
Abstract
Protein kinase CK2 (CK2) is a highly conserved and ubiquitous serine/threonine kinase. It is a multifunctional and pleiotropic protein kinase implicated in the regulation of cell proliferation, survival, and differentiation. Deregulation of CK2 is observed in a wide variety of tumors. It has been the focus of intensive research efforts to establish the cause–effect relationship between CK2 and neoplastic growth. Here, we further validate the role of CK2 in cancer cell growth using siRNA approach. We also screened a library of more than 200,000 compounds and identified several molecules, which inhibit CK2 with IC50 < 1 μM. The binding mode of a representative compound with maize CK2 was determined. In addition, the cellular activity of the compounds was demonstrated by their inhibition of phosphorylation of PTEN Ser370 in HCT116 cells. Treatment of a variety of cancer cell lines with the newly identified CK2 inhibitor significantly blocked cell growth with IC50s as low as 300 nM.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
CK2 was first discovered in 1954 [1] but it was not until 1977 when its physiological function as a cAMP-independent kinase that phosphorylated β-casein was described [2]. In the intervening 40 years, over 2,000 publications describing its activity and biological functions have appeared. Despite these, definition of its physiological role has been elusive. CK2 is one of the most highly conserved proteins in nature [2–4]. It is essential for survival in yeast and embryonic development in mice [5–7]. CK2 is ubiquitously expressed and detected in both the nucleus and cytoplasm [3]. It is constitutively active in vitro, however, there is increasing evidence that the activity and localization of CK2 are regulated by multiple mechanisms and pathways [3, 8]. For example, inositol-1,3,4,5-phosphate activates regulative CK2 phosphorylation of phosphatidylcholine transfer protein-like protein [8]. In another study, the association of CK2 with the nuclear matrix was shown to be dependent on the cell cycle [9] or apoptosis [10]. TGFβ1 treatment of normal mouse hepatocytes decreased the activity and expression of CK2. The inhibition of CK2 activity then led to the stabilization of IκB protein and shut-off of NFκB activity, contributing to the TGFβ-induced apoptosis in these cells [11]. CK2 activity has been implicated in cell cycle, cell growth, survival [12, 13], chromatin remodeling [14], protein transcription [15, 16], translation, folding, and degradation [17]. To date, there are more than 300 CK2 substrates identified in nearly all signaling, regulatory, and metabolic pathways [18, 19].
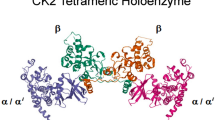
Protein kinase CK2 is a hetero-tetramer composed of two catalytic (α and α′) and two regulatory (β) subunits. In vitro, equal molar amounts of catalytic subunits, at 1 nmol quantity, were able to reconstitute with the regulatory subunits to form the hetero-tetramer [20]. The activity of the catalytic subunits is stimulated by the regulatory subunits [21], however, the extent of the stimulation is substrate dependent [22]. It has been proposed that in addition to many functions of the hetero-tetramer, individual α, α′, and β subunits have independent activities in vivo [23–26].
There is a growing body of evidence suggesting a role for CK2 in cancer [27–29]. The expression levels and activities of CK2 subunits are increased 3–5 fold in many tumors and tumor cell lines [17, 30–33]. Overexpression of CK2α led to increased c-myc expression in lymphoma cells [33], NFκB activation in NIH3T3 cells [17], and protected PC3 cells from etoposide induced apoptosis [34]. Overexpression of CK2β in 3t3-L1 fibroblasts led to an increased growth rate [35]. In transgenic mice, CK2α overexpression co-operated with c-myc or loss (or mutation) of p53 at the lpr locus to promote tumorigenesis [36]. Transgenic expression of CK2α under the MMTV promoter resulted in late onset of adenocarcinomas with increased c-myc and β-catenin expression [36].
Recent work from several groups has sought to abrogate CK2 activity to further delineate its role in cell growth and survival. Methods used included overexpression of dominant negative forms [17], siRNA or antisense oligonucleotides, and the small molecule inhibitors 4,5,6,7-tetrabromobenzimidazole (TBB) and DNB. Direct injection of CK2α antisense DNA into PC3-LN4 xenograft tumors induced cell death in a dose and time dependent manner [34]. The antisense also reduced viability of PC3-LN4 and ALVA-41 in vitro while having a marginal effect on the benign BPH-1 or normal PrEC prostate cells [37]. Antisense to CK2α inhibited growth of squamous carcinoma cells Ca9-22 [38] while antisense to both CK2α and α′ enhanced apoptosis in HeLa cells [39]. siRNA knockdown of CK2α plus α′ has demonstrated a role for CK2 in protection against apoptosis caused by 6-thioguanine [40], TRAIL [41], and ionizing radiation [42]. Several inhibitors have also been used extensively to probe the biological role of CK2 [43–47]. These inhibitors disrupt growth and promote cell death in several cancer cell lines. In some instances, effects have been reversed by re-expression of CK2α [48].
In this study, we further validated the role of CK2 in cancer using siRNA approach. To confirm the knockdown of CK2, the levels of CK2α, α′, and β protein were assessed. The phosphorylation of tumor suppressor PTEN at Ser370 (p-PTEN Ser370) was followed as a cellular marker of CK2 activity in HCT116 cells. The tumor suppressor PTEN is phosphorylated at two residues by CK2, Ser370, and Ser385. This primes PTEN for further phosphorylation by GSK3, leading to its stabilization but also inhibiting the lipid phosphatase activity. The Ser385 site is also phosphorylated by CK1, however, to date only CK2 has been identified as a Ser370 kinase [49]. Therefore, we sought to validate PTEN Ser370 as a suitable biomarker for following the endogenous CK2 kinase activity in cell lines used in this study. siRNA knockdown of CK2α and α′ led to reduced cell growth of HCT116 and HeLa cells, accompanied by a reduction in the level of CK2 subunits and decreased phosphorylation of PTEN Ser370. Several novel and specific inhibitors of CK2 were also identified in a high-throughput screen using a fluorescence resonance transfer assay. These molecules were also capable of reducing cell viability and PTEN Ser370 phosphorylation in multiple cancer cell lines. In addition, the crystal structure of maize CK2 with one of these hit molecules, compound 1, is presented.
Materials and methods
Cancer cell lines and antibodies
Cell lines (HCT116, HeLa, A375, A549, Mia PaCa-2, LOX-IMVI, Malme-3, Malme-3M, and SK-MEL-28) were obtained from American Type Culture Collection (ATCC) and maintained by following ATCC recommendations. Mouse Anti-CK2α mAb, Rabbit anti-CK2α′ polyclonal antibody, and mouse anti-CK2β mAb were purchased from BD Bioscience, Abcam, and Calbiochem, respectively. Rabbit anti-p-PTEN Ser370 polyclonal antibody was obtained from Biosource International. IRDye800 goat-anti-rabbit and IRDye700 goat-anti-mouse antibodies (LiCor) were used as secondary antibodies for western blot analyses.
Small interfering RNA knockdown studies
All small interfering RNAs (siRNA) used in this study were ON-TARGETPlusTM Smartpool siRNAs from Dharmacon. Cells were grown to subconfluency in 24-well plates and transfected with DharmaFECT1 by following the manufacturer’s protocol. Equal molar amounts of non-targeting negative control siRNA or CK2α and CK2α′-specific siRNAs were used for each transfection reaction. After transfection, cells were harvested for western blot analyses and assayed for viability using Alamar Blue.
Western blot analysis
Upon siRNA treatment as indicated in figure legends, cells were harvested and lysed immediately with 1× Cellular Extraction Buffer (Biosource) supplemented with 1 mM sodium orthovanadate, 4 mM PNPP, and 1 mM DTT. Protein concentrations in lysates were determined and 30 μg were resolved on 4–20% or 10% Tris–Glycine gels (Invitrogen) under denaturing conditions and transferred to nitrocellulose membranes. The membranes were blocked with Odyssey Blocking Buffer and developed with antibodies as indicated. The membrane was scanned with the Odyssey system (LiCor) and protein expression levels were determined by quantitation of specific band intensities using the Odyssey software.
Cell viability assays
Seventy-two hours post-transfection with siRNA, cells were trypsinized and harvested in 1 ml growth medium. One hundred microliters of cell suspension from each sample was transferred to 96-well plates and incubated with 20 μl Alamar Blue and 80 μl of growth media for 4 h. Cell viability was assessed by the reduction of Alamar Blue (Biosource), which was measured spectrophotometrically.
To test the impact of CK2 inhibitors on cell viability, cells were seeded in 96-well flat-bottom microtiter plates. After 24 h, the cells were exposed to various concentrations of compound and cultured for 3 days. After the addition of 20 μl/well of Alamar Blue, the cells were incubated for an additional 4 h at 37°C. Thereafter, the plate was read on a Victor plate reader to determine the IC50 value.
Reagents for hCK2α and hCK2α′ biochemical assays
The peptide substrate Arg-Arg-Arg-Ala-Asp-Asp-Ser-Asp-Asp-Asp-Asp-Asp (P-147) was purchased from Biomol Research Laboratories Inc. 6-His tagged active recombinant human full length CK2α (14-445) and CK2α′ (14-689) (hCK2α and hCK2α′) were from Upstate. A custom biotinylated peptide substrate biotinyl-Arg-Arg-Arg-Ala-Asp-Asp-Ser-Asp-Asp-Asp-Asp-Asp was from Biomol Research Laboratories Inc. Streptavidin-dylight was from Pierce Biotechnology and Eu-labeled-anti-rabbit IgG was from Perkin Elmer.
33P radioactive assay
33.2 ng of hCK2α or hCK2α′ enzyme was pre-incubated with 20 μM of peptide substrate for 15 min at room temperature in assay buffer (20 mM Hepes pH 7.0, 60 mM NaCl, 0.1 mM EDTA, 0.01% Triton X-100, 5 mM MgCl2, 0.1 mM DTT). The phosphorylation reaction was started by adding ATP (0.075 μCi of 33P/uM ATP). The reaction was incubated at room temperature for 90 min, and stopped by the addition of 1.5% phosphoric acid. A multiscreen HTS vacuum manifold apparatus from Millipore was used to capture protein on PVDF membrane Multiscreen HV plates. Microscint 20 from Perkin Elmer was added for scintillation read on a Topcount instrument. The reaction was determined to be linear with respect to time and enzyme concentration. To determine Ki and Ki′ for compounds, ATP and inhibitors concentrations were varied. Ki, Ki′, and Kmapp values were determined from global fitting of the data using Grafit software (Eritreus). For inhibition studies, IC50s were measured at 20 μM ATP. Time dependence was determined by a shift in IC50 when the enzyme was pre-incubated with compound for 15 min and when the reaction was initiated with enzyme.
HTRF (homogeneous time resolved florescence) assay
The homogenous time resolved fluorescence (HTRF) CK2 kinase assay was carried out in 384-well black plate (Corning) at a final volume of 35 μl. hCK2α/hCK2α′ from Upstate was diluted in assay buffer to give a final assay concentration of 0.016 ng/μl. Test compound was diluted in 100% DMSO and 0.5 μl was added to 14.5 μl of enzyme. Enzyme and compound were allowed to equilibrate at room temperature for 15 min. The reaction was started with the addition of 10 μl of detection mixture, which gives final assay concentrations of 15 μM ATP, 3.125 nM primary antibody, 15 nM biotinylated peptide substrate, 15 nM streptavidin-dylight (Pierce Technology), and 0.4 nM Eu-labeled-anti-rabbit IgG antibody (Perkin Elmer). Reactions were allowed to proceed for 60 min at room temperature before the addition of 10 μl of stop solution (EDTA 20 mM final). Fluorescence resonance energy transfer (FRET) was measured between streptavidin-dylight that bound to the biotinylated peptide and the Eu-labeled-anti-Rabbit IgG that bound to phosphorylated peptide. An analyst GT (Multiscreen reader) from Molecular Devices was used for ratiometric reading at emission wavelengths of 615 and 655 nm, following excitation at 340 nm. The reaction was determined to be linear with respect to time and enzyme concentration.
Protein purification, crystallization, and structure determination
The maize CK2α (zmCK2α) gene was amplified from a cDNA library (Biochain, Inc.) and cloned into the pET11a vector (EMD Biosciences). Expression was induced in Escherichia coli with IPTG and cells grown for 6 h. Cell pellets were lysed in 50 mM Tris–HCl pH 8.5, 10 mM DTT, 200 mM NaCl with a microfluidizer, clarified by centrifugation at 40,000g for 1 h and applied to an SP Sepharose column (GE Healthcare). The column was developed with a gradient of 100–1,000 mM NaCl in 25 mM Tris pH 8.5, 5 mM DTT. Protein eluted at ~300 mM NaCl. The sample was diluted to 100 mM NaCl and applied to Q sepharose chromatography in the same buffer. The Q column was developed with a gradient of 100–1,000 mM NaCl. Protein was eluted at ~500 mM NaCl, and was mixed with 1/3 volume of 4 M ammonium sulfate and applied to a Butyl Sepharose column. The column was developed with a linear gradient from 1 to 0 M ammonium sulfate in 25 mM Tris pH 8.5, 5 mM DTT. Protein was pooled, concentrated, and subjected to size exclusion chromatography on a Superdex 200 column equilibrated in 25 mM Tris pH 8.4, 400 mM NaCl, 1 mM DTT. Fractions were pooled and concentrated to 10 mg/ml. Protein identity was verified by SDS-PAGE and LC/MS. Biochemical assays showed that the protein is active. Crystals of zmCK2α were obtained in 100 mM Hepes pH 7.5, 10% ethylene glycol, 6% PEG 8,000. Co-crystals with compound 1 were obtained by overnight soaking with 3 mM ligand. Diffraction data were collected at the Advanced Light Source in Berkeley, CA, and structure determination was done using the CCP4 suite of programs [50].
Results
CK2 expression in cancer cell lines
We investigated the protein levels of both CK2α and CK2α′ isoforms in a variety of cancer cell lines. Malme-3 human skin fibroblast cells were used as a normal cell control. The cancer cell lines expressed higher levels of CK2α and CK2α′ relative to the normal fibroblast cells, with the exception of Mia PaCa-2 (Fig. 1, top panel). We also investigated the p-PTEN Ser370 levels in those cell lysates, since CK2 is reported to be the major kinase involved in the phosphorylation of PTEN at this site. More intense signals of p-PTEN Ser370 were detected in cancer cells expressing high levels of CK2 isoforms (Fig. 1, middle panel). Malme-3 human skin fibroblast cells also showed comparable p-PTEN Ser370 level despite the low level of CK2 expression in this cell line, possibly due to the involvement of additional kinase(s) in PTEN phosphorylation or a decreased phosphatase activity in this type of cells. Altogether, these data suggest that HeLa, HCT116, and A549 are suitable cellular models to study the role of CK2 in cancer cells.

CK2α and CK2α′ protein expression in various cancer cell lines. Top panel: western blot for CK2α and CK2α′. Middle panel: corresponding western blot for p-PTEN Ser370. Bottom panel: corresponding western blot for α-actinin as a loading control. 30 μg protein was loaded for each sample
Knockdown of CK2 proteins affects cancer cell viability
To evaluate the involvement of CK2 in cancer cell proliferation, siRNA was used to deplete CK2 in several cancer cell lines. HCT116 cells were transiently transfected with non-targeting negative control siRNA or siRNA directed against CK2α and CK2α′ twice, on day 1 and day 4. Seventy-two hours after the second transfection, lysates were prepared and analyzed by standard western blotting with anti-CK2 antibodies. As shown in Fig. 2a, the levels of CK2α and CK2α′ were drastically reduced by the siRNA (>95%), whereas the level of α-actinin was unchanged. Western blotting experiments were also performed to determine whether targeted knockdown of CK2 proteins affected phosphorylation of PTEN at Ser370. p-PTEN Ser370 signal is significantly lower in CK2 siRNA-transfected HCT116 cells (Fig. 2a), validating CK2 as the major kinase to phosphorylate PTEN Ser370 in this cell line. We next evaluated the impact of CK2 depletion on the proliferation of HCT116 cells with Alamar Blue assay. While transfection with individual CK2 subunit (CK2α or CK2α′) siRNA had minor effects on cell growth, transfection with both siRNA inhibited cell proliferation of HCT116 by >50% (Fig. 2b).

Knockdown of CK2α and CK2α′ protein by siRNA in cancer cells. a Western blot analyses of CK2α, CK2α′, and p-PTEN Ser370 levels in non-targeting negative control siRNA and CK2α/CK2α′ specific siRNAs transfected HCT116 cells. b Knockdown of CK2α and CK2α′ protein inhibits HCT116 growth. c Western blot analyses of CK2α, CK2α′, and CK2β protein levels in lysates from HeLa and A549 cells treated with non-targeting negative control siRNA (Ctrl siRNA), siRNA directed against CK2 catalytic subunits (CK2α/α′ siRNA) or transfection reagent only (Mock). Total siRNA concentration in all of transfection reactions described above was 100 nM
We also optimized transfection conditions in both A549 and HeLa cells. Transfection with 100 nM of siRNAs (50 nM CK2α siRNA plus 50 nM CK2α′ siRNA) downregulated CK2α by 90% and CK2α′ by >95% (Fig. 2c, top panel) in both cell lines. Remarkably, reduction of CK2β protein expression was also observed in both cell lines transfected with siRNAs targeting CK2α/α′ (Fig. 2c, middle panel). Depletion of CK2 proteins did not exert significant effect on A549 cell growth but reduced HeLa cell growth by 40% (Table 1). Surprisingly, the CK2α subunit protein expression is slightly lower in mock and control siRNA-treated HeLa cells (Fig. 2c, middle panel), compared to untreated HeLa cells (Fig. 1). It suggests DharmaFECT1 transfection reagent might affect CK2α protein expression or stability in a cellular context dependent manner.
Compound library screening
An HTRF assay was developed to carry out single point screening. The high-throughput screening was performed on an Evolution P3 robot from Perkin Elmer. Data from 1 of 16 independent test sets performed to screen the entire library of 222,800 compounds are shown in Fig. 3. One thousand four hundred and two compounds with >75% inhibition (at concentration of 40 μg/ml) were defined as primary hits. Dose–response curves identified 696 compounds with IC50 < 10 μM. Of those, 455 were ATP competitive and 438 were time independent. Fifty-eight discreet chemical classes were selected for more in-depth studies to confirm potency and purity.

High-throughput screen. Scatter plot representative of data from a screen of 222,800 compounds at concentration of 40 μg/ml. 100% kinase activity is represented by the activity in the absence of inhibitors. Hits were defined as compounds that reduce the kinase activity by >50%
Enzyme characterization
The apparent Km’s for ATP for human and maize CK2α and α′ subunits were determined using a radioactive format assay and standard Michaelis–Menten kinetic plots. The Km ATPapp was essentially the same for both isoforms at 12 and 10 μM for human CK2α and α′, respectively. hCK2α was used to determine the Ki, Ki′, and reversibility of compounds. The Ki for TBB was determined to be 2 μM, consistent with previous reports [51–53].
To ascertain the molecular basis for the activity and specificity of hit molecules, we determined the co-crystal structure of compound 1 with zmCK2α (Fig. 4). Maize CK2α is now established to be a good surrogate for human CK2α [54]. A very recent study showed that these two enzymes bind emodin in two different orientations [55] but it remains to be seen whether this is a general phenomenon or is restricted to highly symmetric ligands such as emodin [55]. It is worth noting that compound 1 is an asymmetric molecule. We independently verified that the Km ATPapp for zmCK2α, 15 μM, is similar to that for hCK2α, 12 μM, in our biochemical assays and shows the same inhibition profiles for TBB and compound 1 (data not shown).

Co-crystal structures of compound 1 and maize CK2α. a Chemical structure of compounds 1 and 2. The chiral carbon is indicated by the red asterisk. b Co-crystal structure of compound 1 in complex with maize CK2α. The compound binds at the ATP-binding site. Nitrogen and oxygen atoms are colored in blue and red, respectively. Carbon atoms are colored in cyan (compound) and green (protein). Ordered water molecules are shown as magenta spheres, and hydrogen bonds are indicated by the dashed yellow lines. The protein surface is rendered in white. c Structural statistics
The structure was determined to 1.8 Å resolution, with a crystallographic R factor of 20.6% (R free = 25.2%). The electron density was unambiguous for the entire complex, and particularly for the bound ligand, surrounding residues, and bound water molecules at the active site. As shown in Fig. 4b, compound 1 binds CK2α at the ATP binding site, supporting the conclusion that this inhibitor is ATP competitive. Binding is mediated by both hydrophobic contacts and hydrophilic interactions, as observed for many ATP-competitive kinase inhibitors [56, 57]. The isoquinoline ring occupies approximately the same position bound by the adenosine ring of ATP, and makes a single hydrogen bond to the hinge. As observed for many CK2α inhibitors, and in contrast with the majority of characterized inhibitors of many other kinases, the bulk of the hydrogen bonding interactions are with the phosphate binding region of the ATP binding site. In compound 1, these hydrogen bonds are mediated primarily by the triazole ring, in the form of water-bridged bonds to Glu81 (which forms the conserved kinase salt-bridge to Lys68) and to the backbone amide of Asp175 of the DFG loop. The primary amine attached to the chiral carbon (indicated by the red asterisk in Fig. 4b) picks up additional hydrogen bonding interactions with the side chains of Asn161 and Asp168 in the ribose binding pocket. The phenyl ring does not appear to make critical interactions, but probably contributes some weak hydrophobic contacts to the glycine rich-loop.
Cellular activities of CK2 inhibitors
Once identified, CK2 inhibitor compound 1 was fully characterized in cellular assays. Since p-PTEN Ser370 as CK2 substrate is validated in HCT116 cells by siRNA approach, p-PTEN Ser370 western blot using compound 1 treated HCT116 lysates was performed and p-PTEN Ser370 signal was quantified using the Odyssey software. As shown in Fig. 5a, b, CK2 kinase activity to phosphorylate PTEN at Ser370 was inhibited by compound 1 in a dose-dependent manner, with IC50 at 100 nM. Moreover, HCT116 cells cultured for 72 h in the presence of increasing concentrations of the compound displayed a dose-dependent growth inhibition (Fig. 5c). Overall, compound 1 significantly inhibited growth of all CK2 expressing cancer cell lines used in our study, including HCT116, A375, HeLa, LOX-IMVI, A549, SK-MEL-28, and Malme-3M, with IC50 of 0.14, 0.28, 0.3, 0.4, 0.7, 0.7, and 2.01 μM, respectively.

Cellular activities of compound 1 in HCT116. a Compound 1 reduces phosphorylation of PTEN at Ser370 in HCT116. b Quantitation of the data shown in a. c Compound 1 inhibits HCT116 cell growth. Alamar Blue viability assay was performed as described in “Material and methods” section
Discussion
CK2 is receiving increasing interest from the pharmaceutical industry as a target for therapeutic intervention [46, 47, 58–60]. We have generated a drug discovery platform for identification and optimization of small molecule inhibitors of CK2. This platform includes a high-throughput enzyme assay, cellular assays including a phospho-specific biomarker in HCT116 cells and a tool CK2 inhibitor of a novel compound class with utility for target and assay validation.
Many factors may have contributed to the recent interest in CK2 as a cancer target. The level of understanding of the cellular regulation and role of CK2 in cell growth and survival has expanded [40, 61]. Convincing evidence for CK2 as an oncogene is supplied by transgenic studies where it co-operates in tumorigenesis with abrogation of important tumor suppressors [62]. Recently, a cyclic peptide that inhibits CK2 phosphorylation of HPV-16 E7 has entered clinical trials for treatment of cervical cancers, which could produce interesting proof of concept data [63].
Attempts to validate CK2 kinase as a target at the molecular level have met with some difficulties. Each experimental approach has limitations. However, when the body of evidence is taken together a role of CK2 in tumorigenesis is convincing. Studies with siRNA and antisense are made difficult by the long half life of the protein. Knocking down CK2α and α′ subunits to undetectable levels slows cell growth [37–39, 64]. However, the level and distribution of CK2β subunit change significantly following removal of CK2α subunit [39]. This clouds the interpretation that CK2 kinase activity is primarily responsible for the biological effects observed. Overexpression of kinase inactive mutants has also shown to abrogate the kinase activity and lead to inhibition of cell growth [64]. However, overexpression of kinase inactive mutants not only competes with wild-type kinase for substrate but also alters the balance of CK2 subunits and the cellular localization of the kinase. Therefore, it is difficult to attribute the biological effects of these mutants solely to its inhibitory activity on endogenous CK2 kinase. There have been many experiments using the small molecule inhibitors of CK2, TBB, and its analogs [44, 65]. Typically these molecules inhibit cell growth and lead to cell death. Recent work from Pagano and Pinna has shown that, when tested against a panel of 30 kinases, TBB and DMAT are fairly specific [47]. In addition to CK2, these two compounds inhibit Pim1 and Pim3 [47]. The CK2 inhibitors have a potent effect in cells, causing apoptosis in many cell lines at micromolar concentrations. The difference in cancer cell sensitivity to the small molecule CK2 inhibitor or the CK2 siRNA has led to the proposal that the potency of inhibitor in those cells may be largely due to off-target effects. Experiments where the effects of the compound are reversed by ectopic expression of the CK2α subunit support, but do not prove definitively, the hypothesis that the growth inhibition and cell death observed in these studies are due to inhibition of CK2 [48]. Alternative explanations for differences between abrogation of CK2 activity by molecular or chemical means include (i) the presence of residual CK2 activity in the experiments using siRNA, antisense, or dominant negatives, which allow cells to survive, (ii) the long duration required for complete knockdown of CK2 allows the cells to adapt to the loss of this critical activity, or (iii) inhibition of the CK2αα′β holoenzyme is far more effective than manipulation of the individual subunit.
In this study, we found that significant knockdown of CK2α and α′ protein levels in HeLa, A549, and HCT116 cells required the continued presence of the CK2 siRNA for a minimum of 72 h. The strongest effect was observed in HCT116, where >95% reduction in CK2α and α′ led to 50% reduction in growth over the 72 h period, consistent with other studies [37, 38]. The observation that knockdown of CK2α/α′ by siRNA negatively affects the protein level of CK2β subunit confirms the stability of CK2β is related to its ability to assemble with CK2α/α′ to form stable tetrameric complexes [39].
We have also identified a potent (IC50 = 40 nM) small molecule inhibitor of CK2 of a completely novel chemical class, compound 1. Compound 1 has a different specificity profile compared to TBB. At 10 μM, it inhibits 13 out of 80 kinases tested at >80% inhibition, including CDK2, FLT3, GSK3-alpha, GSK3-beta, HIPK1, IKK1, IRAK4, KDR, LOK, MAPK2, p70S6K, PKA, PKC-theta, and Rse. Compound 1 does not inhibit the Pim kinases. Compound 1 has very favorable physico-chemical properties when compared with other commercially available CK2 inhibitors. It represents an excellent starting point for the generation of bio-available, specific CK2 inhibitors for testing in models of human disease. The crystal structure of this compound bound to the catalytic site of maize CK2α will facilitate structure directed modifications to improve potency and selectivity.
Compound 1 can also be used as a tool for biological studies. It inhibits cell growth with IC50s in the single digit micromolar range. This inhibition of cell growth correlates with the inhibition of phosphorylation of the CK2 specific substrate PTEN Ser370 in HCT116 cells. Ser370 in PTEN may act as useful biomarker to monitor CK2 activity in certain cell types, as demonstrated by the response of this phosphorylation to CK2α and α′ knockdown by siRNA in HCT116 cells. This is the first cellular biomarker of CK2 to be validated with the siRNA approach. Miyata and Nishida [66] recently described the use of an antibody to CDC37 Ser13 to follow CK2 activity. The validation of this phospho site of CDC37 as a cellular biomarker for CK2 was its decrease upon TBB treatment and increase in response to overexpression of CK2α or β [66].
The utility of CK2 inhibitors for clinical indications still remains unknown, and will await proof of concept studies first in animal models and subsequently in man. The tools required to facilitate generation of small molecule compounds that target this mechanism are now available.
Abbreviations
- CK2:
-
Protein kinase CK2
- siRNA:
-
Small interfering RNA
- TBB:
-
4,5,6,7-Tetrabromobenzotriazole
- DRB:
-
5,6-Dichloro-β-ribofuranosylbenzimidazole
- HTS:
-
High throughput screening
- HTRF:
-
Homogenous time resolved fluorescence
- FRET:
-
Fluorescence resonance energy transfer
- HDAC:
-
Histone deacetylase
- ATP:
-
Adenosine-5′-triphosphate
- EDTA:
-
Ethylenediaminetetraacetic acid
- Eu:
-
Europium
- μCi:
-
Micro Curie
- Ki:
-
Dissociation constant
- Km:
-
Substrate concentration that results in half-maximal velocity for the enzymatic reaction
- zmCK2α:
-
Maize protein kinase CK2α
References
Burnett G, Kennedy EP (1954) The enzymatic phosphorylation of proteins. J Biol Chem 211:969–980
Tuazon PT, Traugh JA (1991) Casein kinase I and II—multipotential serine protein kinases: structure, function, and regulation. Adv Second Messenger Phosphoprotein Res 23:123–164
Pinna LA (1990) Casein kinase 2: an ‘eminence grise’ in cellular regulation? Biochim Biophys Acta 1054:267–284
Buchou T, Vernet M, Blond O et al (2003) Disruption of the regulatory beta subunit of protein kinase CK2 in mice leads to a cell-autonomous defect and early embryonic lethality. Mol Cell Biol 23:908–915
Lou DY, Dominguez I, Toselli P, Landesman-Bollag E, O’Brien C, Seldin DC (2008) The alpha catalytic subunit of protein kinase CK2 is required for mouse embryonic development. Mol Cell Biol 28:131–139
Poole A, Poore T, Bandhakavi S, McCann RO, Hanna DE, Glover CV (2005) A global view of CK2 function and regulation. Mol Cell Biochem 274:163–170
Seldin DC, Lou DY, Toselli P, Landesman-Bollag E, Dominguez I (2008) Gene targeting of CK2 catalytic subunits. Mol Cell Biochem 316:141–147
Solyakov L, Cain K, Tracey BM et al (2004) Regulation of casein kinase-2 (CK2) activity by inositol phosphates. J Biol Chem 279:43403–43410
Wang H, Yu S, Davis AT, Ahmed K (2003) Cell cycle dependent regulation of protein kinase CK2 signaling to the nuclear matrix. J Cell Biochem 88:812–822
Guo C, Yu S, Davis AT, Wang H, Green JE, Ahmed K (2001) A potential role of nuclear matrix-associated protein kinase CK2 in protection against drug-induced apoptosis in cancer cells. J Biol Chem 276:5992–5999
Cavin LG, Romieu-Mourez R, Panta GR et al (2003) Inhibition of CK2 activity by TGF-beta1 promotes IkappaB-alpha protein stabilization and apoptosis of immortalized hepatocytes. Hepatology 38:1540–1551
Kato T Jr, Delhase M, Hoffmann A, Karin M (2003) CK2 is a C-terminal IkappaB kinase responsible for NF-kappaB activation during the UV response. Mol Cell 12:829–839
Ahmad KA, Wang G, Unger G, Slaton J, Ahmed K (2008) Protein kinase CK2—a key suppressor of apoptosis. Adv Enzyme Regul 48:179–187
Canton DA, Litchfield DW (2006) The shape of things to come: an emerging role for protein kinase CK2 in the regulation of cell morphology and the cytoskeleton. Cell Signal 18:267–275
Gu L, Husain-Ponnampalam R, Hoffmann-Benning S, Henry RW (2007) The protein kinase CK2 phosphorylates SNAP190 to negatively regulate SNAPC DNA binding and human U6 transcription by RNA polymerase III. J Biol Chem 282:27887–27896
Guo C, Davis AT, Yu S, Tawfic S, Ahmed K (1999) Role of protein kinase CK2 in phosphorylation nucleosomal proteins in relation to transcriptional activity. Mol Cell Biochem 191:135–142
Romieu-Mourez R, Landesman-Bollag E, Seldin DC, Sonenshein GE (2002) Protein kinase CK2 promotes aberrant activation of nuclear factor-kappaB, transformed phenotype, and survival of breast cancer cells. Cancer Res 62:6770–6778
Meggio F, Pinna LA (2003) One-thousand-and-one substrates of protein kinase CK2? FASEB J 17:349–368
Filhol O, Cochet C (2009) Cellular functions of Protein kinase CK2: a dynamic affair. Cell Mol Life Sci. doi:10.1007/s00018-009-9151-1
Jakobi R, Traugh JA (1992) Characterization of the phosphotransferase domain of casein kinase II by site-directed mutagenesis and expression in Escherichia coli. J Biol Chem 267:23894–23902
Grankowski N, Boldyreff B, Issinger OG (1991) Isolation and characterization of recombinant human casein kinase II subunits alpha and beta from bacteria. Eur J Biochem 198:25–30
Bodenbach L, Fauss J, Robitzki A et al (1994) Recombinant human casein kinase II. A study with the complete set of subunits (alpha, alpha′ and beta), site-directed autophosphorylation mutants and a bicistronically expressed holoenzyme. Eur J Biochem 220:263–273
Filhol O, Cochet C, Wedegaertner P, Gill GN, Chambaz EM (1991) Coexpression of both alpha and beta subunits is required for assembly of regulated casein kinase II. Biochemistry 30:11133–11140
Bibby AC, Litchfield DW (2005) The multiple personalities of the regulatory subunit of protein kinase CK2: CK2 dependent and CK2 independent roles reveal a secret identity for CK2beta. Int J Biol Sci 1:67–79
Bjorling-Poulsen M, Siehler S, Wiesmuller L, Meek D, Niefind K, Issinger OG (2005) The ‘regulatory’ beta-subunit of protein kinase CK2 negatively influences p53-mediated allosteric effects on Chk2 activation. Oncogene 24:6194–6200
Vilk G, Derksen DR, Litchfield DW (2001) Inducible expression of the regulatory protein kinase CK2beta subunit: incorporation into complexes with catalytic CK2 subunits and re-examination of the effects of CK2beta on cell proliferation. J Cell Biochem 84:84–99
Seldin DC, Landesman-Bollag E, Farago M, Currier N, Lou D, Dominguez I (2005) CK2 as a positive regulator of Wnt signalling and tumourigenesis. Mol Cell Biochem 274:63–67
Guerra B, Issinger OG (2008) Protein kinase CK2 in human diseases. Curr Med Chem 15:1870–1886
Sarno S, Pinna LA (2008) Protein kinase CK2 as a druggable target. Mol Biosyst 4:889–894
Munstermann U, Fritz G, Seitz G, Lu YP, Schneider HR, Issinger OG (1990) Casein kinase II is elevated in solid human tumours and rapidly proliferating non-neoplastic tissue. Eur J Biochem 189:251–257
Prowald K, Fischer H, Issinger OG (1984) Enhanced casein kinase II activity in human tumour cell cultures. FEBS Lett 176:479–483
Siemer S, Ornskov D, Guerra B, Boldyreff B, Issinger OG (1999) Determination of mRNA, and protein levels of p53, MDM2 and protein kinase CK2 subunits in F9 cells after treatment with the apoptosis-inducing drugs cisplatin and carboplatin. Int J Biochem Cell Biol 31:661–670
Channavajhala P, Seldin DC (2002) Functional interaction of protein kinase CK2 and c-Myc in lymphomagenesis. Oncogene 21:5280–5288
Unger GM, Davis AT, Slaton JW, Ahmed K (2004) Protein kinase CK2 as regulator of cell survival: implications for cancer therapy. Curr Cancer Drug Targets 4:77–84
Li D, Dobrowolska G, Aicher LD et al (1999) Expression of the casein kinase 2 subunits in Chinese hamster ovary and 3T3 L1 cells provides information on the role of the enzyme in cell proliferation and the cell cycle. J Biol Chem 274:32988–32996
Landesman-Bollag E, Song DH, Romieu-Mourez R et al (2001) Protein kinase CK2: signaling and tumorigenesis in the mammary gland. Mol Cell Biochem 227:153–165
Slaton JW, Unger GM, Sloper DT, Davis AT, Ahmed K (2004) Induction of apoptosis by antisense CK2 in human prostate cancer xenograft model. Mol Cancer Res 2:712–721
Faust RA, Tawfic S, Davis AT, Bubash LA, Ahmed K (2000) Antisense oligonucleotides against protein kinase CK2-alpha inhibit growth of squamous cell carcinoma of the head and neck in vitro. Head Neck 22:341–346
Seeber S, Issinger OG, Holm T, Kristensen LP, Guerra B (2005) Validation of protein kinase CK2 as oncological target. Apoptosis 10:875–885
Yamane K, Kinsella TJ (2005) Casein kinase 2 regulates both apoptosis and the cell cycle following DNA damage induced by 6-thioguanine. Clin Cancer Res 11:2355–2363
Izeradjene K, Douglas L, Delaney A, Houghton JA (2005) Casein kinase II (CK2) enhances death-inducing signaling complex (DISC) activity in TRAIL-induced apoptosis in human colon carcinoma cell lines. Oncogene 24:2050–2058
Yamane K, Kinsella TJ (2005) CK2 inhibits apoptosis and changes its cellular localization following ionizing radiation. Cancer Res 65:4362–4367
Sandholt IS, Olsen BB, Guerra B, Issinger OG (2009) Resorufin: a lead for a new protein kinase CK2 inhibitor. Anticancer Drugs 20:238–248
Pagano MA, Meggio F, Ruzzene M, Andrzejewska M, Kazimierczuk Z, Pinna LA (2004) 2-Dimethylamino-4,5,6,7-tetrabromo-1H-benzimidazole: a novel powerful and selective inhibitor of protein kinase CK2. Biochem Biophys Res Commun 321:1040–1044
Meggio F, Pagano MA, Moro S et al (2004) Inhibition of protein kinase CK2 by condensed polyphenolic derivatives, An in vitro and in vivo study. Biochemistry 43:12931–12936
Nie Z, Perretta C, Erickson P et al (2008) Structure-based design and synthesis of novel macrocyclic pyrazolo[1, 5-a] [1, 3, 5]triazine compounds as potent inhibitors of protein kinase CK2 and their anticancer activities. Bioorg Med Chem Lett 18:619–623
Pagano MA, Bain J, Kazimierczuk Z et al (2008) The selectivity of inhibitors of protein kinase CK2. An update. Biochem J 415:353–365
Vilk G, Saulnier RB, St Pierre R, Litchfield DW (1999) Inducible expression of protein kinase CK2 in mammalian cells. Evidence for functional specialization of CK2 isoforms. J Biol Chem 274:14406–14414
Al-Khouri AM, Ma Y, Togo SH, Williams S, Mustelin T (2005) Cooperative phosphorylation of the tumor suppressor phosphatase and tensin homologue (PTEN) by casein kinases and glycogen synthase kinase 3beta. J Biol Chem 280:35195–35202
Collaborative Computational Project, Number 4 (1994) The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 50:760–763
Borowski P, Deinert J, Schalinski S et al (2003) Halogenated benzimidazoles and benzotriazoles as inhibitors of the NTPase/helicase activities of hepatitis C and related viruses. Eur J Biochem 270:1645–1653
Sarno S, de Moliner E, Ruzzene M et al (2003) Biochemical and three-dimensional-structural study of the specific inhibition of protein kinase CK2 by [5-oxo-5, 6-dihydroindolo-(1, 2-a)quinazolin-7-yl]acetic acid (IQA). Biochem J 374:639–646
Zien P, Abramczyk O, Domanska K, Bretner M, Szyszka R (2003) TBBz but not TBBt discriminates between two molecular forms of CK2 in vivo and its implications. Biochem Biophys Res Commun 312:623–628
Litchfield DW (2003) Protein kinase CK2: structure, regulation and role in cellular decisions of life and death. Biochem J 369:1–15
Raaf J, Klopffleisch K, Issinger OG, Niefind K (2008) The catalytic subunit of human protein kinase CK2 structurally deviates from its maize homologue in complex with the nucleotide competitive inhibitor emodin. J Mol Biol 377:1–8
Sarno S, Salvi M, Battistutta R, Zanotti G, Pinna LA (2005) Features and potentials of ATP-site directed CK2 inhibitors. Biochim Biophys Acta 1754:263–270
Cherry M, Williams DH (2004) Recent kinase and kinase inhibitor X-ray structures: mechanisms of inhibition and selectivity insights. Curr Med Chem 11:663–673
Ahmad KA, Wang G, Slaton J, Unger G, Ahmed K (2005) Targeting CK2 for cancer therapy. Anticancer Drugs 16:1037–1043
Bretner M, Najda-Bernatowicz A, Lebska M, Muszynska G, Kilanowicz A, Sapota A (2008) New inhibitors of protein kinase CK2, analogues of benzimidazole and benzotriazole. Mol Cell Biochem 316:87–89
Pagano MA, Andrzejewska M, Ruzzene M et al (2004) Optimization of protein kinase CK2 inhibitors derived from 4,5,6,7-tetrabromobenzimidazole. J Med Chem 47:6239–6247
Pagano MA, Cesaro L, Meggio F, Pinna LA (2006) Protein kinase CK2: a newcomer in the ‘druggable kinome’. Biochem Soc Trans 34:1303–1306
Scaglioni PP, Yung TM, Cai LF et al (2006) A CK2-dependent mechanism for degradation of the PML tumor suppressor. Cell 126:269–283
Perera Y, Farina HG, Hernandez I et al (2008) Systemic administration of a peptide that impairs the protein kinase (CK2) phosphorylation reduces solid tumor growth in mice. Int J Cancer 122:57–62
Lebrin F, Chambaz EM, Bianchini L (2001) A role for protein kinase CK2 in cell proliferation: evidence using a kinase-inactive mutant of CK2 catalytic subunit alpha. Oncogene 20:2010–2022
Pagano MA, Poletto G, Di Maira G et al (2007) Tetrabromocinnamic acid (TBCA) and related compounds represent a new class of specific protein kinase CK2 inhibitors. Chembiochem 8:129–139
Miyata Y, Nishida E (2008) Evaluating CK2 activity with the antibody specific for the CK2-phosphorylated form of a kinase-targeting cochaperone Cdc37. Mol Cell Biochem 316:127–134
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zhu, D., Hensel, J., Hilgraf, R. et al. Inhibition of protein kinase CK2 expression and activity blocks tumor cell growth. Mol Cell Biochem 333, 159–167 (2010). https://doi.org/10.1007/s11010-009-0216-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11010-009-0216-0