
Abstract
CK2 is a ubiquitous and pleiotropic Ser-/Thr-targeting acidophilic protein kinase. CK2 plays an important role in the aberrant proliferation of malignant cancer cells. Because of constitutive activity of CK2, its inhibitors have been widely used to analyze the physiological function of CK2 in cellular systems. In addition, CK2 inhibitors are regarded as promising cancer chemotherapeutic candidates. Recently, several commonly used CK2 inhibitors have been shown to suppress DYRK (dual-specificity tyrosine-phosphorylation-regulated protein kinase) family protein kinases. Thus, the results obtained with conventional CK2 inhibitors should be carefully interpreted considering their effects on DYRKs. In this chapter, after an introductory section on CK2 and its inhibitors, the structures and activation mechanism of DYRK family protein kinases are portrayed. DYRK1A is one of the pivotal factors encoded in Down’s syndrome critical region on human chromosome 21, and dysregulation of DYRK1A may be a molecular basis of various phenotypes observed in Down’s syndrome patients. Substrates, physiological function, binding partners, regulatory mechanisms, and CK2 inhibitor sensitivities of DYRK1A are described in detail. Finally, the biological and clinical importance of CK2 and DYRK1A as therapeutic targets will be discussed.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others

Keywords
- CK2
- DYRK1A
- Phosphorylation
- Protein kinase
- Inhibitor
- Down’s syndrome
- TBB
- Cancer chemotherapeutics
- NFAT
- Leukemia
1 CK2 and Its Inhibitors
1.1 Protein Kinase CK2
CK2 (previously misleadingly called as “casein kinase 2”) is a ubiquitous serine-/threonine-specific protein kinase [1–6] which was discovered many decades ago as a major responsible enzyme for protein phosphorylation. CK2 is a tetrameric enzyme consisting of two α- and/or α′-catalytic subunits and two accessory β-subunits that dimerize to bring two catalytic subunits together. CK2 preferentially phosphorylates Ser/Thr residues that are followed by a cluster of acidic amino acids; thus, CK2 is categorized in the “acidophilic” protein kinase class. This is a sharp contrast to other classical AGC group “basophilic” protein kinases, including PKA and PKC. Many signaling protein kinases belonging to the CMGC group such as mitogen-activated protein kinases (MAP kinases) and cyclin-dependent kinases (CDKs) specifically phosphorylate Ser/Thr residues followed by a Pro residue; thus, they are categorized as “Pro-directed” protein kinases. CK2 shares a common root with CMGC group protein kinases in the phylogenic tree (Fig. 1), but the amino acid sequence of the catalytic domain of CK2 is only distantly related to other CMGC kinases. CK2 plays essential and pivotal roles in many physiological systems by phosphorylating a wide variety of more than 300 cellular proteins [7]. In fact, phospho-proteomic analyses suggest that CK2 alone may be responsible for more than 20 % of the eukaryotic protein phosphorylation sites [8, 9]. However, even after the long history of CK2 studies, it remains unclear how CK2 activity is regulated in cells. CK2 activity is readily detectable in extracts of cells and tissues without any stimulation. In contrast to many other protein kinases that require activating low-molecular-weight factors, no pivotal factor that directly activates CK2 in cells is revealed to date. Signaling protein kinases are often positively or negatively controlled by binding to regulatory partner subunits. The β-subunit of CK2 confers substrate specificity in CK2 holoenzyme and CK2β is required for optimal phosphorylation of certain substrates [4, 10, 11]. However, the catalytic CK2α/α′ alone seems to be able to phosphorylate most of CK2 substrates. Many signaling protein kinases are regulated by phosphorylation with upstream “kinase kinases.” Several protein kinases, including Cdc2, Src, ERK2, and Akt, have been reported to phosphorylate CK2 [12–15], but none of these kinases seems to be absolutely required for CK2 activity. All the evidence suggests that the regulatory mechanism of CK2 activity is different from that of other well-characterized signaling protein kinases whose activities are responsive to cellular stimulations. Therefore, CK2 has been often suggested to be “constitutively active.” On the other hand, CK2 activity is upregulated in highly malignant cancer cells [16, 17]. CK2 activity is intimately related to the cell cycle [18–20] and modulated during the circadian rhythm [21–23]. In addition, CK2 plays an important role in mediating Wnt/β-catenin signaling [24]. These facts altogether indicate that there should be a precise control mechanism for CK2 in cells, despite its constitutive activity when isolated.

A phylogenic tree of CMGC group Pro-directed protein kinases and CK2α. Similarity of amino acid sequences of the catalytic domains of indicated kinases was analyzed using the fixed distance scale UMGMA method. CK2 and DYRK family protein kinases are colored
1.2 CK2 Inhibitors
CK2 inhibitors have been widely used both in vivo and in vitro to examine the physiological role of CK2. CK2 activity has long been known to be strongly inhibited by polyamines such as heparin, and thus the heparin sensitivity has been regarded as a Merkmal for CK2 [25], but high molecular weights and the highly negatively charged nature of such polyanions limit their applicability to intact cells and bodies. In fact, heparin does not enter into the cells and also it shows many physiological effects on cells other than CK2 inhibition: therefore, it can only be used as a specific CK2 inhibitor in in vitro studies. Several protein kinase inhibitors, including emodin [26], quercetin [27], and apigenin [28, 29] suppress CK2 activity (Table 1); however, they are rather classified as general ATP-competitive ligands that inhibit a large number of protein kinases other than CK2. One of the first clues of a highly specific CK2 inhibitor came from a finding that DRB (5,6-dichloro-1-β-d-ribofuranosylbenzimidazole), an inhibitor of eukaryotic mRNA transcription, inhibits in a parallel manner the activity of CK2 [30]. A screening of derivatives of DRB identified several compounds that showed higher specificity for CK2 among other protein kinases known those days. The most widely used inhibitor for CK2 in the scientific literatures may be 4,5,6,7-tetrabromobenzotriazole (TBB), which was described as a potent and specific inhibitor for CK2 from yeast and mammalian sources in 1995 [31]. As of June 2014, more than 70 publications using TBB as a CK2 inhibitor are included in the PubMed database, and TBB still continues to be used as a specific CK2 inhibitor. TBB strongly inhibits CK2 (K i = 120 nM) but not another ubiquitous acidophilic kinase CK1. This is an advantage of TBB, because some previous CK2 inhibitors such as DRB showed almost comparable inhibition of CK1.
Recently, because many more kinases are available to examine the effects and specificities of protein kinase inhibitors, it is now apparent that the specificities of some of conventional CK2 inhibitors seem to be inadequate for cell biological and clinical purposes. Notably, many of the commonly used CK2 inhibitors, including DRB, TBB, TBBz (4,5,6,7-tetrabromo-1H-benzimidazole), IQA ([5-oxo-5,6-dihydro-indolo(1,2-a)quinazolin-7-yl]acetic acid), and DMAT (2-dimethylamino-4,5,6,7-tetrabromo-1H-benzimidazole), were revealed to inhibit DYRK family protein kinases as well (Table 1). Therefore, the results obtained with these conventional CK2 inhibitors should be carefully reinterpreted considering their effects on DYRK family protein kinases. In the following sections, a concise description on DYRK family protein kinases will be presented to be of help for those who are interested in and investigating CK2.
2 DYRK Family Protein Kinases
2.1 Structures of DYRK Family Protein Kinases
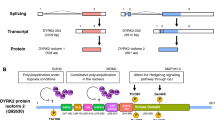
DYRK (dual-specificity tyrosine-phosphorylation-regulated protein kinase) family protein kinases belong to the CMGC protein kinase group that also includes MAP kinases and CDKs (Fig. 1). In mammalian species, the DYRK family consists of five members, including DYRK1A, DYRK1B, DYRK2, DYRK3, and DYRK4. According to the amino acid sequence similarity, DYRK1A and DYRK1B are categorized into “type 1” DYRKs, whereas DYRK2, DYRK3, and DYRK4 are “type 2” DYRKs. All DYRK family protein kinases share a homologous protein kinase catalytic domain in the center of their amino acid sequences (Fig. 2). The amino acid identity in the kinase domain is highest between DYRK1A and DYRK1B (85 %) and then between DYRK2 and DYRK3 (79 %). On the other hand, the amino acid identities between type 1 and type 2 DYRKs in the catalytic domain do not exceed 50 % (Fig. 2). All DYRKs possess characteristic N-terminal and C-terminal domains. DYRK1A and DYRK1B share two regions in their N- and C-terminal domains with amino acid identities of 71 % and 34 %, respectively. On the other hand, all the type 2 DYRKs share in their N-terminal domain a homologous region that is not observed in type 1 DYRKs. Finally, DYRK2 and DYRK3 have in common a similar C-terminal domain with 41 % amino acid identity (Fig. 2). DYRK1A is ubiquitously expressed in many species in multiple tissues and rich in testis and heart [32]. On the other hand, other DYRKs expressed mostly in testis [32]. DYRK1A has been most intensively studied so far; therefore, this chapter focuses mostly on DYRK1A. The detailed description of other members of DYRKs can be obtained in several review articles [33–36].

A schematic illustration of the DYRK family protein kinase structures. Amino acid identities in the N-terminal, kinase catalytic, and C-terminal domains of the five DYRK members with amino acid numbers are shown
2.2 Activation Mechanism of DYRK1A
Many CMGC group protein kinases contain a phosphorylation site in the activation loop region between the protein kinase subdomains VII and VIII. Phosphorylation of the activation loop by an upstream kinase is essential for the optimal activity of these kinases. For example, conventional MAP kinases, including ERK1/2, JNK|SAPK, and p38, encode a TXY motif in the activation loop, and phosphorylation of both the Thr and Tyr residues in the motif by upstream activating kinases, MAP kinase kinases (MAPK kinase, or MEK), activates the MAP kinases. DYRK family protein kinases contain a YXY motif in the corresponding region of the activation loop. Instead of being phosphorylated by an activating upstream kinase, DYRK1A phosphorylates the Tyr in the motif by a cis-autophosphorylation mechanism [37]. This autophosphorylation is required for the full activity of DYRK1A; a replacement of Tyr in the region by a non-phosphorylatable amino acid significantly obstructs the protein kinase activity of DYRK1A. In addition, it was proposed that the autophosphorylation of DYRKs occurs during the polypeptide translation and plays an important role in switching the kinase from the autophosphorylating Tyr-kinase into a Ser/Thr kinase toward exogenous substrates [38]. A recent analysis modified the scheme and it is proposed that DYRK1A is in a structural equilibrium between the Tyr-autophosphorylating kinase and the Ser-/Thr-targeting kinase and that the Tyr-autophosphorylation stabilizes the conformation suitable for the Ser-/Thr-targeting kinase activity in the equilibrium [39]. The involvement of molecular chaperones has been reported in the maturation process of GSK3β [40]. Similarly, molecular chaperones may play a role in the conformational regulation of DYRKs.
Just like CK2, it seems that DYRK1A does not require a cofactor, upstream activating kinases, or an associating subunit for its activity, and it is “constitutively active” after translation and Tyr-autophosphorylation. A possibility cannot be excluded, however, that phosphorylation of DYRK1A by other kinases modulates its activity. In addition, there should be binding partners of DYRK1A that control the activity, stability, and localization of DYRK1A in cells (see Sect. 4).
2.3 Substrates for DYRK1A
Substrate specificities of DYRKs have been determined using peptide-based in vitro assays [32, 41, 42]. Although there may be slight specificity differences between the members, DYRKs in common preferentially phosphorylate Ser/Thr residues followed by a Pro. Therefore, DYRKs can be categorized into the Pro-directed protein kinases as many other CMGC group kinases. In addition, DYRK1A efficiently phosphorylates Ser/Thr residues with Arg at the –3 position and Pro at the –2 position, resulting in the optimal recognition sequence of RPX[ST]P [41]. This is strikingly different from the phosphorylation consensus sequence of CK2, [ST]XX[DEpSpTpY]; therefore, most of CK2 phosphorylation sites in proteins might not be phosphorylated by DYRK1A, except in rare cases that a sequence fulfills the optimal substrate consensus for both CK2 and DYRK1A.
The number of reported DYRK1A substrates has been rapidly increasing in the last decade. The list of DYRK1A substrates includes eIF2B [43], microtubule-binding protein tau [43–46], splicing factor SF3b1|SAP155 [47], caspase 9 [48, 49], glycogen synthase [50], CRY2 [51], cyclin L2 [52], DSCR1|RCAN1 [53], and several transcription factors, including STAT3 [37, 54], CREB [55], FKHR [56], Gli1 [57], and NFAT [58, 59]. The best characterized DYRK1A substrate, NFAT (nuclear factor of activated T cells), locates in the cytosol when phosphorylated and hence transcriptionally inactive. After dephosphorylation by a Ca2+-activated phosphatase calcineurin, NFAT translocates to the nucleus and cooperates with multiple transcription factors to regulate the target gene expression. Nuclear NFAT protein is phosphorylated by several protein kinases including DYRK1A and translocates back to the cytosol [58, 59]. Therefore, DYRK1A is a negative regulator of NFAT-dependent signaling process.
2.4 Physiological Function of DYRK1A
Among the five members of DYRKs, DYRK1A has attracted most extensive attention, not only because of its ubiquitous expression but also because of its pivotal role in pleiotropic phenotypes observed in Down’s syndrome caused by chromosome 21 trisomy. The analysis of partial trisomy cases suggested that Down’s syndrome critical region (DSCR) in chromosome 21 between 21q22.1 and 21q22.3 is responsible for Down’s syndrome caused by the trisomy. DSCR includes 33 genes, and DYRK1A is one of them [60, 61]. In fact, DYRK1A is overexpressed in Down’s syndrome patients [62, 63], and the analyses of mouse models suggested that overexpression of DYRK1A is responsible for at least a part of pleiotropic phenotypes observed in Down’s syndrome patients [64–67], although other genes in chromosome 21 DSCR should also play a role. Minibrain is a mutant of Drosophila that exhibits a marked brain size reduction in the optic lobes and central hemispheres [68]. The responsible gene mnb was identified, which encodes a fruit fly homologue of DYRK1A [68]. These results indicate that DYRK1A plays an important role in developmental and functional regulation of neuronal cells from insects to human. In addition, triplication of DYRK1A is necessary and sufficient in model mice to cause the structural and functional retinal alterations that are also observed in Down’s syndrome children [69]. Gene knockout mice of the major DYRK1A substrate NFAT share various phenotypic alterations in facial structure, social interaction, motor function, and cardiac morphogenesis, with patients as well as model mice of Down’s syndrome [59]. Altogether, DYRK1A is one of the pivotal factors encoded in DSCR and regulates many physiological functions of cells, and dysregulation of DYRK1A leading to NFAT deactivation by hyper-phosphorylation may be a molecular basis of various phenotypes observed in Down’s syndrome patients. The functional importance of phosphorylation of other DYRK1A substrates in Down’s syndrome awaits further experimental evidence.
3 Effect of CK2 Inhibitors on DYRKs (See Table 1)
The human genome encodes more than 500 protein kinases, and recently the effect of conventional CK2 inhibitors has been tested on many more kinases than before. While most of CK2 inhibitors have no impact on most of protein kinases examined, DYRK family protein kinases are often strongly suppressed by conventional CK2 inhibitors (Table 1). This is rather an unexpected result because the amino acid sequences of DYRKs are only distantly related to that of CK2 (Fig. 1).
IQA is an effective and selective CK2 inhibitor (K i = 170 nM), and the structural details of the binding of IQA to CK2 were described by analyzing the CK2-IQA complex [70]. Examination of the effect of 10 μM IQA on a panel of 44 protein kinases indicated that most of the kinases other than CK2 (90 % inhibition) in the panel were not affected by IQA, except only one kinase, DYRK1A (50 % inhibition). Examination of 34 protein kinases for the effect of TBB and its derivatives resulted in a conclusion that TBB and 2-dimethylamino TBB (with better efficiency in CK2 inhibition) significantly inhibited DYRK1A, but not other kinases [71]. Pagano et al. examined a panel of 70 protein kinases for their sensitivity toward several conventional CK2 inhibitors [72]. While most of protein kinases tested were insensitive to 10 μM DMAT (93 % inhibition of CK2), it strongly inhibited the activity of DYRK1A (95 % inhibition), DYRK2 (94 % inhibition), and DYRK3 (97 % inhibition). Several other protein kinases, including PKD1, PIM1/2/3, and HIPK2/3, were also inhibited by DMAT. The effect of TBB on the same expanded set of 70 protein kinases was also examined, and DYRK and PIM kinases were strongly inhibited [72]. Several other newly developed TBB derivatives also significantly inhibited DYRK1A, whereas there are some compounds that showed only a modest inhibition of DYRK1A remaining sufficient efficacy in CK2 inhibition. Specific inhibition of CK2 without touching DYRK1A activity may be achieved if carefully selected compounds are used with appropriate concentrations.
Leucettines were originally developed as specific CLK and DYRK inhibitors, and leucettine L41 effectively suppressed all the members of DYRKs (IC50 for DYRK1A = 10 nM), while many other Pro-directed kinases, including CDKs and ERKs, were not affected [73]. However, the activity of CK2 was strongly suppressed by leucettine L41 (Table 1). Binding proteins for immobilized leucettine L41 were analyzed, and both DYRK1A and CK2 were found to specifically bind to the inhibitor [73]. These results further implicate the significant overlap of the binding and inhibiting spectrum of inhibitors for CK2 and DYRK1A.
All of these analyses suggested that CK2 and DYRKs may share a common structural factor in the ATP binding pocket, which may accept a similar array of ATP-analogous competitive inhibitors. The amino acid sequence alignment of the kinase catalytic domains of CK2α and DYRK1A is shown in Fig. 3. Both of the sequences have in common many characteristic amino acid motifs conserved in most of Ser/Thr kinases. In addition, there are several conserved amino acids between CK2α and DYRK1A, but it remains unclear which of them rule the sensitivity to shared inhibitors for both kinases.

Alignment of amino acid sequences of the kinase catalytic domains of CK2α and DYRK1A. Identical amino acids are shown by “|” and similar amino acids are shown by “+.” Amino acids conserved in most of Ser/Thr kinases are indicated by “*”
4 Functional Regulation of DYRK1A by Cellular Binding Partners
4.1 WDR68
To unveil the physiological function and regulation, the identification of cellular binding partners for DYRK1A should be of potential importance. Several studies revealed that a WD40-repeat protein WDR68 is a major binding partner for DYRK1A and DYRK1B, but not for class 2 DYRKs [50, 74–76]. The amino acid sequence of WDR68 is extremely conserved among species from plant to human, and all known mammalian WDR68 sequences encode exactly the same 342 amino acids. WDR68 is essential for cell proliferation and cell survival in mammalian cultured cells [75]. WDR68 is one of the components of a ubiquitin-conjugating enzyme DDB1-CUL4 complex [77], thus also called as DDB1-CUL4-associated factor 7 (DCAF7). The analysis of its amino acid sequence revealed that WDR68 contains five WD40-repeats; however, our computational structural analysis indicates that WDR68 forms a seven-bladed β-propeller ring [78]. All of these facts suggest that WDR68 plays a fundamental biological role in cells possibly by facilitating protein–protein interactions. Cellular localization of WDR68 is ubiquitous both in the cytoplasm and nucleus [75]. The cellular localization of WDR68 seems to be critical for its proper function, since mislocalization of WDR68 resulted in the developmental malformation of craniofacial structure in zebrafish [79]. Overexpression of DYRK1A induced nuclear accumulation of WDR68 [75]. Moreover, the molecular chaperone TRiC|CCT was essential for the DYRK1A-binding and nuclear accumulation of WDR68 [78]. The balance between cytoplasmic and nuclear WDR68 distribution may be precisely controlled, and DYRK1A should be a pivotal factor for the normal distribution and function of WDR68 in cells. The dysregulation of cellular localization of WDR68 by overexpressed DYRK1A might be a part of the molecular mechanism underlining the pleiotropic pathological alterations observed in Down’s syndrome patients.
4.2 14-3-3
The 14-3-3 proteins are a family of regulatory molecules and participate in a wide range of cellular processes through binding to hundreds of structurally and functionally diverse proteins [80, 81]. 14-3-3 proteins recognize a sequence motif containing phospho-Ser/Thr in target proteins. 14-3-3 proteins were identified as binding partners of DYRK1A by a yeast two-hybrid screening [82, 83]. In yeast, a DYRK1A homologue Yak1p and a 14-3-3 homologue Bmh1/2p have been also shown to be associated together [84, 85]. 14-3-3 binds to either the N-terminal region of DYRK1A in a phosphorylation-independent manner or to the PEST domain near the C-terminal region after autophosphorylation of DYRK1A [82, 83]. The binding of 14-3-3 modestly increased DYRK1A kinase activity in vitro, and the inhibition of the 14-3-3 binding to DYRK1A by a small peptide decreased DYRK1A kinase activity. These results indicate that the 14-3-3 binding facilitates the protein kinase activity of DYRK1A.
4.3 REST|NRSF
REST (RE1-silencing transcription factor)|NRSF (neuron-restrictive silencer factor) plays a pivotal role in neuronal differentiation process by modulating transcription of its target genes by binding to a specific DNA element, the repressor element 1 (RE1)|neuron-restrictive silencer element (NRSE) [86, 87]. Target proteins for REST, including ion channels, synaptic proteins, and neurotransmitter receptors, have fundamental functions in neuronal cells. REST acts as a repressor of neuronal differentiation and activates proliferation. REST is induced during normal aging in human neurons, but is lost in Alzheimer’s disease patients [88]. REST levels are closely correlated with cognitive preservation during aging, suggesting an important role of REST in neuroprotection [88]. DYRK1A overexpression reduces REST protein levels by facilitating its ubiquitination and degradation [89]. DYRK1A interacts with a SWI/SNF complex that is known to bind to REST [89]. REST stability is regulated by phosphorylation-dependent ubiquitination by an E3 ligase; however, it remains unclear if DYRK1A directly phosphorylates REST. Altogether, the DYRK1A function in neural cell differentiation may in part be ascribed to its REST level regulation. On the other hand, REST can activate DYRK1A transcription via a RE1|NRSE site in the human DYRK1A promoter [90], suggesting a negative feedback loop mechanism that precisely controls the expression levels of REST and DYRK1A. Dysregulation of the DYRK1A–REST combination may result in developmental as well as functional defects of neural system observed in Down’s syndrome patients.
5 CK2 and DYRK1A as Therapeutic Targets
5.1 CK2 Inhibitors as Cancer Chemotherapeutic Agents
Early observations suggested that CK2 could be activated by growth factors such as epidermal growth factor and insulin-like growth factor [91–93]; however, recent analyses indicated that this might not generally be the case [94, 95]. On the other hand, CK2 activity is recognized to be higher in rapidly proliferating cells [16, 17], and exogenous overexpression of CK2 in transgenic mice is tumorigenic [96]. CK2 is thus implicated to play an important role in supporting the malignant growth of cancer cells and tumors. There are many known CK2 substrates that are involved in cell growth and proliferation; however, the detailed molecular mechanism of CK2-mediated cell proliferation is not yet fully revealed. CK2-dependent phosphorylation of Cdc37, a kinase-targeting co-chaperone for Hsp90, is essential for the folding and function of many Cdc37/Hsp90 client signaling kinases, including Cdk4 and Raf1 [97–100]. Therefore, phosphorylation of Cdc37 alone might significantly contribute to the important role of CK2 in cell growth and proliferation. CK2 is regarded as a promising molecular target for cancer chemotherapy, and many new-generation CK2 inhibitors have been recently developed. CX-4945 [Silmitasertib®] (5-((3-chlorophenyl)amino)benzo[c][2,6]naphthyridine-8-carboxylic acid) is a potent (Ki < 1 nM) and orally available ATP-competitive inhibitor with unprecedented specificity for CK2α and CK2α′ catalytic subunits [101, 102]. CX-4945 has antiproliferative activity in multiple cancer cell lines, shows antitumor efficacy in mouse models, and is under clinical trials for cancer chemotherapy [103, 104]. In phase I clinical trials, CX-4945 induced stable disease in 20 % of patients with different solid tumors, having promising pharmacodynamic and safety profiles. A combinatorial treatment with CK2 inhibitors and other chemotherapeutic agents might be also a valid therapeutic option.
5.2 Down’s Syndrome, Leukemia, and DYRK1A Inhibitors
Involvement of DYRK1A in tumorigenesis is rather complex. Adult Down’s syndrome individuals overexpressing DYRK1A show reduced tendency of most of malignant solid tumors of epithelial origin [105, 106]. This suggests that DYRK1A may have a tumor-suppressive function, but the molecular basis for this observation is not yet completely understood. It should be pointed out that DYRK family protein kinases have been proposed to play a role in promoting cell apoptosis [36], which is intimately involved in the exclusion of cancer cells in the body. On the contrary, children with Down’s syndrome have a markedly increased risk of developing both acute megakaryoblastic leukemia and acute lymphoblastic leukemia as compared with children who do not have Down’s syndrome [105, 106]. Therefore, DYRK1A could be proleukemic in children and antitumorigenic in adults.
The molecular mechanism behind this paradox is only partially figured out. In general, activation of the NFAT pathway is considered cancer promoting through several mechanisms [107]. The NFAT family proteins were originally discovered in T cells and shown to facilitate T cell activation and proliferation. In addition, NFAT pathway can enhance angiogenesis by activating transcription of VEGF (vascular endothelial growth factor). As describe in Sects. 2.3 and 2.4, increased expression of DYRK1A suppresses NFAT function by the phosphorylation-dependent nuclear export of NFAT. DYRK1A, in cooperation with another protein DSCR1|RCAN1 encoded in DSCR, suppresses VEGF-dependent endothelial cell proliferation [108]. The suppression of tumor angiogenesis by the DYRK1A-provoked NFAT inhibition may explain the lower rate of epithelial cancers in Down’s syndrome adults. Angiogenesis is critical for the growth and expansion of cancer cells in solid tumor, whereas most of leukemic cells in blood do not rely on angiogenesis for proliferation. This could be one of the reasons why leukemia is increased, while solid tumors are decreased in Down’s syndrome patients. Obviously, contributions of other DYRK1A substrates and other proteins encoded in chromosome 21 DSCR cannot be ruled out in the trisomy-related tumor suppression.
In any case, cell biological analyses with specific DYRK1A inhibitors may shed new light on the molecular basis of Down’s syndrome and tumorigenesis as well. In addition, DYRK1A inhibition may have a therapeutic benefit. Recently, several specific DYRK1A inhibitors have been identified [109]. Harmine (7-methoxy-1-methyl-9H-pyrido[3,4-b]indole), originally isolated from South American vine, was reported to suppress the activity of DYRK1A [110], though it also shows a potent inhibitory effect on monoamine oxidase A [111]. Malinge et al. suggested that DYRK1A inhibitors may be clinically useful in the context of Down’s syndrome-related acute megakaryoblastic leukemia by demonstrating that harmine inhibited the growth of megakaryoblastic leukemic cell lines with trisomy 21 [112]. Since some of conventional CK2 inhibitors have potent inhibitory activity on DYRK1A (see Sect. 3), one may consider the CK2 inhibitors as lead compounds for the development of DYRK1A-specific inhibitors that have no impact on CK2 activity.
Down’s syndrome is caused by congenital alteration by the chromosome trisomy, and the therapeutic aim should be the modulation of DYRK1A from 1.5-fold increase back to the normal level [111]. This should be quite challenging having only a narrow therapeutic range for the DYRK1A inhibitors, but certainly worth the further investigation.
Conclusion
CK2 is a ubiquitous and constitutively active protein kinase implicated in the malignant proliferation of cancer cells. Low-molecular-weight specific inhibitors for CK2 have been developed both for biological and clinical applications. Recent studies revealed that many conventional CK2 inhibitors including DMAT and TBB also suppress the activity of DYRK family protein kinases; therefore, the alterations in tissues and cells observed with these drugs should not be ascribed solely to CK2. New-generation highly specific CK2 inhibitors such as CX-4945 have been recently developed and tested for clinical applications as cancer chemotherapeutic agents. DYRK1A plays a pivotal role in Down’s syndrome, and DYRK1A-dependent phosphorylation of NFAT is a key event that causes various phenotypes and also low incidence of solid tumors in Down’s syndrome patients. Specific inhibitors for DYRK1A may also have biological and clinical importance.
References
Olsen BB, Guerra B, Niefind K, Issinger OG (2010) Structural basis of the constitutive activity of protein kinase CK2. Methods Enzymol 484:515–529
Niefind K, Issinger OG (2010) Conformational plasticity of the catalytic subunit of protein kinase CK2 and its consequences for regulation and drug design. Biochim Biophys Acta 1804:484–492
Olsten ME, Litchfield DW (2004) Order or chaos? An evaluation of the regulation of protein kinase CK2. Biochem Cell Biol 82:681–693
Filhol O, Martiel J-L, Cochet C (2004) Protein kinase CK2: a new view of an old molecular complex. EMBO Rep 5:351–355
Pinna LA (2003) The raison d'être of constitutively active protein kinases: the lesson of CK2. Acc Chem Res 36:378–384
Litchfield DW (2003) Protein kinase CK2: structure, regulation and role in cellular decisions of life and death. Biochem J 369:1–15
Meggio F, Pinna LA (2003) One-thousand-and-one substrates of protein kinase CK2? FASEB J 17:349–368
Salvi M, Sarno S, Cesaro L, Nakamura H, Pinna LA (2009) Extraordinary pleiotropy of protein kinase CK2 revealed by weblogo phosphoproteome analysis. Biochim Biophys Acta 1793:847–859
Pagano MA, Cesaro L, Meggio F, Pinna LA (2006) Protein kinase CK2: a newcomer in the “druggable kinome”. Biochem Soc Trans 34:1303–1306
Bolanos-Garcia VM, Fernandez-Recio J, Allende JE, Blundell TL (2006) Identifying interaction motifs in CK2β—a ubiquitous kinase regulatory subunit. Trends Biochem Sci 31:654–661
Bibby AC, Litchfield DW (2005) The multiple personalities of the regulatory subunit of protein kinase CK2: CK2 dependent and CK2 independent roles reveal a secret identity for CK2β. Int J Biol Sci 1:67–79
Bosc DG, Slominski E, Sichler C, Litchfield DW (1995) Phosphorylation of casein kinase II by p34cdc2. Identification of phosphorylation sites using phosphorylation site mutants in vitro. J Biol Chem 270:25872–25878
Donella-Deana A, Cesaro L, Sarno S, Ruzzene M, Brunati AM, Marin O, Vilk G, Doherty-Kirby A, Lajoie G, Litchfield DW, Pinna LA (2003) Tyrosine phosphorylation of protein kinase CK2 by Src-related tyrosine kinases correlates with increased catalytic activity. Biochem J 372:841–849
Ji H, Wang J, Nika H, Hawke D, Keezer S, Ge Q, Fang B, Fang X, Fang D, Litchfield DW, Aldape K, Lu Z (2009) EGF-induced ERK activation promotes CK2-mediated disassociation of α-catenin from β-catenin and transactivation of β-catenin. Mol Cell 36:547–559
Nguyen le XT, Mitchell BS (2013) Akt activation enhances ribosomal RNA synthesis through casein kinase II and TIF-IA. Proc Natl Acad Sci U S A 110:20681–20686
Ahmad KA, Wang G, Slaton J, Unger G, Ahmed K (2005) Targeting CK2 for cancer therapy. Anticancer Drugs 16:1037–1043
Xu X, Landesman-Bollag E, Channavajhala PL, Seldin DC (1999) Murine protein kinase CK2: gene and oncogene. Mol Cell Biochem 191:65–74
Marshak DR, Russo GL (1994) Regulation of protein kinase CKII during the cell division cycle. Cell Mol Biol Res 40:513–517
Lorenz P, Pepperkok R, Pyerin W (1994) Requirement of casein kinase 2 for entry into and progression through early phases of the cell cycle. Cell Mol Biol Res 40:519–527
Homma MK, Homma Y (2008) Cell cycle and activation of CK2. Mol Cell Biochem 316:49–55
Allada R, Meissner RA (2005) Casein kinase 2, circadian clocks, and the flight from mutagenic light. Mol Cell Biochem 274:141–149
Tsuchiya Y, Akashi M, Matsuda M, Goto K, Miyata Y, Node K, Nishida E (2009) Involvement of the protein kinase CK2 in the regulation of mammalian circadian rhythms. Sci Signal 2:ra26
Lin JM, Kilman VL, Keegan K, Paddock B, Emery-Le M, Rosbash M, Allada R (2002) A role for casein kinase 2α in the Drosophila circadian clock. Nature 420:816–820
Seldin DC, Landesman-Bollag E, Farago M, Currier N, Lou D, Dominguez I (2005) CK2 as a positive regulator of Wnt signalling and tumourigenesis. Mol Cell Biochem 274:63–67
Tuazon PT, Traugh JA (1990) Casein kinase I and II—multipotential serine protein kinases: structure, function and regulation. Adv Second Messenger Phosphoprotein Res 23:123–164
Yim H, Lee YH, Lee CH, Lee SK (1999) Emodin, an anthraquinone derivative isolated from the rhizomes of Rheum palmatum, selectively inhibits the activity of casein kinase II as a competitive inhibitor. Planta Med 65:9–13
Davies SP, Reddy H, Caivano M, Cohen P (2000) Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J 351:95–105
Hessenauer A, Montenarh M, Götz C (2003) Inhibition of CK2 activity provokes different responses in hormone-sensitive and hormone-refractory prostate cancer cells. Int J Oncol 22:1263–1270
Zhao M, Ma J, Zhu HY, Zhang XH, Du ZY, Xu YJ, Yu XD (2011) Apigenin inhibits proliferation and induces apoptosis in human multiple myeloma cells through targeting the trinity of CK2, Cdc37 and Hsp90. Mol Cancer 10:104
Zandomeni R, Zandomeni MC, Shugar D, Weinmann R (1986) Casein kinase type II is involved in the inhibition by 5,6-dichloro-1-β-D-ribofuranosylbenzimidazole of specific RNA polymerase II transcription. J Biol Chem 261:3414–3419
Szyszka R, Grankowski N, Felczak K, Shugar D (1995) Halogenated benzimidazoles and benzotriazoles as selective inhibitors of protein kinases CK I and CK II from Saccharomyces cerevisiae and other sources. Biochem Biophys Res Commun 208:418–424
Becker W, Weber Y, Wetzel K, Eirmbter K, Tejedor FJ, Joost H-G (1998) Sequence characteristics, subcellular localization, and substrate specificity of DYRK-related kinases, a novel family of dual specificity protein kinases. J Biol Chem 273:25893–25902
Mercer SE, Friedman E (2006) Mirk/Dyrk1B: a multifunctional dual-specificity kinase involved in growth arrest, differentiation, and cell survival. Cell Biochem Biophys 45:303–315
Aranda S, Laguna A, de la Luna S (2011) DYRK family of protein kinases: evolutionary relationships, biochemical properties, and functional roles. FASEB J 25:449–462
Becker W (2012) Emerging role of DYRK family protein kinases as regulators of protein stability in cell cycle control. Cell Cycle 11:3389–3394
Yoshida K (2008) Role for DYRK family kinases on regulation of apoptosis. Biochem Pharmacol 76:1389–1394
Wiechmann S, Czajkowska H, de Graaf K, Grötzinger J, Joost HG, Becker W (2003) Unusual function of the activation loop in the protein kinase DYRK1A. Biochem Biophys Res Commun 302:403–408
Lochhead PA, Sibbet G, Morrice N, Cleghon V (2005) Activation-loop autophosphorylation is mediated by a novel transitional intermediate form of DYRKs. Cell 121:925–936
Walte A, Rüben K, Birner-Gruenberger R, Preisinger C, Bamberg-Lemper S, Hilz N, Bracher F, Becker W (2013) Mechanism of dual specificity kinase activity of DYRK1A. FEBS J 280:4495–4511
Lochhead PA, Kinstrie R, Sibbet G, Rawjee T, Morrice N, Cleghon V (2006) A chaperone-dependent GSK3β transitional intermediate mediates activation-loop autophosphorylation. Mol Cell 24:627–633
Himpel S, Tegge W, Frank R, Leder S, Joost HG, Becker W (2000) Specificity determinants of substrate recognition by the protein kinase DYRK1A. J Biol Chem 275:2431–2438
Campbell LE, Proud CG (2002) Differing substrate specificities of members of the DYRK family of arginine-directed protein kinases. FEBS Lett 510:31–36
Woods YL, Cohen P, Becker W, Jakes R, Goedert M, Wang X, Proud CG (2001) The kinase DYRK phosphorylates protein-synthesis initiation factor eIF2Bepsilon at Ser539 and the microtubule-associated protein tau at Thr212: potential role for DYRK as a glycogen synthase kinase 3-priming kinase. Biochem J 355:609–615
Ryoo SR, Jeong HK, Radnaabazar C, Yoo JJ, Cho HJ, Lee HW, Kim IS, Cheon YH, Ahn YS, Chung SH, Song WJ (2007) DYRK1A-mediated hyperphosphorylation of Tau. A functional link between Down syndrome and Alzheimer disease. J Biol Chem 282:34850–34857
Liu F, Li B, Tung EJ, Grundke-Iqbal I, Iqbal K, Gong CX (2007) Site-specific effects of tau phosphorylation on its microtubule assembly activity and self-aggregation. Eur J Neurosci 26:3429–3436
Kimura R, Kamino K, Yamamoto M, Nuripa A, Kida T, Kazui H, Hashimoto R, Tanaka T, Kudo T, Yamagata H, Tabara Y, Miki T, Akatsu H, Kosaka K, Funakoshi E, Nishitomi K, Sakaguchi G, Kato A, Hattori H, Uema T, Takeda M (2007) The DYRK1A gene, encoded in chromosome 21 Down syndrome critical region, bridges between beta-amyloid production and tau phosphorylation in Alzheimer disease. Hum Mol Genet 16:15–23
de Graaf K, Czajkowska H, Rottmann S, Packman LC, Lilischkis R, Lüscher B, Becker W (2006) The protein kinase DYRK1A phosphorylates the splicing factor SF3b1/SAP155 at Thr434, a novel in vivo phosphorylation site. BMC Biochem 7:7
Laguna A, Aranda S, Barallobre MJ, Barhoum R, Fernández E, Fotaki V, Delabar JM, de la Luna S, de la Villa P, Arbonés ML (2008) The protein kinase DYRK1A regulates caspase-9-mediated apoptosis during retina development. Dev Cell 15:841–853
Seifert A, Allan LA, Clarke PR (2008) DYRK1A phosphorylates caspase 9 at an inhibitory site and is potently inhibited in human cells by harmine. FEBS J 275:6268–6280
Skurat AV, Dietrich AD (2004) Phosphorylation of Ser640 in muscle glycogen synthase by DYRK family protein kinases. J Biol Chem 279:2490–2498
Kurabayashi N, Hirota T, Sakai M, Sanada K, Fukada Y (2010) DYRK1A and glycogen synthase kinase 3β, a dual-kinase mechanism directing proteasomal degradation of CRY2 for circadian timekeeping. Mol Cell Biol 30:1757–1768
de Graaf K, Hekerman P, Spelten O, Herrmann A, Packman LC, Büssow K, Müller-Newen G, Becker W (2004) Characterization of cyclin L2, a novel cyclin with an arginine/serine-rich domain: phosphorylation by DYRK1A and colocalization with splicing factors. J Biol Chem 279:4612–4624
Jung MS, Park JH, Ryu YS, Choi SH, Yoon SH, Kwen MY, Oh JY, Song WJ, Chung SH (2011) Regulation of RCAN1 protein activity by Dyrk1A protein-mediated phosphorylation. J Biol Chem 286:40401–40412
Matsuo R, Ochiai W, Nakashima K, Taga T (2001) A new expression cloning strategy for isolation of substrate-specific kinases by using phosphorylation site-specific antibody. J Immunol Methods 247:141–151
Yang EJ, Ahn YS, Chung KC (2001) Protein kinase Dyrk1 activates cAMP response element-binding protein during neuronal differentiation in hippocampal progenitor cells. J Biol Chem 276:39819–39824
Woods YL, Rena G, Morrice N, Barthel A, Becker W, Guo S, Unterman TG, Cohen P (2001) The kinase DYRK1A phosphorylates the transcription factor FKHR at Ser329 in vitro, a novel in vivo phosphorylation site. Biochem J 355:597–607
Mao J, Maye P, Kogerman P, Tejedor FJ, Toftgard R, Xie W, Wu G, Wu D (2002) Regulation of Gli1 transcriptional activity in the nucleus by Dyrk1. J Biol Chem 277:35156–35161
Gwack Y, Sharma S, Nardone J, Tanasa B, Iuga A, Srikanth S, Okamura H, Bolton D, Feske S, Hogan PG, Rao A (2006) A genome-wide Drosophila RNAi screen identifies DYRK-family kinases as regulators of NFAT. Nature 441:646–650
Arron JR, Winslow MM, Polleri A, Chang CP, Wu H, Gao X, Neilson JR, Chen L, Heit JJ, Kim SK, Yamasaki N, Miyakawa T, Francke U, Graef IA, Crabtree GR (2006) NFAT dysregulation by increased dosage of DSCR1 and DYRK1A on chromosome 21. Nature 441:595–600
Galceran J, de Graaf K, Tejedor FJ, Becker W (2003) The Mnb/Dyrk1A protein kinase: genetic and biochemical properties. J Neural Transm Suppl 67:139–148
Hämmerle B, Elizalde C, Galceran J, Becker W, Tejedor FJ (2003) The Mnb/Dyrk1A protein kinase: neurobiological functions and Down syndrome implications. J Neural Transm Suppl 67:129–137
Guimerá J, Casas C, Estivill X, Pritchard M (1999) Human minibrain homologue (MNBH/DYRK1): characterization, alternative splicing, differential tissue expression, and overexpression in Down syndrome. Genomics 57:407–418
Dowjat WK, Adayev T, Kuchna I, Nowicki K, Palminiello S, Hwang YW, Wegiel J (2007) Trisomy-driven overexpression of DYRK1A kinase in the brain of subjects with Down syndrome. Neurosci Lett 413:77–81
Altafaj X, Dierssen M, Baamonde C, Martí E, Visa J, Guimerà J, Oset M, González JR, Flórez J, Fillat C, Estivill X (2001) Neurodevelopmental delay, motor abnormalities and cognitive deficits in transgenic mice overexpressing Dyrk1A (minibrain), a murine model of Down’s syndrome. Hum Mol Genet 10:1915–1923
Ahn KJ, Jeong HK, Choi HS, Ryoo SR, Kim YJ, Goo JS, Choi SY, Han JS, Ha I, Song WJ (2006) DYRK1A BAC transgenic mice show altered synaptic plasticity with learning and memory defects. Neurobiol Dis 22:463–472
Martínez de Lagrán M, Altafaj X, Gallego X, Martí E, Estivill X, Sahún I, Fillat C, Dierssen M (2004) Motor phenotypic alterations in TgDyrk1a transgenic mice implicate DYRK1A in Down syndrome motor dysfunction. Neurobiol Dis 15:132–142
Galdzicki Z, Siarey R, Pearce R, Stoll J, Rapoport SI (2001) On the cause of mental retardation in Down syndrome: extrapolation from full and segmental trisomy 16 mouse models. Brain Res Rev 35:115–145
Tejedor F, Zhu XR, Kaltenbach E, Ackermann A, Baumann A, Canal I, Heisenberg M, Fischbach KF, Pongs O (1995) Minibrain: a new protein kinase family involved in postembryonic neurogenesis in Drosophila. Cell 14:287–301
Laguna A, Barallobre MJ, Marchena MA, Mateus C, Ramírez E, Martínez-Cue C, Delabar JM, Castelo-Branco M, de la Villa P, Arbonés ML (2013) Triplication of DYRK1A causes retinal structural and functional alterations in Down syndrome. Hum Mol Genet 22:2775–2784
Sarno S, de Moliner E, Ruzzene M, Pagano MA, Battistutta R, Bain J, Fabbro D, Schoepfer J, Elliott M, Furet P, Meggio F, Zanotti G, Pinna LA (2003) Biochemical and three-dimensional-structural study of the specific inhibition of protein kinase CK2 by [5-oxo-5,6-dihydroindolo-(1,2-a)quinazolin-7-yl]acetic acid (IQA). Biochem J 374:639–646
Pagano MA, Andrzejewska M, Ruzzene M, Sarno S, Cesaro L, Bain J, Elliott M, Meggio F, Kazimierczuk Z, Pinna LA (2004) Optimization of protein kinase CK2 inhibitors derived from 4,5,6,7-tetrabromobenzimidazole. J Med Chem 47:6239–6247
Pagano MA, Bain J, Kazimierczuk Z, Sarno S, Ruzzene M, Di Maira G, Elliott M, Orzeszko A, Cozza G, Meggio F, Pinna LA (2008) The selectivity of inhibitors of protein kinase CK2: an update. Biochem J 415:353–365
Tahtouh T, Elkins JM, Filippakopoulos P, Soundararajan M, Burgy G, Durieu E, Cochet C, Schmid RS, Lo DC, Delhommel F, Oberholzer AE, Pearl LH, Carreaux F, Bazureau JP, Knapp S, Meijer L (2012) Selectivity, cocrystal structures, and neuroprotective properties of leucettines, a family of protein kinase inhibitors derived from the marine sponge alkaloid leucettamine B. J Med Chem 55:9312–9330
Mazmanian G, Kovshilovsky M, Yen D, Mohanty A, Mohanty S, Nee A, Nissen RM (2010) The zebrafish dyrk1b gene is important for endoderm formation. Genesis 48:20–30
Miyata Y, Nishida E (2011) DYRK1A binds to an evolutionarily conserved WD40-repeat protein WDR68 and induces its nuclear translocation. Biochim Biophys Acta 1813:1728–1739
Morita K, Lo Celso C, Spencer-Dene B, Zouboulis CC, Watt FM (2006) HAN11 binds mDia1 and controls GLI1 transcriptional activity. J Dermatol Sci 44:11–20
Jin J, Arias EE, Chen J, Harper JW, Walter JC (2006) A family of diverse Cul4-Ddb1-interacting proteins includes Cdt2, which is required for S phase destruction of the replication factor Cdt1. Mol Cell 23:709–721
Miyata Y, Shibata T, Aoshima M, Tsubata T, Nishida E (2014) The molecular chaperone TRiC/CCT binds to the Trp-Asp 40 (WD40) repeat protein WDR68 and promotes its folding, protein kinase DYRK1A binding, and nuclear accumulation. J Biol Chem 289:33320–33332
Wang B, Doan D, Roman Petersen Y, Alvarado E, Alvarado G, Bhandari A, Mohanty A, Mohanty S, Nissen RM (2013) Wdr68 requires nuclear access for craniofacial development. PLoS One 8:e54363
Darling DL, Yingling J, Wynshaw-Boris A (2005) Role of 14-3-3 proteins in eukaryotic signaling and development. Curr Top Dev Biol 68:281–315
Morrison DK (2009) The 14-3-3 proteins: integrators of diverse signaling cues that impact cell fate and cancer development. Trends Cell Biol 19:16–23
Alvarez M, Altafaj X, Aranda S, de la Luna S (2007) DYRK1A autophosphorylation on serine residue 520 modulates its kinase activity via 14-3-3 binding. Mol Biol Cell 18:1167–1178
Kim D, Won J, Shin DW, Kang J, Kim YJ, Choi SY, Hwang MK, Jeong BW, Kim GS, Joe CO, Chung SH, Song WJ (2004) Regulation of Dyrk1A kinase activity by 14-3-3. Biochem Biophys Res Commun 323:499–504
Moriya H, Shimizu-Yoshida Y, Omori A, Iwashita S, Katoh M, Sakai A (2001) Yak1p, a DYRK family kinase, translocates to the nucleus and phosphorylates yeast Pop2p in response to a glucose signal. Genes Dev 15:1217–1228
Lee P, Paik SM, Shin CS, Huh WK, Hahn JS (2011) Regulation of yeast Yak1 kinase by PKA and autophosphorylation-dependent 14-3-3 binding. Mol Microbiol 79:633–646
Negrini S, Prada I, D’Alessandro R, Meldolesi J (2013) REST: an oncogene or a tumor suppressor? Trends Cell Biol 23:289–295
Qureshi IA, Gokhan S, Mehler MF (2010) REST and CoREST are transcriptional and epigenetic regulators of seminal neural fate decisions. Cell Cycle 9:4477–4486
Lu T, Aron L, Zullo J, Pan Y, Kim H, Chen Y, Yang TH, Kim HM, Drake D, Liu XS, Bennett DA, Colaiácovo MP, Yankner BA (2014) REST and stress resistance in ageing and Alzheimer’s disease. Nature 507:448–454
Lepagnol-Bestel AM, Zvara A, Maussion G, Quignon F, Ngimbous B, Ramoz N, Imbeaud S, Loe-Mie Y, Benihoud K, Agier N, Salin PA, Cardona A, Khung-Savatovsky S, Kallunki P, Delabar JM, Puskas LG, Delacroix H, Aggerbeck L, Delezoide AL, Delattre O, Gorwood P, Moalic JM, Simonneau M (2009) DYRK1A interacts with the REST/NRSF-SWI/SNF chromatin remodelling complex to deregulate gene clusters involved in the neuronal phenotypic traits of Down syndrome. Hum Mol Genet 18:1405–1414
Lu M, Zheng L, Han B, Wang L, Wang P, Liu H, Sun X (2011) REST regulates DYRK1A transcription in a negative feedback loop. J Biol Chem 286:10755–10763
Sommercorn J, Mulligan JA, Lozeman FJ, Krebs EG (1987) Activation of casein kinase II in response to insulin and to epidermal growth factor. Proc Natl Acad Sci U S A 84:8834–8838
Ackerman P, Osheroff N (1989) Regulation of casein kinase II activity by epidermal growth factor in human A-431 carcinoma cells. J Biol Chem 264:11958–11965
Klarlund JK, Czech MP (1988) Insulin-like growth factor I and insulin rapidly increase casein kinase II activity in BALB/c 3T3 fibroblasts. J Biol Chem 263:15872–15875
Litchfield DW, Dobrowolska G, Krebs EG (1994) Regulation of casein kinase II by growth factors: a reevaluation. Cell Mol Biol Res 40:373–381
Miyata Y, Nishida E (2007) Analysis of the CK2-dependent phosphorylation of serine 13 in Cdc37 using a phospho-specific antibody and phospho-affinity gel electrophoresis. FEBS J 274:5690–5703
Seldin DC, Leder P (1995) Casein kinase IIα transgene-induced murine lymphoma: relation to theileriosis in cattle. Science 267:894–897
Miyata Y (2013) The pivotal role of CK2 in the kinome-targeting Hsp90 chaperone machinery. The Wiley-IUBMB series on biochemistry and molecular biology: protein kinase CK2. John Wiley & Sons, Inc. (Hoboken, NJ, U.S.A.)
Miyata Y (2009) CK2: the kinase controlling the Hsp90 chaperone machinery. Cell Mol Life Sci 66:1840–1849
Miyata Y, Nishida E (2004) CK2 controls multiple protein kinases by phosphorylating a kinase-targeting molecular chaperone Cdc37. Mol Cell Biol 24:4065–4074
Miyata Y, Nishida E (2004) Supervision of multiple signaling protein kinases by the CK2-Cdc37 couple, a possible novel cancer therapeutic target. Ann N Y Acad Sci 1030:150–157
Siddiqui-Jain A, Drygin D, Streiner N, Chua P, Pierre F, O’Brien SE, Bliesath J, Omori M, Huser N, Ho C, Proffitt C, Schwaebe MK, Ryckman DM, Rice WG, Anderes K (2010) CX-4945, an orally bioavailable selective inhibitor of protein kinase CK2, inhibits prosurvival and angiogenic signaling and exhibits antitumor efficacy. Cancer Res 70:10288–10298
Battistutta R, Cozza G, Pierre F, Papinutto E, Lolli G, Sarno S, O’Brien SE, Siddiqui-Jain A, Haddach M, Anderes K, Ryckman DM, Meggio F, Pinna LA (2011) Unprecedented selectivity and structural determinants of a new class of protein kinase CK2 inhibitors in clinical trials for the treatment of cancer. Biochemistry 50:8478–8488
Pierre F, Chua PC, O’Brien SE, Siddiqui-Jain A, Bourbon P, Haddach M, Michaux J, Nagasawa J, Schwaebe MK, Stefan E, Vialettes A, Whitten JP, Chen TK, Darjania L, Stansfield R, Bliesath J, Drygin D, Ho C, Omori M, Proffitt C, Streiner N, Rice WG, Ryckman DM, Anderes K (2011) Pre-clinical characterization of CX-4945, a potent and selective small molecule inhibitor of CK2 for the treatment of cancer. Mol Cell Biochem 356:37–43
Kim J, Kim SH (2012) Druggability of the CK2 inhibitor CX-4945 as an anticancer drug and beyond. Arch Pharm Res 35:1293–1296
Hasle H, Clemmensen IH, Mikkelsen M (2000) Risks of leukaemia and solid tumours in individuals with Down’s syndrome. Lancet 355:165–169
Nižetić D, Groet J (2012) Tumorigenesis in Down’s syndrome: big lessons from a small chromosome. Nat Rev Cancer 12:721–732
Müller MR, Rao A (2010) NFAT, immunity and cancer: a transcription factor comes of age. Nat Rev Immunol 10:645–656
Baek KH, Zaslavsky A, Lynch RC, Britt C, Okada Y, Siarey RJ, Lensch MW, Park IH, Yoon SS, Minami T, Korenberg JR, Folkman J, Daley GQ, Aird WC, Galdzicki Z, Ryeom S (2009) Down’s syndrome suppression of tumour growth and the role of the calcineurin inhibitor DSCR1. Nature 459(7250):1126–1130
Becker W, Sippl W (2011) Activation, regulation, and inhibition of DYRK1A. FEBS J 278:246–256
Göckler N, Jofre G, Papadopoulos C, Soppa U, Tejedor FJ, Becker W (2009) Harmine specifically inhibits protein kinase DYRK1A and interferes with neurite formation. FEBS J 276:6324–6337
Becker W, Soppa U, Tejedor FJ (2014) DYRK1A: a potential drug target for multiple Down syndrome neuropathologies. CNS Neurol Disord Drug Targets 13:26–33
Malinge S, Bliss-Moreau M, Kirsammer G, Diebold L, Chlon T, Gurbuxani S, Crispino JD (2012) Increased dosage of the chromosome 21 ortholog Dyrk1a promotes megakaryoblastic leukemia in a murine model of Down syndrome. J Clin Invest 122:948–962
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2015 Springer International Publishing Switzerland
About this chapter
Cite this chapter
Miyata, Y. (2015). CK2 Inhibitors and the DYRK Family Protein Kinases. In: Ahmed, K., Issinger, OG., Szyszka, R. (eds) Protein Kinase CK2 Cellular Function in Normal and Disease States. Advances in Biochemistry in Health and Disease, vol 12. Springer, Cham. https://doi.org/10.1007/978-3-319-14544-0_19
Download citation
DOI: https://doi.org/10.1007/978-3-319-14544-0_19
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-14543-3
Online ISBN: 978-3-319-14544-0
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)