1 via ERK and PI3 K/Akt signaling. These results shed some light on the mechanisms whereby acteoside protects against β-amyloid-induced cytotoxicity (Wang et al. 2012).
It is well established that pharmacological inhibitors of classical HDACs are protective in various cell culture and in vivo models of neurodegenerative disease but the protective effects of targeted therapeutic of neurotoxic HDAC(s) inhibitors are still unclear. IGF-1 promotes neuronal survival by suppressing the neurotoxic activity of HDAC3. It is well accepted that GSK3 β promotes neurodegeneration both in experimental models and human pathologies by activating downstream mechanism. Studies suggested that HDAC3 represents a downstream effector of GSK3 β neurotoxicity (Bardai and D'Mello 2011). Effects of BDNF on neuronal survival, synaptic activity, learning, memory, and neurogenesis are well documented. BDNF exerted a novel protective effect in astrocytes through TrkB-T1, involving calcium-Src-ERK and Akt pathways, and a potent antioxidant action, probably through induction of Nrf2. Whereas, BDNF-treated astrocytes release protective factors that prevent death and oxidative stress in neurons. We hope that this work will encourage further studies on glial cells as therapeutic targets for neurodegenerative disorders (Saba et al. 2018). This review also deals with the role of PI3K/Akt and Erk signaling in aberrant protein clearance through autophagy.
Future Prospective
In the next year, the characterization of both the newly identified and novel targets of Trk and p75NTR will allow us to identify the mechanisms used by these receptors to develop, maintain, and repair the nervous system. Co-application of agents capable of inhibiting the ERK pathway and activating PI3-K/Akt pathway or using multi-functional drug capable of regulating these pathways simultaneously might have therapeutic significances in the treatment of NDs. Moreover, based on the findings discussed in the review, ERK activation should also be taken into account for experimental animals designing and therapeutic approaches. Since, in different pathological conditions chronic activation of ERK occurs, future studies should be aimed on underlying molecular mechanism that will help to elucidate their pathogenesis and may provide the molecular basis of pharmacological intervention in neurodegenerative disorders.
References
Abdulkareem IH, Blair M (2013) Phosphatase and tensin homologue deleted on chromosome 10 Nigerian medical journal. J Niger Med Assoc 54:79
Ahn NG, Seger R, Bratlien R, Diltz C, Tonks N, Krebs E (1991) Multiple components in an epidermal growth factor-stimulated protein kinase cascade. In vitro activation of a myelin basic protein/microtubule-associated protein 2 kinase. J Biol Chem 266:4220–4227
Alessandrini A, Namura S, Moskowitz MA, Bonventre JV (1999) MEK1 protein kinase inhibition protects against damage resulting from focal cerebral ischemia. Proc Natl Acad Sci 96:12866–12869
Alessi DR, Saito Y, Campbell DG, Cohen P, Sithanandam G, Rapp U, Ashworth A, Marshall CJ, Cowley S (1994) Identification of the sites in MAP kinase kinase-1 phosphorylated by p74raf-1. EMBO J 13:1610–1619
Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings B (1996a) Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J 15:6541–6551
Alessi DR, Barry Caudwell F, Andjelkovic M, Hemmings BA, Cohen P (1996b) Molecular basis for the substrate specificity of protein kinase B; comparison with MAPKAP kinase-1 and p70 S6 kinase. FEBS Lett 399:333–338
Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P (1997) Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase Bα. Curr Biol 7:261–269
Altiok S, Batt D, Altiok N, Papautsky A, Downward J, Roberts TM, Avraham H (1999) Heregulin induces phosphorylation of BRCA1 through phosphatidylinositol 3-kinase/AKT in breast cancer cells. J Biol Chem 274:32274–32278
Andreadi CK, Howells LM, Atherfold PA, Manson MM (2006) Involvement of Nrf2, p38, B-Raf, and nuclear factor-κB, but not phosphatidylinositol 3-kinase, in induction of hemeoxygenase-1 by dietary polyphenols. Mol Pharmacol 69:1033–1040
Apostol BL et al (2005) Mutant huntingtin alters MAPK signaling pathways in PC12 and striatal cells: ERK1/2 protects against mutant huntingtin-associated toxicity. Hum Mol Genet 15:273–285
Aroeira RI, Sebastião AM, Valente CA (2015) BDNF, via truncated TrkB receptor, modulates GlyT1 and GlyT2 in astrocytes. Glia 63:2181–2197
Bae JH, Schlessinger J (2010) Asymmetric tyrosine kinase arrangements in activation or autophosphorylation of receptor tyrosine kinases. Mol Cell 29:443–448
Bardai FH, D'Mello SR (2011) Selective toxicity by HDAC3 in neurons: regulation by Akt and GSK3β. J Neurosci 31:1746–1751
Bayascas JR, Alessi DR (2005) Regulation of Akt/PKB Ser473 phosphorylation. Mol Cell 18:143–145
Bi G et al (2018) Therapeutic effect of transmembrane TAT-tCNTF via Erk and Akt activation using in vitro and in vivo models of Alzheimer’s disease. Int J Clin Exp Pathol 11:1855–1865
Binder DK, Scharfman HE (2004) Mini review. Growth Factors 22:123–131
Bodai L, Marsh JL (2012) A novel target for Huntington’s disease: ERK at the crossroads of signaling: the ERK signaling pathway is implicated in Huntington’s disease and its upregulation ameliorates pathology. Bioessays 34:142–148
Bohush A, Niewiadomska G, Filipek A (2018) Role of mitogen activated protein kinase signaling in Parkinson’s disease. Int J Mol Sci 19:2973
Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME (1999) Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and-independent mechanisms. Science 286:1358–1362
Borsello T, Forloni G (2007) JNK signalling: a possible target to prevent neurodegeneration. Curr Pharm Des 13:1875–1886
Brazil DP, Park J, Hemmings BA (2002) PKB binding proteins: getting in on the Akt. Cell 111:293–303
Brazil DP, Yang Z-Z, Hemmings BA (2004) Advances in protein kinase B signalling: AKTion on multiple fronts. Trends Biochem Sci 29:233–242
Brunet A, Bonni A, Zigmond MJ, Lin MZ, Juo P, Hu LS, Anderson MJ, Arden KC, Blenis J, Greenberg ME (1999) Akt promotes cell survival by phosphorylating and inhibiting a Forkhead transcription factor. Cell 96:857–868
Brunet A, Datta SR, Greenberg ME (2001) Transcription-dependent and-independent control of neuronal survival by the PI3K–Akt signaling pathway. Curr Opin Neurobiol 11:297–305
Calabrese V, Cornelius C, Dinkova-Kostova AT, Calabrese EJ, Mattson MP (2010) Cellular stress responses, the hormesis paradigm, and vitagenes: novel targets for therapeutic intervention in neurodegenerative disorders. Antioxid Redox Signal 13:1763–1811
Calabresi P, Gubellini P, Picconi B, Centonze D, Pisani A, Bonsi P, Greengard P, Hipskind RA, Borrelli E, Bernardi G (2001) Inhibition of mitochondrial complex II induces a long-term potentiation of NMDA-mediated synaptic excitation in the striatum requiring endogenous dopamine. J Neurosci 21:5110–5120
Cao Q et al (2017) Amentoflavone protects dopaminergic neurons in MPTP-induced Parkinson's disease model mice through PI3K/Akt and ERK signaling pathways. Toxicol Appl Pharmacol 319:80–90
Casarejos MJ, Perucho J, Gomez A, Muñoz MP, Fernandez-Estevez M, Sagredo O, Fernandez Ruiz J, Guzman M, de Yebenes JG, Mena MA (2013) Natural cannabinoids improve dopamine neurotransmission and tau and amyloid pathology in a mouse model of tauopathy. J Alzheimers Dis 35:525–539
Cavanaugh JE (2004) Role of extracellular signal regulated kinase 5 in neuronal survival. Eur J Biochem 271:2056–2059
Chen W-F et al (2017) Neuroprotective properties of icariin in MPTP-induced mouse model of Parkinson's disease: involvement of PI3K/Akt and MEK/ERK signaling pathways. Phytomedicine 25:93–99
Choi Y, Zhang J, Murga C, Yu H, Koller E, Monia BP, Gutkind JS, Li W (2002) PTEN, but not SHIP and SHIP2, suppresses the PI3K/Akt pathway and induces growth inhibition and apoptosis of myeloma cells. Oncogene 21:5289–5300
Chuderland D, Konson A, Seger R (2008) Identification and characterization of a general nuclear translocation signal in signaling proteins. Mol Cell 31:850–861
Cohen P (1999) The development and therapeutic potential of protein kinase inhibitors. Curr Opin Chem Biol 3:459–465
Colucci-D'Amato L, Perrone-Capano C, di Porzio U (2003) Chronic activation of ERK and neurodegenerative diseases. Bioessays 25:1085–1095
Cui W, Li W, Han R, Mak S, Zhang H, Hu S, Rong J, Han Y (2011a) PI3-K/Akt and ERK pathways activated by VEGF play opposite roles in MPP+-induced neuronal apoptosis. Neurochem Int 59:945–953
Cui W, Li W, Zhao Y, Mak S, Gao Y, Luo J, Zhang H, Liu Y, Carlier PR, Rong J, Han Y (2011b) Preventing H2O2-induced apoptosis in cerebellar granule neurons by regulating the VEGFR-2/Akt signaling pathway using a novel dimeric antiacetylcholinesterase bis (12)-hupyridone. Brain Res 1394:14–23
Cuny G (2009) Kinase inhibitors as potential therapeutics for acute and chronic neurodegenerative conditions. Curr Pharm Des 15:3919–3939
Dagda RK, Zhu J, Chu CT (2009) Mitochondrial kinases in Parkinson’s disease: converging insights from neurotoxin and genetic models. Mitochondrion 9:289–298
Deak M, Clifton AD, Lucocq JM, Alessi DR (1998) Mitogen-and stress-activated protein kinase-1 (MSK1) is directly activated by MAPK and SAPK2/p38, and may mediate activation of CREB. EMBO J 17:4426–4441
Deshmukh M, Johnson EM Jr (1998) Evidence of a novel event during neuronal death: development of competence-to-die in response to cytoplasmic cytochrome c. Neuron 21:695–705
Dhanasekaran N, Reddy EP (1998) Signaling by dual specificity kinases. Oncogene 17:1447–1455
Dudek H, Datta SR, Franke TF, Birnbaum MJ, Yao R, Cooper GM, Segal RA, Kaplan DR, Greenberg ME (1997) Regulation of neuronal survival by the serine-threonine protein kinase Akt. Science 275:661–665
Dyson JM, O'Malley CJ, Becanovic J, Munday AD, Berndt MC, Coghill ID, Nandurkar HH, Ooms LM, Mitchell CA (2001) The SH2-containing inositol polyphosphate 5-phosphatase, SHIP-2, binds filamin and regulates submembraneous actin. J Cell Biol 155:1065–1080
Elstner M, Morris CM, Heim K, Bender A, Mehta D, Jaros E, Klopstock T, Meitinger T, Turnbull DM, Prokisch H (2011) Expression analysis of dopaminergic neurons in Parkinson’s disease and aging links transcriptional dysregulation of energy metabolism to cell death. Acta Neuropathol 122:75–86
Faden AI, Wu J, Stoica BA, Loane DJ (2016) Progressive inflammation-mediated neurodegeneration after traumatic brain or spinal cord injury. Br J Pharmacol 173:681–691
Fayard E, Tintignac LA, Baudry A, Hemmings BA (2005) Protein kinase B/Akt at a glance. J Cell Sci 118:5675–5678
Forlenza OV, Torres CA, Talib LL, de Paula VJ, Joaquim HP, Diniz BS, Gattaz WF (2011) Increased platelet GSK3B activity in patients with mild cognitive impairment and Alzheimer’s disease. J Psychiatr Res 45:220–224
Fukunaga R, Hunter T (1997) MNK1, a new MAP kinase-activated protein kinase, isolated by a novel expression screening method for identifying protein kinase substrates. EMBO J 16:1921–1933
Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, Franke TF, Papapetropoulos A, Sessa WC (1999) Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature 399:597–601
Gerfen CR, Miyachi S, Paletzki R, Brown P (2002) D1 dopamine receptor supersensitivity in the dopamine-depleted striatum results from a switch in the regulation of ERK1/2/MAP kinase. J Neurosci 22:5042–5054
Gingras A-C, Kennedy SG, O’Leary MA, Sonenberg N, Hay N (1998) 4E-BP1, a repressor of mRNA translation, is phosphorylated and inactivated by the Akt (PKB) signaling pathway. Genes Dev 12:502–513
Glass CK, Saijo K, Winner B, Marchetto MC, Gage FH (2010) Mechanisms underlying inflammation in neurodegeneration. Cell 140:918–934
Gines S, Ivanova E, Seong I-S, Saura CA, MacDonald ME (2003) Enhanced Akt signaling is an early pro-survival response that reflects N-methyl-D-aspartate receptor activation in Huntington's disease knock-in striatal cells. J Biol Chem 278:50514–50522
van Golen CM, Schwab TS, Ignatoski KW, Ethier SP, Feldman EL (2001) PTEN/MMAC1 overexpression decreases insulin-like growth factor-I-mediated protection from apoptosis in neuroblastoma cells. Cell Growth Differ 12:371–378
Gómez N, Cohen P (1991) Dissection of the protein kinase cascade by which nerve growth factor activates MAP kinases. Nature 353:170–173
Gómez-Santos C, Ferrer I, Reiriz J, Viñals F, Barrachina M, Ambrosio S (2002) MPP+ increases α-synuclein expression and ERK/MAP-kinase phosphorylation in human neuroblastoma SH-SY5Y cells. Brain Res 935:32–39
Gonzalez FA, Raden DL, Davis RJ (1991) Identification of substrate recognition determinants for human ERK1 and ERK2 protein kinases. J Biol Chem 266:22159–22163
González-Polo RA, Soler G, Alvarez A, Fabregat I, Fuentes JM (2003) Vitamin E blocks early events induced by 1-methyl-4-phenylpyridinium (MPP+) in cerebellar granule cells. J Neurochem 84:305–315
Gotoh Y, Nishida E (1995) Activation mechanism and function of the MAP kinase cascade. Mol Reprod Dev 42:486–492
Grazia Spillantini M, Crowther RA, Jakes R, Hasegawa M (1998) Goedert M alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with Lewy bodies. In: Proceedings of the National Academy of Science. pp 6469–6473
Griffin RJ, Moloney A, Kelliher M, Johnston JA, Ravid R, Dockery P, O'Connor R, O'Neill C (2005) Activation of Akt/PKB, increased phosphorylation of Akt substrates and loss and altered distribution of Akt and PTEN are features of Alzheimer’s disease pathology. J Neurochem 93:105–117
Guyton KZ, Liu Y, Gorospe M, Xu Q, Holbrook NJ (1996) Activation of mitogen-activated protein kinase by ho role in cell survival following oxidant injury. J Biol Chem 271:4138–4142
Hanada M, Feng J, Hemmings BA (2004) Structure, regulation and function of PKB/AKT—a major therapeutic target. Biochim Biophys Acta 1697:3–16
Harris CA, Johnson EM (2001) BH3-only Bcl-2 family members are coordinately regulated by the JNK pathway and require Bax to induce apoptosis in neurons. J Biol Chem 276:37754–37760
Hashimoto M, Bar-on P, Ho G, Takenouchi T, Rockenstein E, Crews L, Masliah E (2004) β-Synuclein regulates Akt activity in neuronal cells a possible mechanism for neuroprotection in Parkinson′ s disease. J Biol Chem 279:23622–23629
Heras-Sandoval D, Pérez-Rojas JM, Hernández-Damián J, Pedraza-Chaverri J (2014) The role of PI3K/AKT/mTOR pathway in the modulation of autophagy and the clearance of protein aggregates in neurodegeneration. Cell Signal 26:2694–2701
Hu M, Li F, Wang W (2018) Vitexin protects dopaminergic neurons in MPTP-induced Parkinson’s disease through Pi3K/Akt signaling pathway. Drug Des Devel Ther 12:565
Hernandez F, Nido JD, Avila J, Villanueva N (2009) GSK3 inhibitors and disease. Mini Rev Med Chem 9:1024–1029
Hoover BR, Reed MN, Su J, Penrod RD, Kotilinek LA, Grant MK, Pitstick R, Carlson GA, Lanier LM, Yuan LL, Ashe KH, Liao D (2010) Tau mislocalization to dendritic spines mediates synaptic dysfunction independently of neurodegeneration. Neuron 68:1067–1081
Huang S-H, Wang J, Sui WH, Chen B, Zhang XY, Yan J, Geng Z, Chen ZY (2013) BDNF-dependent recycling facilitates TrkB translocation to postsynaptic density during LTP via a Rab11-dependent pathway. J Neurosci 33:9214–9230
Humbert S et al (2002) The IGF-1/Akt pathway is neuroprotective in Huntington's disease and involves Huntingtin phosphorylation by Akt. Dev Cell 2:831–837
Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M (1999) Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev 13:76–86
Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, Huang Q, Qin J, Su B (2006) SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 127:125–137
Jaiswal M, Sandoval H, Zhang K, Bayat V, Bellen H (2012) Probing mechanisms that underlie human neurodegenerative diseases in Drosophila. Annu Rev Genet 46:371–396
Jakel RJ, Townsend JA, Kraft AD, Johnson JA (2007) Nrf2-mediated protection against 6-hydroxydopamine. Brain Res 1144:192–201
Jerónimo-Santos A, Fonseca-Gomes J, Guimarães DA, Tanqueiro SR, Ramalho RM, Ribeiro JA, Sebastião AM, Diógenes MJ (2015) Brain-derived neurotrophic factor mediates neuroprotection against A β-induced toxicity through a mechanism independent on adenosine 2A receptor activation. Growth Factors 33:298–308
Jiao S, Li Z (2011) Nonapoptotic function of BAD and BAX in long-term depression of synaptic transmission. Neuron 70:758–772
Kang SS, Kwon T, Do SI (1999) Akt protein kinase enhances human telomerase activity through phosphorylation of telomerase reverse transcriptase subunit. J Biol Chem 274:13085–13090
Kanninen K, Malm TM, Jyrkkänen HK, Goldsteins G, Keksa-Goldsteine V, Tanila H, Yamamoto M, Ylä-Herttuala S, Levonen AL, Koistinaho J (2008) Nuclear factor erythroid 2-related factor 2 protects against beta amyloid. Mol Cell Neurosci 39:302–313
Kaplan DR, Miller FD (2000) Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol 10:381–391
Kase MS, Persons A, Napier C (2017) Effects of chronic pramipexole on AMPA receptor trafficking and Akt/GSK-3β signaling in a rat model of Parkinson’s disease. The FASEB Journal 31.1_supplement lb587-lb587
Kim EK, Choi E-J (2010) Pathological roles of MAPK signaling pathways in human diseases. Biochim Biophys Acta 1802:396–405
Kitagishi Y, Nakanishi A, Ogura Y, Matsuda S (2014) Dietary regulation of PI3K/AKT/GSK-3β pathway in Alzheimer’s disease. Alzheimers Res Ther 6:35
Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, Kim M, Nishito Y, Iemura SI, Natsume T, Ueno T, Kominami E, Motohashi H, Tanaka K, Yamamoto M (2010) The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol 12:213–223
Kulich SM, Chu CT (2001) Sustained extracellular signal-regulated kinase activation by 6-hydroxydopamine: implications for Parkinson’s disease. J Neurochem 77:1058–1066
Kuruvilla R, Ye H, Ginty DD (2000) Spatially and functionally distinct roles of the PI3-K effector pathway during NGF signaling in sympathetic neurons. Neuron 27:499–512
Kyriakis JM, App H, X-f Z, Banerjee P, Brautigan DL, Rapp UR, Avruch J (1992) Raf-1 activates MAP kinase-kinase. Nature 358:417–421
Kyriakis J, Force T, Rapp U, Bonventre J, Avruch J (1993) Mitogen regulation of c-Raf-1 protein kinase activity toward mitogen-activated protein kinase-kinase. J Biol Chem 268:16009–16019
Lange-Carter CA, Pleiman CM, Gardner AM, Blumer KJ, Johnson GL (1993) A divergence in the MAP kinase regulatory network defined by MEK kinase and Raf. Science 260:315–319
Langston J, Langston E, Irwin I (1984) MPTP-induced parkinsonism in human and non-human primates—clinical and experimental aspects. Acta Neurol Scand Suppl 100:49–54
Lebrun B, Bariohay B, Moyse E, Jean A (2006) Brain-derived neurotrophic factor (BDNF) and food intake regulation: a minireview. Auton Neurosci 126:30–38
Lefloch R, Pouysségur J, Lenormand P (2008) Single and combined silencing of ERK1 and ERK2 reveals their positive contribution to growth signaling depending on their expression levels. Mol Cell Biol 28:511–527
Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Münch AE, Chung WS, Peterson TC, Wilton DK, Frouin A, Napier BA, Panicker N, Kumar M, Buckwalter MS, Rowitch DH, Dawson VL, Dawson TM, Stevens B, Barres BA (2017) Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541:481–487
Liu D, Yang X, Songyang Z (2000) Identification of CISK, a new member of the SGK kinase family that promotes IL-3-dependent survival. Curr Biol 10:1233–1236
Luttrell LM, Daaka Y, Lefkowitz RJ (1999) Regulation of tyrosine kinase cascades by G-protein-coupled receptors. Curr Opin Cell Biol 11:177–183
Malagelada C, Jin ZH, Greene LA (2008) RTP801 is induced in Parkinson's disease and mediates neuron death by inhibiting Akt phosphorylation/activation. J Neurosci 28:14363–14371
Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC (2002) Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell 10:151–162
Mazzoni IE, Saıd FA, Aloyz R, Miller FD, Kaplan D (1999) Ras regulates sympathetic neuron survival by suppressing the p53-mediated cell death pathway. J Neurosci 19:9716–9727
Menzies FM, Fleming A, Caricasole A, Bento CF, Andrews SP, Ashkenazi A, Füllgrabe J, Jackson A, Jimenez Sanchez M, Karabiyik C, Licitra F, Lopez Ramirez A, Pavel M, Puri C, Renna M, Ricketts T, Schlotawa L, Vicinanza M, Won H, Zhu Y, Skidmore J, Rubinsztein DC (2017) Autophagy and neurodegeneration: pathogenic mechanisms and therapeutic opportunities. Neuron 93:1015–1034
Morris ME, Iansek R, Matyas TA, Summers JJ (1994) Ability to modulate walking cadence remains intact in Parkinson’s disease. J Neurol Neurosurg Psychiatry 57:1532–1534
Morrison BE, Majdzadeh N, Zhang X, Lyles A, Bassel-Duby R, Olson EN, D'Mello SR (2006) Neuroprotection by histone deacetylase-related protein. Mol Cell Biol 26:3550–3564
Murray B, Alessandrini A, Cole AJ, Yee AG, Furshpan EJ (1998) Inhibition of the p44/42 MAP kinase pathway protects hippocampal neurons in a cell-culture model of seizure activity. Proc Natl Acad Sci 95:11975–11980
Nah J, Yuan J, Jung Y-K (2015) Autophagy in neurodegenerative diseases: from mechanism to therapeutic approach. Mol Cells 38:381–389
Nakaso K, Tajima N, Horikoshi Y, Nakasone M, Hanaki T, Kamizaki K, Matsura T (2014) The estrogen receptor β-PI3K/Akt pathway mediates the cytoprotective effects of tocotrienol in a cellular Parkinson's disease model. Biochim Biophys Acta (BBA)-Mol Basis Dis 1842:1303–1312
Naor Z, Benard O, Seger R (2000) Activation of MAPK cascades by G-protein-coupled receptors: the case of gonadotropin-releasing hormone receptor. Trends Endocrinol Metab 11:91–99
Nataraj J, Manivasagam T, Thenmozhi AJ, Essa MM (2017) Neurotrophic effect of asiatic acid, a triterpene of centella asiatica against chronic 1-methyl 4-phenyl 1, 2, 3, 6-tetrahydropyridine hydrochloride/probenecid mouse model of Parkinson’s disease: the role of MAPK, PI3K-Akt-GSK3β and mTOR signalling pathways. Neurochem Res 42:1354–1365
Nishimoto S, Nishida E (2006) Mapk signalling: Erk5 versus erk1/2. EMBO Rep 7:782–786
Northwood IC, Gonzalez FA, Wartmann M, Raden DL, Davis RJ (1991) Isolation and characterization of two growth factor-stimulated protein kinases that phosphorylate the epidermal growth factor receptor at threonine 669. J Biol Chem 266:15266–15276
Nuytemans K, Theuns J, Cruts M, Van Broeckhoven C (2010) Genetic etiology of Parkinson disease associated with mutations in the SNCA, PARK2, PINK1, PARK7, and LRRK2 genes: a mutation update. Hum Mutat 31:763–780
Orike N, Middleton G, Borthwick E, Buchman V, Cowen T, Davies AM (2001) Role of PI 3-kinase, Akt and Bcl-2–related proteins in sustaining the survival of neurotrophic factor–independent adult sympathetic neurons. J Cell Biol 154:995–1006
Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB (1999) NF-κB activation by tumour necrosis factor requires the Akt serine–threonine kinase. Nature 401:82–85
Pap M, Cooper GM (1998) Role of glycogen synthase kinase-3 in the phosphatidylinositol 3-kinase/Akt cell survival pathway. J Biol Chem 273:19929–19932
Paradis S, Ruvkun G (1998) C. elegans Akt/PKB transduces insulin receptor-like signals from AGE-1 PI3K to the DAF-16 transcription factor. In: East Coast Worm Meeting
Parrish AB, Freel CD, Kornbluth S (2013) Cellular mechanisms controlling caspase activation and function. Cold Spring Harb Perspect Biol 5:a008672
Pariyar R et al (2017) Sulfuretin attenuates MPP+-induced neurotoxicity through Akt/GSK3β and ERK signaling pathways. Int J Mol Sci 18(12):2753
Payne DM, Rossomando AJ, Martino P, Erickson AK, Her JH, Shabanowitz J, Hunt DF, Weber MJ, Sturgill TW (1991) Identification of the regulatory phosphorylation sites in pp42/mitogen-activated protein kinase (MAP kinase). EMBO J 10:885–892
Petit-Paitel A, Brau F, Cazareth J, Chabry J (2009) Involvment of cytosolic and mitochondrial GSK-3β in mitochondrial dysfunction and neuronal cell death of MPTP/MPP+-treated neurons. PLoS One 4:e5491
Pettmann B, Henderson CE (1998) Neuronal cell death. Neuron 20:633–647
Quesada A, Lee BY, Micevych PE (2008) PI3 kinase/Akt activation mediates estrogen and IGF-1 nigral DA neuronal neuroprotection against a unilateral rat model of Parkinson's disease. Dev Neurobiol 68:632–644
Rai SN et al (2017) Mucuna pruriens protects against MPTP intoxicated neuroinflammation in Parkinson’s disease through NF-κB/pAKT signaling pathways. Front Aging Neurosci 9:421. https://doi.org/10.3389/fnagi.2017.00421
Ramalingam M, Kim S-J (2017) Protective effects of activated signaling pathways by insulin on C6 glial cell model of MPP+-induced Parkinson’s disease. J Recept Signal Transduct 37:100–107
Rane SG (1999) Ion channels as physiological effectors for growth factor receptor and Ras/ERK signaling pathways. Adv Second Messenger Phosphoprotein Res:107–130
Read DE, Gorman AM (2009) Involvement of Akt in neurite outgrowth. Cell Mol Life Sci 66:2975–2984
Reichardt LF (2006) Neurotrophin-regulated signalling pathways. Philos Trans R Soc Lond B Biol Sci 361:1545–1564
Reszka AA, Seger R, Diltz CD, Krebs EG, Fischer EH (1995) Association of mitogen-activated protein kinase with the microtubule cytoskeleton. Proc Natl Acad Sci 92:8881–8885
Romashkova JA, Makarov SS (1999) NF-κB is a target of AKT in anti-apoptotic PDGF signalling. Nature 401:86–90
van Roon-Mom WM, Pepers BA, AC't Hoen P, Verwijmeren CA, den Dunnen JT, Dorsman JC, van Ommen GB (2008) Mutant huntingtin activates Nrf2-responsive genes and impairs dopamine synthesis in a PC12 model of Huntington’s disease. BMC Mol Biol 9:84
Ross CA, Akimov SS (2014) Human-induced pluripotent stem cells: potential for neurodegenerative diseases. Hum Mol Genet 23:R17–R26
Russo C, Dolcini V, Salis S, Venezia V, Zambrano N, Russo T, Schettini G (2002) Signal transduction through tyrosine-phosphorylated C-terminal fragments of amyloid precursor protein via an enhanced interaction with Shc/Grb2 adaptor proteins in reactive astrocytes of Alzheimer’s disease brain. J Biol Chem 277:35282–35288
Saba J, Turati J, Ramírez D, Carniglia L, Durand D, Lasaga M, Caruso C (2018) Astrocyte truncated‐TrkB mediates BDNF antiapoptotic effect leading to neuroprotection. J Neurochem. https://doi.org/10.1111/jnc.14476
Schlessinger J (2000) Cell signaling by receptor tyrosine kinases. Cell 103:211–225
Shaughnessy DT, McAllister K, Worth L, Haugen AC, Meyer JN, Domann FE, van Houten B, Mostoslavsky R, Bultman SJ, Baccarelli AA, Begley TJ, Sobol RW, Hirschey MD, Ideker T, Santos JH, Copeland WC, Tice RR, Balshaw DM, Tyson FL (2014) Mitochondria, energetics, epigenetics, and cellular responses to stress. Environ Health Perspect 122:1271–1278
Sheikh S, Haque E, Mir SS (2013) Neurodegenerative diseases: multifactorial conformational diseases and their therapeutic interventions. J Neurodegener Dis 2013
Shin S, Dimitri CA, Yoon S-O, Dowdle W, Blenis J (2010) ERK2 but not ERK1 induces epithelial-to-mesenchymal transformation via DEF motif-dependent signaling events. Mol Cell 38:114–127
Simonian N, Coyle J (1996) Oxidative stress in neurodegenerative diseases. Annu Rev Pharmacol Toxicol 36:83–106
Slater A, Stefan C, Nobel I, Orrenius S (1996) Intracellular redox changes during apoptosis. Cell Death Differ 3:57–62
Song G, Ouyang G, Bao S (2005) The activation of Akt/PKB signaling pathway and cell survival. J Cell Mol Med 9:59–71
Stanciu M, DeFranco DB (2002) Prolonged nuclear retention of activated extracellular signal-regulated protein kinase promotes cell death generated by oxidative toxicity or proteasome inhibition in a neuronal cell line. J Biol Chem 277:4010–4017
Stanciu M, Wang Y, Kentor R, Burke N, Watkins S, Kress G, Reynolds I, Klann E, Angiolieri MR, Johnson JW, DeFranco DB (2000) Persistent activation of ERK contributes to glutamate-induced oxidative toxicity in a neuronal cell line and primary cortical neuron cultures. J Biol Chem 275:12200–12206
Stephens L, Anderson K, Stokoe D, Erdjument-Bromage H, Painter GF, Holmes AB, Gaffney PRJ, Reese CB, McCormick F, Tempst P, Coadwell J, Hawkins PT (1998) Protein kinase B kinases that mediate phosphatidylinositol 3, 4, 5-trisphosphate-dependent activation of protein kinase B. Science 279:710–714
Sturgill TW, Ray LB, Erikson E, Maller JL (1988) Insulin-stimulated MAP-2 kinase phosphorylates and activates ribosomal protein S6 kinase II. Nature 334:715–718
Sturla L-M, Cowan CW, Guenther L, Castellino RC, Kim JY, Pomeroy SL (2005) A novel role for extracellular signal-regulated kinase 5 and myocyte enhancer factor 2 in medulloblastoma cell death. Cancer Res 65:5683–5689
Subramaniam S, Unsicker K (2006) Extracellular signal-regulated kinase as an inducer of non-apoptotic neuronal death. Neuroscience 138:1055–1065
Subramaniam S, Unsicker K (2010) ERK and cell death: ERK1/2 in neuronal death. FEBS J 277:22–29
Sugino T, Nozaki K, Takagi Y, Hattori I, Hashimoto N, Moriguchi T, Nishida E (2000) Activation of mitogen-activated protein kinases after transient forebrain ischemia in gerbil hippocampus. J Neurosci 20:4506–4514
Tejeda GS, Ayuso-Dolado S, Arbeteta R, Esteban-Ortega GM, Vidaurre OG, Díaz-Guerra M (2016) Brain ischaemia induces shedding of a BDNF-scavenger ectodomain from TrkB receptors by excitotoxicity activation of metalloproteinases and γ-secretases. J Pathol 238:627–640
Tsirigotis M, Baldwin RM, Tang MY, Lorimer IA, Gray DA (2008) Activation of p38MAPK contributes to expanded polyglutamine-induced cytotoxicity. PloS one 3:e2130
Vaillant A, Mazzoni I, Tudan C, Boudreau M, Kaplan D, Miller F (1999) Depolarization and neurotrophins converge on the phosphatidylinositol 3-kinase–Akt pathway to synergistically regulate neuronal survival. J Cell Biol 146:955–966
Vanhaesebroeck B, Alessi DR (2000) The PI3K–PDK1 connection: more than just a road to PKB. Biochem J 346(Pt 3):561–576
Vargas MR, Johnson DA, Sirkis DW, Messing A, Johnson JA (2008) Nrf2 activation in astrocytes protects against neurodegeneration in mouse models of familial amyotrophic lateral sclerosis. J Neurosci 28:13574–13581
Vidaurre ÓG et al (2013) Imbalance of neurotrophin receptor isoforms TrkB-FL/TrkB-T1 induces neuronal death in excitotoxicity. Cell Death Dis 3:e256
Von Kriegsheim A et al (2009) Cell fate decisions are specified by the dynamic ERK interactome. Nat Cell Biol 11:1458–1464
Wang RM, Zhang QG, Li CH, Zhang GY (2005) Activation of extracellular signal-regulated kinase 5 may play a neuroprotective role in hippocampal CA3/DG region after cerebral ischemia. J Neurosci Res 80:391–399
Wang J, Zhang M, Zhang L, Cai H, Zhou S, Zhang J, Wang Y (2010) Correlation of Nrf2, HO-1, and MRP3 in gallbladder cancer and their relationships to clinicopathologic features and survival. J Surg Res 164:e99–e105
Wang H-Q, Xu Y-X, Zhu C-Q (2012) Upregulation of heme oxygenase-1 by acteoside through ERK and PI3 K/Akt pathway confer neuroprotection against beta-amyloid-induced neurotoxicity. Neurotox Res 21:368–378
Wang T et al (2018) α-Lipoic acid attenuates oxidative stress and neurotoxicity via the ERK/Akt-dependent pathway in the mutant hSOD1 related Drosophila model and the NSC34 cell line of amyotrophic lateral sclerosis. Brain Res Bull 140:299–310
Waskiewicz AJ, Flynn A, Proud CG, Cooper JA (1997) Mitogen-activated protein kinases activate the serine/threonine kinases Mnk1 and Mnk2. EMBO J 16:1909–1920
Wellbrock C, Karasarides M, Marais R (2004) The RAF proteins take centre stage. Nat Rev Mol Cell Biol 5:875–885
Wick A, Wick W, Waltenberger J, Weller M, Dichgans J, Schulz JB (2002) Neuroprotection by hypoxic preconditioning requires sequential activation of vascular endothelial growth factor receptor and Akt. J Neurosci 22:6401–6407
Wortzel I, Seger R (2011) The ERK cascade: distinct functions within various subcellular organelles. Genes Cancer 2:195–209
Wymann MP, Zvelebil M, Laffargue M (2003) Phosphoinositide 3-kinase signalling–which way to target? Trends Pharmacol Sci 24:366–376
Xia Z, Dickens M, Raingeaud J, Davis RJ, Greenberg ME (1995) Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science 270:1326–1331
Xia XG, Harding T, Weller M, Bieneman A, Uney JB, Schulz JB (2001) Gene transfer of the JNK interacting protein-1 protects dopaminergic neurons in the MPTP model of Parkinson’s disease. Proc Natl Acad Sci 98:10433–10438
Xu X, Zhang A, Zhu Y, He W, Di W, Fang Y, and Shi X (2019) MFG‐E8 reverses microglial‐induced neurotoxic astrocyte (A1) via NF‐κB and PI3K‐Akt pathways. J Cell Physiol 234(1):904–914
Yao R, Cooper GM (1995) Requirement for phosphatidylinositol-3 kinase in the prevention of apoptosis by nerve growth factor. Science 267:2003–2006
Yoon S, Seger R (2006) The extracellular signal-regulated kinase: multiple substrates regulate diverse cellular functions. Growth Factors 24:21–44
Yung Y, Yao Z, Hanoch T, Seger R (2017) ERK1b, a 46-kDa ERK isoform that is differentially regulated by MEK. J Biol Chem 292:8854
Zheng C-F, Guan K-L (1993) Properties of MEKs, the kinases that phosphorylate and activate the extracellular signal-regulated kinases. J Biol Chem 268:23933–23939
Zhu J-H, Kulich SM, Oury TD, Chu CT (2002a) Cytoplasmic aggregates of phosphorylated extracellular signal-regulated protein kinases in Lewy body diseases. Am J Pathol 161:2087–2098
Zhu X, Lee H-g, Raina AK, Perry G, Smith MA (2002b) The role of mitogen-activated protein kinase pathways in Alzheimer’s disease. Neurosignals 11:270–281
Zhu J-h, Horbinski C, Guo F, Watkins S, Uchiyama Y, Chu CT (2007) Regulation of autophagy by extracellular signal-regulated protein kinases during 1-methyl-4-phenylpyridinium-induced cell death. Am J Pathol 170:75–86
Zimmermann S, Moelling K (1999) Phosphorylation and regulation of Raf by Akt (protein kinase B). Science 286:1741–1744
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Rai, S.N., Dilnashin, H., Birla, H. et al. The Role of PI3K/Akt and ERK in Neurodegenerative Disorders. Neurotox Res 35, 775–795 (2019). https://doi.org/10.1007/s12640-019-0003-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-019-0003-y