
Abstract
Rheumatoid arthritis (RA) is characterised by severe joint and bone damage due to heightened autoimmune response at the articular sites. Worldwide annual incidence and prevalence rate of RA is 3 cases per 10,000 population and 1%, respectively. Several genetic and environmental (microbiota, smoking, infectious agents) factors contribute to its pathogenesis. Although convention treatment strategies, predominantly Disease Modifying Anti Rheumatic Drugs (DMARDs) and Glucocorticoids (GC), are unchanged as the primary line of treatment; novel strategies consisting of biological DMARDs, are being developed and explored. Personalized approaches using biologicals targetspecific pathways associated with disease progression. However, considering the economic burden and side-effects associated with these, there is an unmet need on strategies for early stratification of the inadequate responders with cDMARDs. As RA is a complex disease with a variable remission rate, it is important not only to evaluate the current status of drugs in clinical practice but also those with the potential of personalised therapeutics. Here, we provide comprehensive data on the treatment strategies in RA, including studies exploring various combination strategies in clinical trials. Our systematic analysis of current literature found that conventional DMARDs along with glucocorticoid may be best suited for early RA cases and a combination of conventional and targeted DMARDs could be effective for treating seronegative patients with moderate to high RA activity. Clinical trials with insufficient responders to Methotrexate suggest that adding biologicals may help in such cases. However, certain adverse events associated with the current therapy advocate exploring novel therapeutic approaches such as gene therapy, mesenchymal stem cell therapy in future.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rheumatoid arthritis (RA) is an autoimmune chronic disease, primarily characterised by synovial inflammation (synovitis), which further leads to cartilage damage and bone erosion. Early symptoms include general malaise, swollen and tender joints and morning stiffness. If untreated, chronic RA can lead to systemic inflammation resulting in abnormalities in heart, liver, intestine, muscle and in some cases can also cause cognitive decline [1]. It usually starts between the age group of 20 to 40 years and its prevalence rate varies from 0.3 to 1% [2]. Several factors are known to contribute to the development of this disease. Approximately, 60% of RA cases are linked to genetic predisposing factors, compounded by environmental factors. Among the contributing genes, single nucleotide polymorphisms (SNPs) in HLA-DRB1 alleles (DRB1*01 and DRB1*04; DQ8) are mostly involved. Exact mechanism of disease development has not been completely understood but pathogenic or non-pathogenic triggering events may be involved, such as bacterial/viral infections, microvascular damage or microtrauma [3]. Thus, presence of auto-reactive B and T cells along with generation of Neo-epitopes leading to loss of tolerance is found [4]. Deregulated factors such as cytokine and other inflammatory molecules along with immune cells like B, T and mast cells eventually accumulate in the synovial sites, causing the damaging effect. Certain biomarkers such as rheumatoid factor (RF), antibodies to citrullinated protein antigen (ACPAs), C-reactive protein CRP (CRP) and erythrocyte sedimentation rate (ESR) are useful in RA diagnosis [5].
The overall aim for treating RA is to achieve disease remission. Although Methotrexate is a staple drug for its treatment, the repertoire of therapeutic options for rheumatologists has increased over the past decade. DMARDs have been effectively utilised to target inflammation and prevent further joint damage. Besides these, Non-steroidal Anti-inflammatory drugs (NSAIDs); cause symptom improvement but no effect on disease progression and Glucocorticoids (non-specific immune suppression but with long term side effects) have also been used. DMARDs include those drugs which target rheumatoid inflammation and effectively control disease progression. Two categories of DMARDs are available for use (i) Synthetic DMARDs and (ii) Biological DMARDs. Synthetic DMARDs can be subdivided further into Conventional and Targeted DMARDs. Conventional DMARDs (cDMARDs) include routinely used drugs such as Methotrexate, Hydroxychloroquine and Sulfadiazine, whereas newer targeted DMARDs include JAK inhibitors (Baricitinib/Tofacitib). Biological DMARDs (bDMARDs) include antibodies against TNF-alpha, TNF-R, IL-6, IL6-R, co stimulatory molecules and B cell depleting antibodies. Importantly, combination therapies of two/three synthetic DMARDs or synthetic and biological DMARDs can also be used [6,7,8,9,10,11]. However, not all patients respond with therapy and multi-refractory (MR) patients showing insufficient response to at least three bDMARDs or bDMARDs with different mechanism of action have also been reported [12]. Further insights are required into biomarker development for early diagnosis of RA and early identification of non-responder population with cDMARDs, so that other treatment arms may be implemented to avoid disease progression.
Treat-to-Target approach is the newer strategy in RA treatment that engages in stringent observation of the disease progression and management if respective therapy is not encouraging [13]. The American College of Rheumatology (ACR), EULAR and the Asia Pacific League of Associations for Rheumatology (APLAR) have employed the treat-to-target approach in their recommendations [14, 15]. At present, RA treatment is focused on reducing disease activity followed by potential remission preventing joint deformities and disease progression [16, 17]. However, advanced approaches emphasise on the importance of attaining at least 50% improvement in disease activity within 3 months of drug administration [18]. In case the desired outcome has not been achieved, modification in treatment strategy is suggested depending upon patient’s situation in order to improve disease management.
Methodology
PubMed and Google scholar sites were used for literature search of peer-reviewed articles. The main keywords used for search were “rheumatoid arthritis and therapy and DMARD”. For including clinical trial data, we have referred to the NIH Clinical Trials website (https://clinicaltrials.gov/). Only Phase 3 and 4 studies with published results were included in the article. Figure of targeted therapy and mechanism of action of cDMARDs was generated originally using Biorender software. Algorithm of European League Against Rheumatology (EULAR) recommendation is adapted from published source. Systematic chart of inclusion and exclusion criteria is mentioned in Fig. 1.

Systematic chart of inclusion and exclusion criteria
Aetiology of rheumatoid arthritis
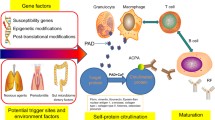
RA is a multifactorial disease and its progression and severity depends on both environmental and genetic factors. Genomic studies have identified more than 100 loci associated with this disease. Out of these, Major Histocompatibility Complex class II (MHC class II) encoding genes such as Human Leukocyte Antigen DR01/04 (HLA DR01/04) are prominently involved [19]. Products of HLADR01/04 (DR1 and DR4) help T-cells in preferential recognition of auto-reactive peptides [20]. Further, genes associated with inflammatory processes are also found associated with higher risk. Genome Wide Association Study (GWAS) have identified involvement of multiple genes like PTPN22, STAT4, PADI4, CTLA4, CD40, TNF in RA. Single Nucleotide Polymorphisms (SNPs) in genes such as HLA-DR, PTPN22, and TRAF1-C5 are associated with 40-fold higher risk. Recently, epigenetic modifications are also found associated with disease etiology [21]. Altered methylome signatures are found in synovial fibroblasts that contribute to their activation. Pathways associated with hypomethylated genes are involved in cell adhesion and migration and trans-endothelial migration, thus leading to a migratory phenotype [22]. In one of the studies, higher expression of polyamine-modulated factor 1-binding protein 1 (PMFBP1) and spermidine/spermine N1-acetyltransferase (SSAT1), hypomethylating factors, is reported in fibroblasts from RA patients [23]. Inhibition of PMFBP1 and SSAT1 downregulates Matrix MetalloProteinase 1 (MMP1), involved in cell migration and invasion, and hence can help in preventing disease progression [23]. In another study, inhibition of Histone Deacetylase (HDAC) leads to increased expression of Tumor Necrosis Factor A (TNFA) induced ICAM-1and VCAM-1 in synovial fibroblasts [24]. ICAM-1 and VCAM-1 level in blood is found to be positively correlated with RA severity [25] role. Comprehensive epigenetic profiling of fibroblasts like synoviocytes from RA patients found changes in expression of genes associated with pathways such as Protein Kinase A signaling, Clathrin-Mediated Endocytosis, role of osteoblasts, osteoclasts and chondrocytes and leukocyte extravasation signaling along with others [26].
Environmental factors such as smoking and infection play an important role as triggers for the disease development. Through epidemiological studies and in vivo models, the role of smoking has been established. In smokers, risk of developing RA is twofold higher than in non-smokers and in female smokers it is 1.3-fold higher than non-smokers [27]. Smoking can activate oxidative stress pathways by inducing free radical generation [28]. It can also induce Fas (CD95) and CD4 T-cell expression that can lead to increased cell death and autoimmune response contributing to synovial inflammation [29]. In smokers, higher expression of inflammatory molecules such as CRP, Fibrinogen, ICAM-1 along with other cytokines is found [27]. Cytokines such as IL-1α, IL-1β, IL-6 and IL-8 can be induced in FLS cells by condensate from cigarette smoke [30]. Smoking can increase the level of monocytes and macrophages in alveoli that can cause heightened inflammatory response and may contribute to disease development [31]. Smokers also show higher level of MMPs in their synovial fluid that contributes to joint destruction [32]. Further, smoking is reported to cause genome-wide methylation pattern in the MHC region [33].
Interestingly, dietary factors may affect RA risk. Studies have linked increased alcohol consumption with lower RA risk [34]. Study in Danish population has shown that consuming alcohol leads to decreased ACPA-positive RA. Certain foods can also increase susceptibility to disease development. Mediterranean diet is known to be the best for RA prevention. Study found that Mediterranean diet caused the decrease in disease activity score [35]. Calorie restriction methods like intermittent fasting also reduces inflammation. Although there is not much evidence regarding association of poultry products and fish with RA risk. In one of the studies, red meat is shown to be associated with RA development [36]. Among other environmental factors, vitamin D is known to be strongly associated with RA risk. Vitamin D plays a major role in regulation of hormones and immune system [37]. Vitamin D being an immune regulator and suppressor of the inflammatory process, it is found to be associated with increased risk or disease severity.
Treatment strategies
Previously until early 1990s, unconfirmed diagnosis combined with the use of NSAIDs as first line therapy, led to chronic RA disease resulting in adverse consequences, with serious joint deformities and disabilities. However, the recent approach to treat any disease is primarily focused on achieving disease remission or reducing the disease activity and therefore multiple strategies, including combination of cDMARDs at the start and switching over to targeted or bDMARDs, are employed to decrease joint damage and related symptoms. In addition, advanced biological treatments that are currently being employed and some combinations under clinical trials, have shown to induce decreased activation of the immune system and inflammation [38]. According to the latest EULAR recommendations (2019), DMARDs are to be used either as monotherapy or in combination with a biological (TNF-inhibitors, IL-inhibitors etc.) depending upon response to respective drugs (Fig. 2, adapted from [39]) In addition, combination therapy with inclusion of NSAIDs and Glucocorticoids in the initial treatment regimen helps in pain management and general anti-inflammatory actions. The key therapeutic DMARD drugs and their mechanism of action are compiled in Table 1.

Flowchart diagram for latest EULAR recommendations for disease management in rheumatoid arthritis
Synthetic DMARDs
Conventional DMARDs
RA being an inflammatory disease, first line of treatment involves cDMARDs along with NSAIDs that alleviate inflammation, pain and control radiographic progression at the joints. cDMARDs include Methotrexate (MTX), Sulfasalazine, Leflunomide, and Hydroxychloroquine. NSAIDs and Glucocorticoids are also given initially to suppress pain and inflammation but they have limited benefit and do not reduce disease progression. Hence these are not categorised under DMARDs.
MTX has been a cardinal drug for RA therapy for more than two decades now. Its mechanism of action has been well reported [6]. It is folate analogue, which interferes with the activity of dihydrofolate reductase, thereby inhibiting nucleotide synthesis and metabolism. It has also been reported to increase adenosine release, thereby causing anti- inflammatory action. It is beneficial as it is reliable, affordable, effective and tolerable at low doses. On comparison with other cDMARDs, MTX is found most effective, however, certain patients are observed to be unresponsive or intolerable to MTX, as a consequence of which Hydroxychloroquine [7], Sulfasalazine and Leflunomide are included in the treatment. Potential side-effects may include ulcers, hepatitis, interstitial pneumonitis, cirrhosis, renal dysfunction and cytopenias. The mechanism of action of these cDMARDs is represented in Fig. 3.

Mechanism of action of csDMARDs. Methotrexate and Sulfasalazine share nearly similar mechanism of action. Although other potential mechanisms such as folate antagonism, adhesion molecules, generation of reactive oxygen species, alteration of cytokine profile and polyamide inhibition have been studies for Methotrexate, adenosine signalling happens to be the most acceptable. Methotrexate blocks 5-aminoamidazole-4-carboxamide ribonucleotide (AICAR) followed by its accumulation further blocking adenosine deaminase. Ent1 is the nucleoside transporter responsible for extracellular export of adenosine. Dephosphorylation of ATP and ADP to AMP takes place by CD39 and converted to adenosine by CD73. Leflunomide is an immunomodulatory drug and acts by inhibiting the enzyme dihydroorotate dehydrogenase (DHODH), which is essential for converting dihydroorotate to orotate. DHODH is one of the essential enzymes responsible for initiating pyrimidine synthesis, nucleic acids formation followed by lymphocyte proliferation. Mechanism of action of HCQ in RA is yet to be fully explored, however, molecular effects of HCQ include lysosomal activity, TLR signalling pathway and autophagy. HCQ interferes with immune activation by inhibiting several innate and adaptive immune processes. Specific molecular targets include TLR-signalling pathways and antigen-presentation cells (APCs). In APCs, HCQ inhibits antigen processing and MHC II presentation to T cells, thereby preventing T-cell activation and differentiation
Currently, MTX is given to every newly diagnosed RA patients but 50% of patients either do not show optimum clinical outcome or show some adverse events. In one of the studies, adverse events associated with MTX were evaluated on 1069 patients. Approximately, 77.5% of patients showed at least one adverse event. Some of the most common adverse events were of gastrointestinal, mucocutaneous, neurological and hematological types. Alcohol consumption and gender were also found to play role in these adverse events [53]. Some studies show that bioavailability and response to MTX is partially determined by microbial composition of gastrointestinal tract of RA patients. Pharmacomicrobiology studies have identified several factors that alter the bioavailability and response to MTX [54]. Insufficient response to MTX has also been observed. Some patients are either unresponsive or show inadequate response within the first 6 months of treatment. Lack of response during the early window of opportunity has been a problem in making suitable therapeutic decisions and some biomarkers have been found to predict this. A multi-centric prospective observational study was conducted to identify baseline predictors of non-response to MTX. 43% (449/1050) of patients were classified as non-responders on the basis of RF-negativity (0.62), higher HAQ (Health Assessment Questionnaire) score (1.64), higher Tender Joint Count (1.06) and lower disease activity (0.29). Non-responders were thereby switched to biologicals (TNF or IL-1 inhibitors). This is the first study emphasising the importance of initial stratification of patients as responders and non-responders, further employing alternative approaches to ameliorate disease progression [55]. Another study highlights a significant correlation between gut microbiota and response to MTX. High-throughput metagenomic sequencing, conducted for drug-naive patients (n = 26), indicated that the microbiota’s metabolic capacity can influence MTX response. Thus, pre-treatment microbiome composition could be used as a predictor of MTX response [56]. Another study conducted for elucidating baseline levels of serum biomarkers representing the multi-biomarker disease activity (MBDA) test observed significant correlation between 4 biomarkers and response to MTX in DMARD-naive early RA patients (n = 298) from SWEFOT trial. Reduced CRP, Leptin and higher levels of Tumor Necrosis Factor Receptor I (TNF-RI) and VCAM1 were found to be independently associated with reduced disease activity after 3 months of MTX therapy. A combination score of these biomarkers was found as a predictor of response to MTX after 3 months [57].
Targeted DMARDs
Targeted DMARDs were developed to specifically target the JAK-STAT pathway, which is a key cytokine mediated pro-inflammatory pathway. Cytokines like IL-6, IFN gamma, GM-CSF have a common mechanism of action after they bind to their respective receptors on cells. This binding activates the binding of JAK kinase to their receptors, which further phosphorylates these receptors, leading to phosphorylation and dimerisation of STAT molecules. This further causes the nuclear localisation of STAT and induction of further transcription of pro-inflammatory genes. Tofacitinib is a pan-JAK inhibitor inhibiting JAK-1/2/3, Baricitinib is a JAK1/2 inhibitor, and Filgotinib and Upadacitinib are selective JAK-1 inhibitors.
Encouraging results have been obtained from such drugs targeting JAK pathways [58, 59]. Tofacitinib with/without MTX was approved by the European Medicines Agency (EMA) in 2016–2017 for patients with moderate to active RA [60]. In addition, Baricitinib has also been approved for patients displaying non-efficacious response to DMARDs [61, 62]. In five rheumatology units including adults with RA initiating Baricitinib, retrospective longitudinal cohort study was conducted. Data from 182 patients found that patients treated with Baricitinib had long-standing and refractory disease. After 6 and 12 months of treatment initiation, high persistence and improvement in disease activity and pain were found [63]. Long-term safety was evaluated in RA patients from the completed extension trial of Baricitinib. Integrated analysis on data from the 3770 active RA patients found no new safety signals [64]. In one of the case studies, 35-year-old man with seronegative RA had bilateral severe non-granulomatous panuveitis which was resistant to steroid treatment, methotrexate, salazosulfapydine, adalimumab and infliximab. Patient was given Baricitinib which decreased the activity of systemic arthritis and also ameliorated the inflammatory activity in seronegative RA. It was found that Baricitinib was not effective only in refractory systemic arthritis but also in uveitis [65].
FL has shown good efficacy and safety in Phase II and III trials, both as a MTX add-on and as mono-therapy, in MTX inadequate responders over 24 weeks [66]. Rapid and significant improvements in disease activity have been found when FL was given alone or in combination with other conventional drugs. Phase 3 randomized, controlled FINCH 3 trial found significant improvement in Health Assessment Questionnaire-Disability Index was seen at week 24 of treatment along with sign and symptoms in active RA patients [67]. Another clinical trial, (DARWIN 3, a long-term, open-label extension study) on 739 patients found FL is well-tolerated in a 4-year safety profile [68]. Using integrated data from 7 trials (NCT01668641, NCT01894516, NCT02889796, NCT02873936, NCT02886728, NCT02065700 and NCT03025308), safety of Filgotinib was evaluated in moderately to severely active rheumatoid arthritis patients. Study on 3691 patients found that over a median period of 1.6 and maximum of 5.6 years of exposure, safety or tolerability of FIL200 and FIL100 were similar. Lower incidence of infections was found with FIL200 [69].
Upadacitinib (UPD) was also evaluated in Phase III study in Methotrexate Inadequate responders (MTX IR), and a significantly higher clinical and functional efficacy was observed with UPD (68–71%) as compared to continued MTX usage (41%) for 14 weeks. In a randomized controlled phase 3 trial (SELECT-COMPARE), long-term safety and efficacy of UPD vs Adalimumab (TNF inhibitor) over 3 years was investigated in patients with active rheumatoid arthritis and inadequate MTX response. Comparative study between UPD and Adalimumab found that herpes zoster, lymphopaenia, hepatic disorder and CPK elevation were higher with UPD. However, UPD showed better clinical response than Adalimumab [70]. UPD vs placebo (PBO) and UPD vs Adalimumab was compared in RA patients who were on stable methotrexate treatment but had an inadequate response. Data from SELECT-COMPARE trial was taken for post-hoc analyses. Analysis found that UPD showed greater efficacy than Adalimumab as evidenced by DAS 28 score based on C-reactive protein [71].
To understand the mode of action of FL, secreted and cell-based biomarkers were studied from blood samples. Longitudinal analysis of these revealed that although lymphoid populations were unchanged, the RA pathophysiology related biomarkers were altered. Various key cytokine and molecules involved in mediating inflammation, leukocyte migration, angiogenesis and matrix adhesion were regulated by FL therapy [66]. Three phase III trials are also reported with FL in case of MTX-IR (Inadequate Responders), MTX-naive, bDMARD-IR patients. In MTX-IR study, FL showed significant improvement in signs and symptoms, physical function and prevented radiographic progression, compared to placebo. Also, its efficacy was found similar to Adalimumab, a TNF alpha inhibitor. In MTX naive patients, it was found that FL in combination with MTX led to significant improvements in signs and symptoms and patient-reported outcomes compared to MTX alone. Here, significant clinical response occurred as early as 2 weeks of treatment initiation [66].
Biological DMARDs
bDMARDs are class of drugs that are produced through living organisms and have a very selective anti-inflammatory mechanism of action and are usually prescribed upon failure of cDMARD therapy. bDMARDs execute their action by inhibiting mechanisms such as cytokine function or B/T cell activation. bDMARDs fall under different categories, like some of them are monoclonal and chimeric humanized fusion antibodies, whereas others are human immunoglobulin fused receptors or fused to inhibitors of certain signaling molecules.
Inflammation and hyperplasia are common manifestations in RA. There are several cytokines like TNF-α, IL-1, IL-7, IL-15, IL-17A, IL-18, IL-21, IL-23, IL-32, and IL-33 that participate in inflammatory process [72]. TNF-alpha and IL-6 are some of the key cytokines involved in RA development and progression. Monocytes, macrophages, B-cells, T-cells and fibroblasts mainly produce TNF-alpha, which is found to play a prominent role in synovitis. Fibroblasts are found to be stimulated by TNF-alpha that leads to expression of ICAM-1 [73]. Inhibiting TNF-alpha leads to reduction in the production of IL-1, IL-6, IL-8 and GM-CSF [74]. IL-6 is glycoprotein and is involved in activation of B-cell differentiation. In RA, IL-6 causes the production of autoantibodies by acting on plasmablasts [75]. IL-6 is also involved in bone destruction by inducing endothelial cells to produce IL-8 and MCP-1. This leads to the activation of adhesion molecules and leukocytes recruitment in joints [76]. Therefore, targeting TNF-alpha and IL-6 along with other cytokines can prove to be very effective in RA treatment.
TNF alpha-inhibitors
TNF alpha-inhibitors are either neutralising monoclonal antibodies (Infliximab, Adalimumab, Certolizumab), soluble TNAF-alpha receptor (Etanercept) or antibody fragments (Certolizumab pegol) [77, 78]. Their mechanism of action primarily involves inhibiting binding of TNF-alpha to their receptors on cells followed by complement-dependent response. TNF alpha inhibitor is clinically used as the second line of treatment if patients fail to respond to synthetic DMARDs. Choice of the inhibitor depends upon several factors including patient response to DMARDs, drug cost and other contributing factors.
Etanercept is a chimeric protein molecule which combines a TNF-receptor 2 subunit with the Fc domain of human IgG1 molecule. The best choice of anti-TNF therapy in RA patients was evaluated in one retrospective study. PubMed, EMBASE and Cochrane Library were searched and 72 randomized controlled trials (RCTs) with total of 28,332 subjects included. This study found that Certolizumab combined DMARDs therapy should not be recommended because of more adverse events and Etanercept monotherapy is the optimal choice for RA patients [79]. To estimate cost effectiveness of treatment with Etanercept in Japanese RA patients, markov modeling was used. This study found that the quality-adjusted life-years for the Etanercept 25 mg was increased by 0.841 compared to placebo group which suggests that maintenance treatment with Etanercept 25 mg is also cost-effective [80].
Insufficient response to TNF alpha inhibitors has also been observed but the reasons are still not completely understood. In one of the studies, potential predictive biomarkers and mechanism of insufficient response to Infliximab was explored. Analysis of differential gene expression was done on Infliximab responders and non-responders using two datasets GSE58795 (responders) and GSE78068 (non-responders). Module associated with nonresponse to Infliximab was identified by co-expression analysis and further enrichment analysis was done on the module genes. Gene signature was developed by least absolute shrink and selection operator (LASSO) regression for predicting therapeutic effect of infliximab in RA. From the two datasets, 46 common genes were obtained in which 25 gene signatures were found to have potential predictive value for infliximab. Derlin-1 (DERL1) was identified as the hub gene which is found to be involved in regulation of autophagy and immune response. Expression of DERL1 was found to increase in synovial tissue of RA patients [81]. Multi-omics approach was used in another study to compare effects of MTX, Infliximab and Tocilizumab on peripheral blood signatures during a longitudinal analysis of patients and healthy controls up to 24 weeks of treatment. Molecular phenotyping revealed better normalization of molecular signatures using Infliximab and Tocilizumab than MTX, further emphasizing the need of personalized therapy in RA [82].
Golimumab (GLM) is a human IgG1κ monoclonal antibody which neutralizes TNFα and prevents inflammation and protects cartilage degradation and bone erosion. Decreased level of serum acute phase reactants and other inflammatory biomarkers were found when given alone or in combination with MTX in phase III clinical trial [83]. Serum level of cytokines such as serum amyloid A, E-selectin, MMPs, IL-6 and TNFR II is found to be reduced in patients treated with GLM. This drug also decreases the B and T cell number and macrophages [84].
However, anti-TNF alpha therapy have side effects like formation of Anti-Drug antibodies or drug-induced sarcoidosis-like disease. Usually the affected organs are lungs, skin, and lymph nodes. Retrospective study was done on RA patients from 2000 to 2021 in which 2492 patients were included. Out of these, 697 patients had received TNF-inhibitor therapy. Four patients in which sarcoidosis were induced by anti-TNF were studied. Patient 1 and 2 was classified as incomplete Heerfordt syndrome and sarcoid-like granulomatosis respectively. Patient 3 and 4 was classified as pulmonary sarcoidosis with hilar adenopathies. Patients 1, 2 and 3 were treated with Etanercept and patient 4 was given Infliximab. Also, they found that upon removal of anti-TNF agent and treatment with glucocorticoid, all of these patients recovered [85].
IL-6/IL-6R inhibitors
IL-6 blockers constitute an important group of bDMARDs which work by either (i) directly neutralising IL-6 (Siltuximab, Sirukumab) (ii) binding to IL-6R and directly blocking the anti-inflammatory signalling mediated by IL-6 (Tocilizumab, Sarilumab).
Tociluzumab is a humanised monoclonal antibody against IL-6R and is most commonly used for RA. Study done to evaluate effectiveness and safety of Toclizumab dose in RA patients revealed low disease activity (LDA) after treatment with Toclizumab. Nationwide cohort data of RA patients was collected in South Korea. 350 patients who were treated with Toclizumab and showed low disease activity were included in this study. Study found that after the achievement of low-disease activity, tapering tocilizumab dose increases the risk of losing LDA, whereas it does not significantly affect the safety [86]. After inadequate response (IR) to janus kinase inhibitors (JAKi) and Tocilizumab, effectiveness and safety of Sarilumab in RA patients was evaluated in a prospective, observational, 24-month single-arm PROSARA study (SARILL09661). Post-baseline effectiveness assessment was documented for 502 patients out of 536 patients. It was found that Sarilumab treatment for 6 months attenuated the disease activity in JAKi-IR, Tocilizumab-IR, bDMARD TH and b/tsDMARD-naïve patients to a similar extent [87]. 40% of patients showed poor clinical response despite targeted biological treatments. In more than 50% of RA patients, CD20+B cells, which are target for Rituximab are found to be either low or absent in joint synovium. This could be one of the reasons for failure of therapy in patients receiving Rituximab. It has been hypothesized that in such patients Tocilizumab could be more effective. 48-week, biopsy-driven, multicenter, open-label, phase 4 randomized controlled trial was done to compare the effect of Tocilizumab with Rituximab in RA patients. These patients had inadequate response to anti-TNF therapy stratified for synovial B cell status. Study found that Tocilizumab was more effective than Rituximab in patients with low or absent B-cell lineage expression signature [88].
B-cell depleting antibodies
Several B-cell-targeted therapies have been investigated in the past, with Rituximab being the only FDA approved for RA patients [89,90,91]. Rituximab is a chimeric monoclonal antibody targeting CD20 surface molecule on B cells. CD20 is a calcium channel being expressed at the pre-B cell stage only.
Rituximab is recommended for RA patients who fail to respond to cDMARDs and at least one anti-TNF therapy. Meta-analysis of clinical studies have shown good efficacy when Rituximab is used in combination with MTX [92]. In one of the retrospective studies, factors associated with Rituximab discontinuation was explored between the time period from 1998 to 2020. Analysis of 404 patients found that overall 31.2% of patients discontinued treatment due to primary inefficacy and patients who had previously failed other bDMARDs showed more chances of drug discontinuation [93]. A randomized, placebo-controlled, investigator-initiated clinical trial (AMARA study) was done in RA patients to check the Rituximab plus Leflunomide treatment. Study found that Rituximab plus Leflunomide treatment showed clinical benefit compared to Leflunomide in secondary endpoints but this combination was associated with more adverse events [94]. However, incomplete B cell depletion is observed with Rituximab treatment as both memory and plasma B cells could be detected after the first infusion [90]. In addition, absence of autoantibodies, high DAS score and failure with other biologicals is found to be associated with reduced response to Rituximab [95].
Co-stimulation blocker/s
This is a new class of molecules which suppresses inflammation upstream to the inflammatory cascade. Abatacept is a chimeric antibody containing the extracellular domain of CTLA-4 fused to Fc portion of human IgG antibody. It neutralises binding of CTLA-4 part to either CD80/CD86 on activated APCs, hereafter interrupting pathways associated with T-cell activation [96, 97]. Abatacept is also found to target B cells, inhibit osteoclast differentiation and reduce expression of MMP1, 3 and 15 in fibroblast-like synoviocytes.
Abatacept has been shown to result in clinically significant disease reduction and is generally well tolerated in RA patients. A multicenter, randomized controlled study of active RA patients (n = 115) concluded significant reduction in disease activity, maintained over a period of 12 months [98]. On the contrary, another multi-centre study concluded that use of Abatacept in combination with MTX had no significant improvement in treatment response in comparison with Abatacept monotherapy [99]. In another study, effectiveness and safety of Abatacept was evaluated in biologic-naïve RA patients over a period of 52 weeks. These patients were having moderate disease activity in the prospective, 5-year, observational study (ORIGAMI study) in Japan. Analysis of 325 patients found that Abatacept significantly improved disease activity, physical disability, and quality of life for up to 52 weeks [100]. In RA patients having MTX background therapy and positive for ACPA, safety and efficacy of Abatacept was evaluated in a post hoc analysis of a randomized, double-blind, placebo-controlled phase 4 study (NCT01758198). This study found that regardless of baseline MTX dose, similar efficacy and safety of Abatacept was observed in biologic naïve ACPA-positive RA patients [101].
RANK-L inhibitor
Receptor Activator of Nuclear Factor Kappa-B Ligand (RANK-L), a TNF superfamily member, binds to RANK and is involved in osteoclast development, survival and activation. Normally produced by osteoblast cells, RANK-L expression is induced in RA joints and immune cells and FLS cells become the main producers. Therefore, neutralisation of RANK-L prevents bone erosion and destruction.
Denosumab is a recently approved human monoclonal antibody, which targets RANK-L. Denosumab is also well tolerated in patients receiving conventional therapy. One of the studies found that Denosumab suppresses joint margin erosion and prevents narrowing of the joint space. Further, cartilage turnover marker, serum Cartilage Oligomeric Matrix Protein (COMP) was found unchanged but bone metabolism marker, C-telopeptide of type I collagen (CTX-I), was found reduced [102]. In a multi-centric observational study, changes in the bone mineral density (BMD) and erosion after Denosumab discontinuation in RA patients was investigated. Primary endpoint was change in lumbar spine (LS) BMD from baseline. Study on 59 patients found that compared to baseline, increased levels of serum C-telopeptide of type I collagen was observed after Denosumab discontinuation. Increased level of CTX-1 is associated with increased bone turnover. There was no significant difference in bone erosion score between on-treatment period and after Denosumab discontinuation that tells that considering patient’s disease activity, denosumab discontinuation could be explored. On the other hand post discontinuation, numerical increase in bone erosion was observed [103]. Safety and efficacy of long-term Denosumab (60 mg dose) was evaluated in a 12 months, randomized, double-blind, placebo-controlled, multicenter phase 3 trial. After Denosumab initiation, BMD consistently increased in all groups irrespective of concomitant glucocorticoid administration. Post-Denosumab treatment, serum C-telopeptide of type 1 collagen was also found to be decreased. Study found that progression of joint destruction was inhibited after Denosumab treatment for up to 36 months. Regarding potential risk of infection there is no clear consensus on Denosumab in patients who are also receiving bDMARDs. In one of the studies, rate of infection in postmenopausal women who were receiving Denosumab and bDMARDs was compared. Similar rate of infections were found in the two groups here (4.5% vs 5%). Osteomyelitis of first metatarsal bone was other adverse events seen in bDMARDs plus Denosumab group [104]. Figure 4 is a comprehensive pictorial representation of key pathways and their targets, either approved or being explored for RA treatment.

Targeted therapy for rheumatoid arthritis (RA). T cells, B cells and macrophages play crucial roles in RA pathogenesis. This figure illustrates clinically approved and promising drugs for therapy and their respective targets. Abbreviations: APC Antigen Presenting Cell; LFA1 Lymphocyte Function-Associated Antigen 1; ICAM1 Intercellular Adhesion Molecule 1; CCL Chemokine Ligand; CXCL C-X-C Motif Chemokine Ligand; IL Interleukins; TNF Tumor Necrosis Factor; VEGF Vascular Endothelial Growth Factor; SDF1 Stromal cell Derived Factor 1; RANK Receptor Activator of Nuclear Factor k B; RANKL Receptor Activator of Nuclear Factor k B Ligand
Several drug combinations are being evaluated for their efficacy and are in different phases of clinical trials. Some of the promising and published results from Phase III and IV studies are compiled and listed in Table 2.
Secukinumab is one such promising monoclonal antibody against IL-17A, which has been tested as a long-term therapy in non-responder RA patients in a phase II study. In this 60-week long study, 237 non-responders with DMARDs and biologicals, were treated with different monthly doses of Secukinumab and significant improvement was observed with 150 mg at 52 weeks. A recent meta-analysis on 1292 non-responders to TNF inhibitor, has also revealed better clinical efficacy with 150 mg Secukimumab at 16 weeks [113]. In bDMARD refractory patients, the proportion of FL receivers (66% for 200 mg and 57.5% for 100 mg) showed a significantly better clinical response (ACR20) as compared to placebo (33%) at week 12. Thus, FL holds great potential as therapy for all types of RA patients. Few TNF inhibitors (Certolizumab Pegol and Adalimumab) in combination with MTX have been evaluated in phase III and IV trials. It is evident that MTX along with ADA offers many benefits over mono therapy. Importantly, in both MTX naive and MTX-IR patients, greater efficacy (ACR50 response) was achieved despite reported MTX-related toxicity, which remained stable [111]. In addition, comparative trial between Certolizumab Pegol in combination with MTX and Adalimumab with MTX has completed phase IV trials. The study involves 915 patients receiving 200 ml Certolizumab injections, MTX orally administrated and Adalimumab plus MTX injections. Significant difference between the drug efficacies has not been reported. Certolizumab pegol (CZP) was evaluated in Canadian adults with moderate to severe, active RA in a 2 year prospective, observational study. DAS-28 Scores (DAS28) < 2.6 at week 104 was taken as the primary objective. Improvements in Patients’ assessment of Arthritis Pain (PtAAP), fatigue, Health Assessment Questionnaire-Disability Index (HAQ-DI), and the proportion of patients achieving minimal clinically important differences (MCID) in HAQ-DI was taken as secondary endpoints. Study found that in Canadian practice, CZP was an effective RA treatment and no new CZP-related safety signals were identified [114]. A phase IV trial of combination of JAK inhibitor, Tofacitinib with MTX has been done. DMARDs have also been combined with glucocorticoids such as Prednisone and have been shown to produce good results.
Although bDMARDs are shown to be effective for RA treatment, but some patients either show inadequate response or do not respond to this line of therapy after some time. In one of the studies, researchers had included 7540 RA patients in which they found 2527 showed response to bDMARDs whereas 5013 were non-responders. The study concluded that non-responders faced a higher economic burden in terms of increased healthcare resource use, direct medical costs etc. [115]. Therefore, it is important to identify early biomarkers which can predict response to bDMARDs therapy. In one study, gene expression classifier was identified to predict response to anti-TNF Infliximab therapy, by training classifier based on published blood gene expression data sets. RA patients were treated with Infliximab and therapy response was assessed after 14–16 months post treatment. Study identified 18 signaling mechanisms associated with higher TNF-mediated inflammatory signals. Mostly these 18 markers in the classifier were found to regulate pathways associated with wounding, which is an inflammation affecting small blood vessels in the skin (FOXA2, ERBB2, IL1, MAP2K3, MST1R, NOS2, NR2F6, PPARG, S100A8) and development of nervous system (FOXA2, MEIS1, NF1, PPARG, norepinephrine, gamma secretase complex) [116]. Another study reported using machine-learning algorithm that rate of remission with TNF alpha inhibitor was 5 times more in T allele carriers of a TLR-9 gene polymorphism (rs352139) (https://pubmed.ncbi.nlm.nih.gov/34635730/). Response to Abatacept (ABA) was studied in a different study to identify responders and non-responders. Here, differential expression of 610 genes (218 genes up-regulated and 392 genes down-regulated) was observed in responders. Gene ontology analysis of 218 genes identified response Interferon type I (Type I IFN) score as a marker for ABA responsiveness in RA patients. It was observed that type I IFN score decreases in ABA treated responders vs non-responders to ABA. Further, higher expression levels of nine genes (BATF2, LAMP3, CD82, CLEC4A, IDO1, STAT1, STAT2 and TNFSF10) was observed in ABA responders [117]. Similarly, type I IFN network genes (LY6E, HERC5, IFI44L, ISG15, MxA, MxB, EPSTI1 and RSAD2) can also be used to discriminate responders and non-responders in Rituximab-treated RA patients. Lower expression levels of these genes were found to be associated with responders [117]. In another study, thirty-two patients who were treated with Anakinara (100 mg/day) (IL-1 receptor antagonist), in combination with MTX were studied to identify responsiveness to treatment. Gene expression profiling of PBMCs isolated from treated patients identified 52 transcripts that can be used to discriminate responders and non-responders to combination therapy of Anakinara and MTX. Study identified 34 genes out of which 56% were found to have role in IL-1β-dependent pathway. Further analysis found that these genes are regulated by transcription factors, not very specific to IL-1-β pathways. Some of the identified transcription factors and their targets are: JUN (BST2), CEBPβ (RUNX1T1, ELF2), HIF1A (EP300), ESR1 (EMP2), CTNNB1 (CDH5, EIFS12), TP52 (CDK8) and STAT3 (LEPR). IL-1β pathway associated genes like co-stimulator ligand F (ICOSLG) and Transthyretin (TTR) were also identified in this study. In one of the studies, Anti-Drug Antibody (ADA) against GLM was explored in RA patients to assess the clinical response. ADA is found to be associated with low drug levels and low response rates. Lower GLM levels were found in non-responder RA patients as compared to responders after 28 weeks of treatment [119]. Some of the common side effects of using bDMARDs are infection of bacteria, fungus or viruses [120]. In few cases, tuberculosis reactivation is also observed [121]. Further, suppression of bone marrow and liver toxicity has been found to be associated with bDMARDs. In certain cases, congestive heart failure and demyelination of nervous system was reported with anti-TNF agents. Side effects associated with IL-6 inhibitors are hyperlipidemia and pancytopenia. Inflammatory bowel disease can be worsened by IL-17 inhibitors. Multifocal leukoencephalopathy has been observed in Rituximab-treated patients [122].
Novel therapeutic approaches
Epigenetic modifications such as DNA methylation and histone modifications are also found associated with RA pathogenesis [123]. Distinct epigenetic clinical markers are currently being explored for early diagnosis and targeted therapy. Presently, inhibitors of DNA Methyltransferase (DNMT) and Histone Deacetylase Inhibitors (HDAC) are available as therapeutic drugs. Preclinical studies have concluded that HDAC inhibitors are involved in reducing inflammation, edema, synovial angiogenesis and joint damage [124,125]. A HDAC inhibitor, Trichostatin A, has been shown to interfere with production of inflammatory mediators in synoviocytes [126]. Also, MI192 is shown to exhibit an inhibitory effect on expression of TNF and IL-6 [127]. Other HDAC inhibitors (Vorinostat, Entinostat) were found to repress NF-kB pathway in synovial fibroblasts thereby downregulating inflammatory cytokines. A case–control study evaluated effect of a DNMT inhibitor, delineating its effect on hypomethylation of a gene (SFRP4) involved in RA pathogenesis [128]. However, epigenetic therapy is still in its nascent stage for RA and clinical trials are required for its validation.
In addition, inhibiting PAD4 activity, which is critical for generation of citrullinated proteins responsible for disease pathogenesis, could also be an effective therapeutic strategy. Though several reversible (streptonigrin, GSK199, GSK484) and irreversible inhibitors (Cl-amidine, F-amidine, YW-356, TDFA, and TCDA) have emerged, their clinical efficacy is yet to be proven in clinical trials. Synthesising novel PAD4 inhibitors which may synergistically and effectively target hypercitrullination and NET (Neutrophil Extracellular Traps) formation might be a good approach in future.
Gene therapy, specific DNA or RNA, is administered using viral vectors to modify expression of the gene of interest. In development and progression of RA, overproduction of inflammatory cytokines by FLS cells play a very important role. Therefore, inhibiting pro-inflammatory cytokines or overexpression of anti-inflammatory cytokines are strategies for RA therapy [3]. In Collagen-Induced Arthritis (CIA) animal models, gene therapy with IL-4 and IL-10 showed protection of joint and reversed degradation of cartilage but clinical trials have not shown much efficacy. In a novel approach, fusion protein of IL-4 and IL-10 (IL4-IL10 FP) was employed for RA therapy. In this synergetic approach, glycosylated IL-4-IL-10 FP showed decreased severity of proteoglycan-induced arthritis (PGIA) in mice [129]. In animal models, administration of the immunosuppressive cytokine IL-35 significantly exacerbated RA progression which could be due to indirect effect of IL-35 on the Th17 [130].
miRNAs also play a key role in the regulation of inflammatory responses and could be effective therapeutic targets in the future. In cells and tissues of RA patients, upregulation of miR-155 and miR-146a was found. Increased expression of miR-155 in RA patients is found to be associated with repression of MMPs. In RA patients, upregulation of miR-146a causes persistent production of TNF-alpha [131]. Inhibition of proliferation, migration and invasion of RA-FLS cells was observed after downregulating miR-135a [132]. In another study, miRNA-21 inhibition in RA-FLSs led to decreased proliferation of RA-FLSs. On the other hand, overexpression of miRNA-21 increased the rate of proliferation of normal FLSs [133]. Expression of MMP-3/13 and IL-1β was found to be inhibited by miR-124a [134]. In FLS cells from RA patients, miR-27a expression was found to be significantly decreased in serum, synovial tissues, and FLS compared to healthy controls. miR-27a targets pro-inflammatory mediators such as Follistatin-Like protein 1 (FSTL1). miR-27a overexpression downregulates expression of MMPs and Rho family proteins [135]. In RA-FLSs, gastric adenocarcinoma predictive long intergenic noncoding RNA is found to be overexpressed. Proliferation and migration of FLSs is found to be negatively regulated by Lowly expressed in rheumatoid fibroblast-like synoviocytes (LEFRS) lncRNA. Overexpression of zinc finger NFX1-type 1 containing 1 antisense RNA 1 (ZFSA1) is also found in RA-FLS. LEFRS positively regulates the invasion and migration of FLSs [136]. In PBMCs of RA patients, Nuclear Enriched Abundant Transcript 1 (NEAT1) is found to be overexpressed. This lncRNA is involved in restrained immune cell differentiation and helps in decreasing inflammation in CIA mice [137].
Mesenchymal stem cell (MSC) therapy could be another therapeutic option for RA treatment because it can exert immunosuppressive functions in both adaptive and innate immune cells. There are 14 MSC-based therapies listed in clinical trials for RA. Reduced erythrocyte sedimentation rate, improvement on DAS28 clinical score and diminished on the serum anti-cyclic citrullinated peptide (anti-CCP) antibody level was found upon intravenous infusion of allogenic bone marrow and umbilical cord-derived MSC in a small group of refractory RA patients. These patients were resistant to the anti-TNF monoclonal antibody therapy [138]. In a study, safety and effectiveness of allogenic UC-MSCs were demonstrated in large number of RA patients. In 172 active RA patients, MSCs and DMARDs were co-administered intravenously which resulted in significant increase in the percentage of regulatory CD4+T cells in the blood. For up to 6 months, significant clinical improvement was also seen [139]. However further clinical studies with conclusive results are required to demonstrate their safety and efficacy.
Summary and conclusion
Management of Rheumatoid Arthritis depends on early diagnosis and identifying various factors to minimise disease progression. The challenge still lies in the early identification of the disease before it has progressed to the clinical stage of joint damage. Efforts are ongoing to develop novel biomarkers for this. Therapeutic approaches, using conventional DMARDs comprising of MTX, Leflunamide, Sulfasalzine and Hydroxycholoroquine, have proven to be very effective. However, there is still a non-responder or insufficient responder population. To overcome this, a combination therapy approach is employed using either two DMARDs as the primary line of action or using a cDMARD and targeted DMARD/bDMARD combination in insufficient responders. Various such novel combinations, are undergoing clinical trials, and hold great promise . More efforts are required to find key biomarkers to stratify patients based on disease severity and responder/non-responder population. Since various adverse events are associated with cDMARDs and bDMARDs both, exploring gene therapy approaches and mesenchymal stem cell-based therapy would be beneficial for RA patients in future. Further, various nanoparticle based formulations of Methotrexate being developed would also help navigate the adverse effects because of targeted release of the drug.
Data availability
Not applicable.
Code availability
BioRender software (free trial version for Figures).
References
Guo Q, Wang Y, Xu D, Nossent J, Pavlos NJ, Xu J (2018) Rheumatoid arthritis: pathological mechanisms and modern pharmacologic therapies. Bone Res 6(1):1–4. https://doi.org/10.1038/s41413-018-0016-9
Smolen JS, Aletaha D, McInnes IB (2016) Rheumatoid arthritis.Erratum. Lancet 388(10055):2023–2038. https://doi.org/10.1016/S0140-6736(16)30173-8. Epub 2016 May. PMID: 27156434.
Yap H-Y, Tee S, Wong M, Chow S-K, Peh S-C, Teow S-Y (2018) Pathogenic role of immune cells in rheumatoid arthritis: implications in clinical treatment and biomarker development. Cells 7(10):161. https://doi.org/10.3390/cells7100161
Kurkó J, Besenyei T, Laki J, Glant TT, Mikecz K, Szekanecz Z (2013) Genetics of rheumatoid arthritis—a comprehensive review. Clin Rev Allergy Immunol 45(2):170–179. https://doi.org/10.1007/s12016-012-8346-7
Jeffery RC (2014) Clinical features of rheumatoid arthritis. Medicine (United Kingdom) 42(5):231–236. https://doi.org/10.1016/j.mpmed.2014.02.011
Friedman B, Cronstein B (2019) Methotrexate mechanism in treatment of rheumatoid arthritis. Jt Bone Spine 86(3):301–307. https://doi.org/10.1016/j.jbspin.2018.07.004
Schrezenmeier E, Dörner T (2020) Mechanisms of action of hydroxychloroquine and chloroquine: implications for rheumatology. Nat Rev Rheumatol 16(3):155–166. https://doi.org/10.1038/s41584-020-0372-x
Dhillon S (2017) Tofacitinib: a review in rheumatoid arthritis. Drugs 77(18):1987–2001. https://doi.org/10.1007/s40265-017-0835-9
Al-Salama ZT, Scott LJ (2018) Baricitinib: a review in rheumatoid arthritis. Drugs 78(7):761–772. https://doi.org/10.1007/s40265-018-0908-4
Zhao S, Mysler E, Moots RJ (2018) Etanercept for the treatment of rheumatoid arthritis. Immunotherapy 10(6):433–445. https://doi.org/10.2217/imt-2017-0155
Radner H, Aletaha D (2015) Anti-TNF therapie in der rheumatoiden Arthritis—ein Überblick. Wien Med Wochenschr 165(1–2):3–9. https://doi.org/10.1007/s10354-015-0344-y
Novella-Navarro M, Plasencia C, Tornero C et al (2020) Clinical predictors of multiple failure to biological therapy in patients with rheumatoid arthritis. Arthritis Res Ther 22(1):1–8. https://doi.org/10.1186/s13075-020-02354-1
van Vollenhoven R (2019) Treat-to-target in rheumatoid arthritis—are we there yet? Nat Rev Rheumatol 15(3):180–186. https://doi.org/10.1038/s41584-019-0170-5
Arora S, Rafiq A, Jolly M (2016) Management of rheumatoid arthritis: review of current guidelines. J Arthrosc Jt Surg 3(2):45–50. https://doi.org/10.1016/j.jajs.2016.07.002
Lau CS, Chia F, Dans L et al (2019) 2018 update of the APLAR recommendations for treatment of rheumatoid arthritis. Int J Rheum Dis 22(3):357–375. https://doi.org/10.1111/1756-185X.13513
Aletaha D, Smolen J, Ward MM (2006) Measuring function in rheumatoid arthritis: identifying reversible and irreversible components. Arthritis Rheum 54(9):2784–2792. https://doi.org/10.1002/art.22052
Smolen JS, Aletaha D, Grisar JC, Stamm TA, Sharp JT (2010) Estimation of a numerical value for joint damage-related physical disability in rheumatoid arthritis clinical trials. Ann Rheum Dis 69(6):1058–1064. https://doi.org/10.1136/ard.2009.114652
Aletaha D, Alasti F, Smolen JS (2016) Optimisation of a treat-to-target approach in rheumatoid arthritis: strategies for the 3-month time point. Ann Rheum Dis 75(8):1479–1485. https://doi.org/10.1136/annrheumdis-2015-208324
van Drongelen V, Holoshitz J (2017) Human leukocyte antigen-disease associations in rheumatoid arthritis. Rheum Dis Clin North Am 43(3):363–376. https://doi.org/10.1016/j.rdc.2017.04.003
Fugger L, Svejgaard A (2000) Association of MHC and rheumatoid arthritis HLA-DR4 and rheumatoid arthritis: studies in mice and men. Arthritis Res 2(3):208–211. https://doi.org/10.1186/ar89
Nemtsova MV, Zaletaev DV, Bure IV et al (2019) Epigenetic changes in the pathogenesis of rheumatoid arthritis. Front Genet 14(10):1–13. https://doi.org/10.3389/fgene.2019.00570
Karouzakis E, Raza K, Kolling C et al (2018) Analysis of early changes in DNA methylation in synovial fibroblasts of RA patients before diagnosis. Sci Rep 8(1):1–6. https://doi.org/10.1038/s41598-018-24240-2
Karouzakis E, Gay RE, Gay S, Neidhart M (2012) Increased recycling of polyamines is associated with global DNA hypomethylation in rheumatoid arthritis synovial fibroblasts. Arthritis Rheum 64(6):1809–1817. https://doi.org/10.1002/art.34340
Ahmed S, Riegsecker S, Beamer M et al (2013) Largazole, a class I histone deacetylase inhibitor, enhances TNF-α-induced ICAM-1 and VCAM-1 expression in rheumatoid arthritis synovial fibroblasts. Toxicol Appl Pharmacol 270(2):87–96. https://doi.org/10.1016/j.taap.2013.04.014
Wang L, Ding Y, Guo X, Zhao Q (2015) Role and mechanism of vascular cell adhesion molecule-1 in the development of rheumatoid arthritis. Exp Ther Med 10(3):1229–1233. https://doi.org/10.3892/etm.2015.2635
Ai R, Laragione T, Hammaker D et al (2018) Comprehensive epigenetic landscape of rheumatoid arthritis fibroblast-like synoviocytes. Nat Commun 9(1):1–1. https://doi.org/10.1038/s41467-018-04310-9
Chang K, Yang SM, Kim SH, Han KH, Park SJ, Il SJ (2014) Smoking and rheumatoid arthritis. Int J Mol Sci 15(12):22279–22295. https://doi.org/10.3390/ijms151222279
Ozguner F, Koyu A, Cesur G (2005) Active smoking causes oxidative stress and decreases blood melatonin levels. Toxicol Ind Health 21(10):21–26. https://doi.org/10.1191/0748233705th211oa
Bijl M, Horst G, Limburg PC, Kallenberg CGM (2001) Effects of smoking on activation markers, Fas expression and apoptosis of peripheral blood lymphocytes. Eur J Clin Invest 31(6):550–553. https://doi.org/10.1046/j.1365-2362.2001.00842.x
Shizu M, Itoh Y, Sunahara R et al (2007) Cigarette smoke condensate upregulates the gene and protein expression of proinflammatory cytokines in human fibroblast-like synoviocyte line. J Interferon Cytokine Res 28(8):509–521. https://doi.org/10.1089/jir.2007.0081
Lee J, Taneja V, Vassallo R (2012) Cigarette smoking and inflammation: cellular and molecular mechanisms. J Dent Res 91(2):142–149. https://doi.org/10.1177/0022034511421200
Xue M, McKelvey K, Shen K et al (2014) Endogenous MMP-9 and not MMP-2 promotes rheumatoid synovial fibroblast survival, inflammation and cartilage degradation. Rheumatol (United Kingdom) 53(12):2270–2279. https://doi.org/10.1093/rheumatology/keu254
Cribbs A, Feldmann M, Oppermann U (2015) Towards an understanding of the role of DNA methylation in rheumatoid arthritis: therapeutic and diagnostic implications. Ther Adv Musculoskelet Dis 7(5):206–219. https://doi.org/10.1177/1759720X15598307
Lu B, Solomon DH, Costenbader KH, Karlson EW (2014) Alcohol consumption and risk of incident rheumatoid arthritis in women: a prospective study. Arthritis Rheumatol 66(8):1998–2005. https://doi.org/10.1002/art.38634
Forsyth C, Kouvari M, D’Cunha NM et al (2018) The effects of the mediterranean diet on rheumatoid arthritis prevention and treatment: a systematic review of human prospective studies. Rheumatol Int 38(5):737–747. https://doi.org/10.1007/s00296-017-3912-1
Khanna S, Jaiswal KS, Gupta B (2017) Managing rheumatoid arthritis with dietary interventions. Front Nutr. https://doi.org/10.3389/fnut.2017.00052
Kostoglou-Athanassiou I, Athanassiou P, Lyraki A, Raftakis I, Antoniadis C (2012) Vitamin D and rheumatoid arthritis. Ther Adv Endocrinol Metab 3(6):181–187. https://doi.org/10.1177/2042018812471070
Smolen JS, Steiner G (2003) Therapeutic strategies for rheumatoid arthritis. Nat Rev Drug Discov 2(6):473–488. https://doi.org/10.1038/nrd1109
Smolen JS, Landewé RBM, Bijlsma JWJ, Burmester GR, Dougados M, Kerschbaumer A et al (2020) EULAR recommendations for the management of rheumatoid arthritis with synthetic and biological disease- modifying antirheumatic drugs: 2019 update. Ann Rheum Dis 79(6):S685–S699
Mazouyès A, Clay M, Bernard AC, Gaudin P, Baillet A (2017) Efficacy of triple association methotrexate, sulfasalazine and hydroxychloroquine in early treatment of rheumatoid arthritis with insufficient response to methotrexate: meta-analysis of randomized controlled trials. Jt Bone Spine 84(5):563–570. https://doi.org/10.1016/j.jbspin.2016.10.010
Herrmann ML, Schleyerbach R, Kirschbaum BJ (2000) Leflunomide: an immunomodulatory drug for the treatment of rheumatoid arthritis and other autoimmune diseases. Immunopharmacology 47(2–3):273–289. https://doi.org/10.1016/S0162-3109(00)00191-0
Hirohata S, Ohshima N, Yanagida T, Aramaki K (2002) Regulation of human B cell function by sulfasalazine and its metabolites. Int Immunopharmacol 2(5):631–640. https://doi.org/10.1016/S1567-5769(01)00186-2
Rainsford KD, Parke AL, Clifford-Rashotte M, Kean WF (2015) Therapy and pharmacological properties of hydroxychloroquine and chloroquine in treatment of systemic lupus erythematosus, rheumatoid arthritis and related diseases. Inflammopharmacology 23(5):231–269. https://doi.org/10.1007/s10787-015-0239-y
Taylor PC, Abdul Azeez M, Kiriakidis S (2017) Filgotinib for the treatment of rheumatoid arthritis. Expert Opin Investig Drugs 26(10):1181–1187. https://doi.org/10.1080/13543784.2017.1372422
Jegatheeswaran J, Turk M, Pope JE (2019) Comparison of Janus kinase inhibitors in the treatment of rheumatoid arthritis: a systemic literature review. Immunotherapy 11(8):737–754. https://doi.org/10.2217/imt-2018-0178
Bernal J, Vela P, Ruiz García V, Burls A, Cabello J, Bort-Martí S (2017) FRI0199 systematic review and meta-analysis on certolizumab pegol for rheumatoid arthritis in adults. Rheum Dis. https://doi.org/10.1136/annrheumdis-2017-eular.6433
Jones G, Wallace T, McIntosh MJ, Brockwell L, Gomez-Reino JJ, Sebba A (2017) Five-year efficacy and safety of tocilizumab monotherapy in patients with rheumatoid arthritis who were methotrexate-and biologic-naive or free of methotrexate for 6 months: the ambition study. J Rheumatol 44(2):142–146. https://doi.org/10.3899/jrheum.160287
Ruscitti P, Cipriani P, Liakouli V et al (2018) The emerging role of IL-1 inhibition in patients affected by rheumatoid arthritis and diabetes. Rev Recent Clin Trials 13(3):210–214. https://doi.org/10.2174/1574887113666180314102651
Blair HA, Deeks ED (2017) Abatacept: a review in rheumatoid arthritis. Drugs 77(11):1221–1233. https://doi.org/10.1007/s40265-017-0775-4
Chiu YG, Ritchlin CT (2017) Denosumab: targeting the RANKL pathway to treat rheumatoid arthritis. Expert Opin Biol Ther 17(1):119–128. https://doi.org/10.1080/14712598.2017.1263614
It T, Effects S. Denosumab (Prolia)
Crotti C, Raimondo MG, Becciolini A, Biggioggero M, Favalli EG (2017) Spotlight on mavrilimumab for the treatment of rheumatoid arthritis: evidence to date. Drug Des Devel Ther 11:211–223. https://doi.org/10.2147/DDDT.S104233
Sherbini AA, Gwinnutt JM, Hyrich KL et al (2022) Rates and predictors of methotrexate-related adverse events in patients with early rheumatoid arthritis: results from a nationwide UK study. Rheumatology. https://doi.org/10.1093/rheumatology/keab917
Yan H, Su R, Xue H, Gao C, Li X, Wang C (2021) Pharmacomicrobiology of methotrexate in rheumatoid arthritis: gut microbiome as predictor of therapeutic response. Front Immunol 12:1–15. https://doi.org/10.3389/fimmu.2021.789334
Sergeant JC, Hyrich KL, Anderson J et al (2018) Prediction of primary non-response to methotrexate therapy using demographic, clinical and psychosocial variables: results from the UK Rheumatoid Arthritis Medication Study (RAMS). Arthritis Res Ther 20(1):1–11. https://doi.org/10.1186/s13075-018-1645-5
Artacho A, Isaac S, Nayak R et al (2021) The pretreatment gut microbiome is associated with lack of response to methotrexate in new-onset rheumatoid arthritis. Arthritis Rheumatol 73(6):931–942. https://doi.org/10.1002/art.41622
Hambardzumyan K, Bolce RJ, Wallman JK, Van Vollenhoven RF, Saevarsdottir S (2019) Serum biomarkers for prediction of response to methotrexate monotherapy in early rheumatoid arthritis: results from the SWEFOT trial. J Rheumatol 46(6):555–563. https://doi.org/10.3899/jrheum.180537
Fragoulis GE, Mcinnes IB, Siebert S (2019) JAK-inhibitors. new players in the field of immune-mediated diseases, beyond rheumatoid arthritis. Rheumatology (United Kingdom) 58:i43–i54
Harigai M (2019) Growing evidence of the safety of JAK inhibitors in patients with rheumatoid arthritis. Rheumatology (United Kingdom) 58(1):i34–i42. https://doi.org/10.1093/rheumatology/key287
Lunzer R (2014) Tofacitinib versus methotrexate in rheumatoid arthritis: kommentar. J Miner 21(4):157. https://doi.org/10.1056/NEJMoa1310476
Fleischmann R, Mysler E, Hall S et al (2017) Efficacy and safety of tofacitinib monotherapy, tofacitinib with methotrexate, and adalimumab with methotrexate in patients with rheumatoid arthritis (ORAL Strategy): a phase 3b/4, double-blind, head-to-head, randomised controlled trial. Lancet 390(10093):457–468. https://doi.org/10.1016/S0140-6736(17)31618-5
Mogul A, Corsi K, McAuliffe L (2019) Baricitinib: the second FDA-approved JAK inhibitor for the treatment of rheumatoid arthritis. Ann Pharmacother 53(9):947–953. https://doi.org/10.1177/1060028019839650
Hernández-Cruz B, Rosas J, Díaz-Torné C et al (2022) Real-world treatment patterns and clinical outcomes of baricitinib in rheumatoid arthritis patients in spain: results of a multicenter, observational study in routine clinical practice (The ORBIT-RA Study). Rheumatol Ther 9(2):589–608. https://doi.org/10.1007/s40744-021-00423-8
Taylor PC, Takeuchi T, Burmester GR et al (2022) Safety of baricitinib for the treatment of rheumatoid arthritis over a median of 4.6 and up to 9.3 years of treatment: final results from long-term extension study and integrated database. Ann Rheum Dis 81(3):335–343. https://doi.org/10.1136/annrheumdis-2021-221276
Kaneko Y, Murakami T, Nishitsuka K, Takakubo Y, Takagi M, Yamashita H (2022) Effectiveness of baricitinib in refractory seronegative rheumatoid arthritis and uveitis: a case report. Front Med 8:1–6. https://doi.org/10.3389/fmed.2021.764067
Westhovens R, Taylor PC, Alten R et al (2017) Filgotinib (GLPG0634/GS-6034), an oral JAK1 selective inhibitor, is effective in combination with methotrexate (MTX) in patients with active rheumatoid arthritis and insufficient response to MTX: results from a randomised, dose-finding study (DARWIN 1). Ann Rheum Dis 76(6):998–1008. https://doi.org/10.1136/annrheumdis-2016-210104
Westhovens R, Rigby WFC, Van Der Heijde D et al (2021) Filgotinib in combination with methotrexate or as monotherapy versus methotrexate monotherapy in patients with active rheumatoid arthritis and limited or no prior exposure to methotrexate: the phase 3, randomised controlled FINCH 3 trial. Ann Rheum Dis 80(6):727–738. https://doi.org/10.1136/annrheumdis-2020-219213
Kavanaugh A, Westhovens RR, Winthrop KL et al (2021) Safety and efficacy of filgotinib: Up to 4-year results from an open-label extension study of phase II rheumatoid arthritis programs. J Rheumatol 48(8):1230–1238. https://doi.org/10.3899/jrheum.201183
Winthrop KL, Tanaka Y, Takeuchi T et al (2022) Integrated safety analysis of filgotinib in patients with moderately to severely active rheumatoid arthritis receiving treatment over a median of 1.6 years. Ann Rheum Dis 81(2):184–192. https://doi.org/10.1136/annrheumdis-2021-221051
Fleischmann R, Mysler E, Bessette L et al (2022) Long-term safety and efficacy of upadacitinib or adalimumab in patients with rheumatoid arthritis: results through 3 years from the SELECT-COMPARE study. RMD Open 8(1):1–12. https://doi.org/10.1136/rmdopen-2021-002012
Conaghan P, Cohen S, Burmester G et al (2022) Benefit-risk analysis of upadacitinib compared with adalimumab in the treatment of patients with moderate-to-severe rheumatoid arthritis. Rheumatol Ther 9(1):191–206. https://doi.org/10.1007/s40744-021-00399-5
Koch AE (2007) The pathogenesis of rheumatoid arthritis. Am J Orthop (Belle Mead NJ) 36(7):5–8
Tilders FJ, DeRuk RH, Van Dam AM, Vincent VA, Schotanus K, Persoons JH (1994) Activation of the hypothalamus-pituitary-adrenal axis by bacterial endotoxins: routes and intermediate signals. Psychoneuroendocrinology 19(2):209–232
Haworth C, Brennan FM, Chantry D, Turner M, Maini RN, Feldmann M (1991) Expression of granulocyte-macrophage colony-stimulating factor in rheumatoid arthritis: regulation by tumor necrosis factor-α. Eur J Immunol 21(10):2575–2579. https://doi.org/10.1002/eji.1830211039
Suematsu S, Matsuda T, Aozasa K et al (1989) IgG1 plasmacytosis in interleukin 6 transgenic mice. Proc Natl Acad Sci USA 86(19):7547–7551. https://doi.org/10.1073/pnas.86.19.7547
Suzuki M, Hashizume M, Yoshida H, Mihara M (2010) Anti-inflammatory mechanism of tocilizumab, a humanized anti-IL-6R antibody: effect on the expression of chemokine and adhesion molecule. Rheumatol Int 30(3):309–315. https://doi.org/10.1007/s00296-009-0953-0
Nestorov I (2005) Clinical pharmacokinetics of TNF antagonists: how do they differ? Semin Arthritis Rheum 34(5):12–18. https://doi.org/10.1016/j.semarthrit.2005.01.004
Voulgari PV, Drosos AA (2006) Adalimumab for rheumatoid arthritis. Expert Opin Biol Ther 6(12):1349–1360. https://doi.org/10.1517/14712598.6.12.1349
He B, Li Y, Luo W et al (2022) The risk of adverse effects of TNF-α inhibitors in patients with rheumatoid arthritis: a network meta-analysis. Front Immunol 13:1–16. https://doi.org/10.3389/fimmu.2022.814429
Hirose T, Kawaguchi I, Murata T, Atsumi T (2022) Cost-effectiveness analysis of etanercept 25 mg maintenance therapy after treatment with etanercept 50 mg for moderate rheumatoid arthritis in the PRESERVE trial in japan. Value Heal Reg Issues 28:105–111. https://doi.org/10.1016/j.vhri.2021.06.012
Cai Y, Xu K, Aihaiti Y et al (2022) Derlin-1, as a potential early predictive biomarker for nonresponse to infliximab treatment in rheumatoid arthritis is related to autophagy. Front Immunol 12:1–15. https://doi.org/10.3389/fimmu.2021.795912
Tasaki S, Suzuki K, Kassai Y et al (2018) Multi-omics monitoring of drug response in rheumatoid arthritis in pursuit of molecular remission. Nat Commun 9(1):2755. https://doi.org/10.1038/s41467-018-05044-4
Pelechas E, Voulgari PV, Drosos AA (2019) Golimumab for rheumatoid arthritis. J Clin Med 8(3):387. https://doi.org/10.3390/jcm8030387
Tanaka Y, Senoo A, Fujii H, Baker D (2016) Evaluation of golimumab for the treatment of patients with active rheumatoid arthritis. Expert Opin Drug Metab Toxicol 12(3):319–326. https://doi.org/10.1517/17425255.2016.1146682
Theunssens X, Bricman L, Dierckx S et al (2022) Anti-TNF induced sarcoidosis-like disease in rheumatoid arthritis patients: review cases from the RA UCLouvain brussels cohort. Rheumatol Ther 9(2):763–770. https://doi.org/10.1007/s40744-022-00424-1
Park JW, Kim MJ, Kim H-A, Kim JH, Lee EB, Shin K (2022) The aftermath of tapering tocilizumab after achieving treatment target in patients with rheumatoid arthritis: a nationwide cohort study. Front Med 9:1–9. https://doi.org/10.3389/fmed.2022.839206
Tony H-P, Feist E, Aries PM et al (2022) Sarilumab reduces disease activity in rheumatoid arthritis patients with inadequate response to janus kinase inhibitors or tocilizumab in regular care in Germany. Rheumatol Adv Pract 6(1):1–7. https://doi.org/10.1093/rap/rkac002
Humby F, Durez P, Buch MH et al (2021) Rituximab versus tocilizumab in anti-TNF inadequate responder patients with rheumatoid arthritis (R4RA): 16-week outcomes of a stratified, biopsy-driven, multicentre, open-label, phase 4 randomised controlled trial. Lancet 397(10271):305–317. https://doi.org/10.1016/S0140-6736(20)32341-2
Cross AH, Stark JL, Lauber J, Ramsbottom MJ, Lyons JA (2006) Rituximab reduces B cells and T cells in cerebrospinal fluid of multiple sclerosis patients. J Neuroimmunol 180(1–2):63–70. https://doi.org/10.1016/j.jneuroim.2006.06.029
Pollastro S, Klarenbeek PL, Doorenspleet ME et al (2019) Non-response to rituximab therapy in rheumatoid arthritis is associated with incomplete disruption of the B cell receptor repertoire. Ann Rheum Dis 78(10):1339–1345. https://doi.org/10.1136/annrheumdis-2018-214898
Oldroyd AGS, Symmons DPM, Sergeant JC et al (2018) Long-term persistence with rituximab in patients with rheumatoid arthritis. Rheumatology (United Kingdom) 57(6):1089–1096. https://doi.org/10.1093/rheumatology/key036
Wang Z, Bao HW, Ji Y, Li Y (2020) A systematic review and meta-analysis of rituximab combined with methotrexate versus methotrexate alone in the treatment of rheumatoid arthritis. Medicine (United States) 99(8):e19193. https://doi.org/10.1097/MD.0000000000019193
Norris-Grey C, Cambridge G, Moore S, Reddy V, Leandro M (2022) Long-term persistence of rituximab in patients with rheumatoid arthritis: an evaluation of the UCL cohort from 1998 to 2020. Rheumatology (Oxford) 61(2):591–596. https://doi.org/10.1093/rheumatology/keab248
Behrens F, Koehm M, Rossmanith T et al (2021) Rituximab plus leflunomide in rheumatoid arthritis: a randomized, placebo-controlled, investigator-initiated clinical trial (AMARA study). Rheumatology 60(11):5318–5328. https://doi.org/10.1093/rheumatology/keab153
Scher JU (2012) B-cell therapies for rheumatoid arthritis. Bull NYU Hosp Jt Dis 70(3):200–203
Cron RQ (2005) Editorial: a signal achievement in the treatment of arthritis. Arthritis Rheum 52(8):2229–2232. https://doi.org/10.1002/art.21206
Pombo-Suarez M, Gomez-Reino JJ (2019) Abatacept for the treatment of rheumatoid arthritis. Expert Rev Clin Immunol 15(4):319–326. https://doi.org/10.1080/1744666X.2019.1579642
Kremer JM, Dougados M, Emery P et al (2005) Treatment of rheumatoid arthritis with the selective costimulation modulator abatacept: twelve-month results of a phase IIb, double-blind, randomized, placebo-controlled trial. Arthritis Rheum 52(8):2263–2271. https://doi.org/10.1002/art.21201
Pascart T, Philippe P, Drumez E et al (2019) Abatacept monotherapy versus abatacept plus methotrexate for treatment-refractory rheumatoid arthritis. Am J Ther 26(3):E358–E363. https://doi.org/10.1097/MJT.0000000000000645
Tamura N, Azuma T, Misaki K et al (2021) Effectiveness and safety of subcutaneous abatacept in biologic-naïve RA patients at Week 52: a japanese multicentre investigational study (ORIGAMI study). Mod Rheumatol 32(3):1–11. https://doi.org/10.1093/mr/roab090
Tanaka Y, Matsubara T, Hashizume K, Amano N, Takeuchi T (2021) Efficacy and safety of abatacept in biologic-naïve patients with active rheumatoid arthritis by background methotrexate dose: post hoc analysis of a randomized, placebo-controlled, phase 4 study. Mod Rheumatol 32(3):1–8. https://doi.org/10.1093/mr/roab029
Takeuchi T, Tanaka Y, Soen S et al (2019) Effects of the anti-RANKL antibody denosumab on joint structural damage in patients with rheumatoid arthritis treated with conventional synthetic disease-modifying antirheumatic drugs (DESIRABLE study): a randomised, double-blind, placebo-controlled phase. Ann Rheum Dis 78(7):899–907. https://doi.org/10.1136/annrheumdis-2018-214827
Tanaka S, Kobayashi M, Saito K, Takita A (2022) Correction to: impact of denosumab discontinuation on changes in bone mineral density and bone erosion in rheumatoid arthritis patients. Mod Rheumatol 32(2):292–295. https://doi.org/10.1093/mr/roab102
Mirzaei A, Jahed SA, Kadijani AA, Zabihiyeganeh M (2021) Risk of infection in postmenopausal women with rheumatoid arthritis and osteoporosis taking denosumab and bDMARDS. Med J Islam Repub Iran 35(1):1–4. https://doi.org/10.47176/mjiri.35.12
Smolen JS, Pangan AL, Emery P et al (2019) Upadacitinib as monotherapy in patients with active rheumatoid arthritis and inadequate response to methotrexate (SELECT-MONOTHERAPY): a randomised, placebo-controlled, double-blind phase 3 study. Lancet 393(10188):2303–2311. https://doi.org/10.1016/S0140-6736(19)30419-2
Strand V, Mysler E, Moots RJ et al (2019) Patient-reported outcomes for tofacitinib with and without methotrexate, or adalimumab with methotrexate, in rheumatoid arthritis: a phase IIIB/IV trial. RMD Open 5(2):1–10. https://doi.org/10.1136/rmdopen-2019-001040
NCT01202760. A Rheumatoid Arthritis Study in Participants [Internet], 2010. http://cochranelibrarywiley.com/o/cochrane/clcentral/articles/205/CN-01524205/frame.html
Weinblatt ME, Baranauskaite A, Dokoupilova E et al (2018) Switching from reference adalimumab to SB5 (Adalimumab Biosimilar) in Patients With rheumatoid arthritis: fifty-two–week phase III randomized study results. Arthritis Rheumatol 70(6):832–840. https://doi.org/10.1002/art.40444
A Study of Certolizumab Pegol as Additional Therapy in Chinese Patients With Active Rheumatoid Arthri- tis (RAPID-C), NCT02151851. 2018.
Study to Assess the Short- and Long-term Efficacy of Certolizumab Pegol Plus Methotrexate Compared to Adalimumab Plus Methotrexate in Subjects With Moderate to Severe Rheumatoid Arthritis (RA) Inadequately Responding to Methotrexate.
Burmester GR, Kaeley GS, Kavanaugh AF et al (2017) Treatment efficacy and methotrexate-related toxicity in patients with rheumatoid arthritis receiving methotrexate in combination with adalimumab. RMD Open 3(2):1–9. https://doi.org/10.1136/rmdopen-2017-000465
Verschueren P, De Cock D, Corluy L et al (2015) Methotrexate in combination with other DMARDs is not superior to methotrexate alone for remission induction with moderate-to-high-dose glucocorticoid bridging in early rheumatoid arthritis after 16 weeks of treatment: the CareRA trial. Ann Rheum Dis 74(1):27–34. https://doi.org/10.1136/annrheumdis-2014-205489
Huang Y, Fan Y, Liu Y, Xie W, Zhang Z (2019) Efficacy and safety of secukinumab in active rheumatoid arthritis with an inadequate response to tumor necrosis factor inhibitors: a meta-analysis of phase III randomized controlled trials. Clin Rheumatol 38(10):2765–2776. https://doi.org/10.1007/s10067-019-04595-1
Bessette L, Haraoui B, Chow A et al (2019) Effectiveness and safety of certolizumab pegol in rheumatoid arthritis patients in canadian practice: 2-year results from the observational FαsT-CAN study. Ther Adv Musculoskelet Dis. https://doi.org/10.1177/1759720x19831151
Strand V, Tundia N, Song Y, Macaulay D, Fuldeore M (2016) Economic burden of non-responders to biologic DMARD treatments in rheumatoid arthritis. Arthritis Rheumatol 68:9–10
Thomson TM, Lescarbeau RM, Drubin DA et al (2015) Blood-based identification of non-responders to anti-TNF therapy in rheumatoid arthritis. BMC Med Genomics. https://doi.org/10.1186/s12920-015-0100-6
Yokoyama-Kokuryo W, Yamazaki H, Takeuchi T et al (2020) Identification of molecules associated with response to abatacept in patients with rheumatoid arthritis. Arthritis Res Ther 22(1):1–8. https://doi.org/10.1186/s13075-020-2137-y
Raterman HG, Vosslamber S, de Ridder S et al (2012) The interferon type I signature towards prediction of non-response to rituximab in rheumatoid arthritis patients. Arthritis Res Ther 14(2):R95. https://doi.org/10.1186/ar3819
Kneepkens EL, Pascual-Salcedo D, Plasencia C et al (2013) Golimumab levels, anti-drug antibodies and clinical response in rheumatoid arthritis Patients at 28 week of follow-up: 1456. Arthritis Rheum 65:S618–S619. https://doi.org/10.1002/art.38216%0A
Benjamin O, Bansal P, Goyal A, Lappin SL (2021) Disease modifying anti-rheumatic drugs (DMARD). StatPearls Publishing, Treasure Island
Cho SK, Kim D, Won S et al (2017) Safety of resuming biologic DMARDs in patients who develop tuberculosis after anti-TNF treatment. Semin Arthritis Rheum 47(1):102–107. https://doi.org/10.1016/j.semarthrit.2017.01.004
Berger JR, Malik V, Lacey S, Brunetta P, Lehane PB (2018) Progressive multifocal leukoencephalopathy in rituximab-treated rheumatic diseases: a rare event. J Neurovirol 24(3):323–331. https://doi.org/10.1007/s13365-018-0615-7
Aslani S, Mahmoudi M, Karami J, Jamshidi AR, Malekshahi Z, Nicknam MH (2016) Epigenetic alterations underlying autoimmune diseases. Autoimmunity 49(2):69–83. https://doi.org/10.3109/08916934.2015.1134511
Nile CJ, Read RC, Akil M, Duff GW, Wilson AG (2008) Methylation status of a single CpG site in the IL6 promoter is related to IL6 messenger RNA levels and rheumatoid arthritis. Arthritis Rheum 58(9):2686–2693. https://doi.org/10.1002/art.23758
Huber LC, Brock M, Hemmatazad H et al (2007) Histone deacetylase/acetylase activity in total synovial tissue derived from rheumatoid arthritis and osteoarthritis patients. Arthritis Rheum 56(4):1087–1093. https://doi.org/10.1002/art.22512
Grabiec AM, Korchynskyi O, Tak PP, Reedquist KA (2012) Histone deacetylase inhibitors suppress rheumatoid arthritis fibroblast-like synoviocyte and macrophage IL-6 production by accelerating mRNA decay. Ann Rheum Dis 71(3):424–431. https://doi.org/10.1136/ard.2011.154211
Choo QY, Ho PC, Tanaka Y, Lin HS (2010) Histone deacetylase inhibitors MS-275 and SAHA induced growth arrest and suppressed lipopolysaccharide-stimulated NF-ΚB p65 nuclear accumulation in human rheumatoid arthritis synovial fibroblastic E11 cells. Rheumatology 49(8):1447–1460. https://doi.org/10.1093/rheumatology/keq108
Miao CG, Huang C, Huang Y et al (2013) MeCP2 modulates the canonical Wnt pathway activation by targeting SFRP4 in rheumatoid arthritis fibroblast-like synoviocytes in rats. Cell Signal 25(3):598–608. https://doi.org/10.1016/j.cellsig.2012.11.023
Steen-Louws C, Hartgring SAY, Popov-Celeketic J et al (2019) IL4-10 fusion protein: a novel immunoregulatory drug combining activities of interleukin 4 and interleukin 10. Clin Exp Immunol 195(1):1–9. https://doi.org/10.1111/cei.13224
Thiolat A, Denys A, Petit M et al (2014) Interleukin-35 gene therapy exacerbates experimental rheumatoid arthritis in mice. Cytokine 69(1):87–93. https://doi.org/10.1016/j.cyto.2014.05.015
Ceribelli A, Nahid MA, Satoh M, Chan EKL (2011) MicroRNAs in rheumatoid arthritis. FEBS Lett 585(23):3667–3674. https://doi.org/10.1016/j.febslet.2011.05.020
Therapy AG, Deviatkin AA, Vakulenko YA, Akhmadishina LV, Tarasov VV (2020) Emerging concepts and challenges in rheumatoid. Biomedicines 8(1):9
Payet M, Dargai F, Gasque P, Guillot X (2021) Epigenetic regulation (Including micro-rnas, dna methylation and histone modifications) of rheumatoid arthritis: a systematic review. Int J Mol Sci 22(22):12170. https://doi.org/10.3390/ijms222212170
Li J, Song Q, Shao L, Zhang LL, Guo XH, Mao YJ (2018) MiR-124A inhibits proliferation and invasion of rheumatoid arthritis synovial fibroblasts. Eur Rev Med Pharmacol Sci 22(14):4581–4588
Ye Y, Gao X, Yang N (2018) LncRNA ZFAS1 promotes cell migration and invasion of fibroblast-like synoviocytes by suppression of miR-27a in rheumatoid arthritis. Hum Cell 31(1):14–21. https://doi.org/10.1007/s13577-017-0179-5
Zou Y, Xu S, Xiao Y et al (2018) Long noncoding RNA LERFS negatively regulates rheumatoid synovial aggression and proliferation. J Clin Invest 128(10):4510–4524. https://doi.org/10.1172/JCI97965
Shui X, Chen S, Lin J, Kong J, Zhou C, Wu J (2019) Knockdown of lncRNA NEAT1 inhibits Th17/CD4+ T cell differentiation through reducing the STAT3 protein level. J Cell Physiol 234(12):22477–22484. https://doi.org/10.1002/jcp.28811
Zhang J, Huang X, Wang H et al (2015) The challenges and promises of allogeneic mesenchymal stem cells for use as a cell-based therapy. Stem Cell Res Ther 6(1):1–7. https://doi.org/10.1186/s13287-015-0240-9
Causton B, Pardo-Saganta A, Gillis J et al (1998) Page 1 of 47 1. 409
Acknowledgements
We acknowledge the Department of Research at Sir Ganga Ram Hospital, New Delhi, 110060, India for providing the infrastructure and facility for preparing the manuscript.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Author information
Authors and Affiliations
Contributions
PP: Manuscript writing & editing, SV: Manuscript writing & editing, S: Manuscript writing & editing, NKG: Editing and Final approval of manuscript, VC: Editing & Final approval of manuscript, SM: Concept & Design, Manuscript writing & editing, and Final approval of manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors have no financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Ethical approval
Not applicable.
Consent to participate
Not applicable.
Consent to publication
Not applicable.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Prasad, P., Verma, S., Surbhi et al. Rheumatoid arthritis: advances in treatment strategies. Mol Cell Biochem 478, 69–88 (2023). https://doi.org/10.1007/s11010-022-04492-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-022-04492-3