
Abstract
Human activities directly and indirectly influence the gene flow of wildlife populations, significantly affecting their population structure. On Bali, Indonesia, long-tailed macaque (Macaca fascicularis) populations are associated with relatively undisturbed forest remnants, providing resources for macaques through human worship practices. To evaluate the long-term impact of this anthropogenic landscape on gene flow in macaques, we measured the microsatellite heterozygosity and genetic distance of 15 populations across the island. We then used assignment tests to measure more contemporary movement between populations. We found significant population structure across the island and found that despite this significant structuring, contemporary macaque dispersal across the island is relatively high, with a number of first generation migrants detected. Moreover, we identified one population in the core of the island that acts as a magnet for migrants, receiving 50 % of the first generation migrants in this analysis. Finally, we used individual-level Bayesian clustering analysis combined with kriging techniques to measure fine-scale genetic structure and identify significant boundaries relative to the landscape. Significant genetic structure suggests that the existence of forested temple sites and long-term co-existence with humans may have contributed to relative isolation between populations, even though macaques are known for their high dispersal abilities. However, more recent changes in land use practices in Bali, such as reallocation of lands for tourism, are influencing the patterns of dispersal and increasing the movement of individuals between novel sites, shifting the population structure of the macaques and potentially reducing island-wide genetic diversity.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Human actions can both directly and indirectly affect genetic structure of wildlife populations by altering the landscape and creating potential corridors and barriers to gene flow. For example, the regional Osceola–Ocala wildlife corridor in Florida, designed to maintain gene flow between wildlife populations through secondary habitat, resulted in black bear (Ursus americanus floridanus) movement in largely one direction. This movement resulted in limited population mixing, but with evidence of successful gene flow determined through assignment tests (Dixon et al. 2006). Human activity, via landscape alterations, can also shift population mating dynamics indirectly. For example, Macdougall-Shackleton et al. (2011) compared the structure of the songbird Melospiza melodia in urban island populations and true island populations and found that within urban islands—created through the development of roads and housing projects—there was a significant increase in homozygosity and inbreeding.
As the increased inbreeding in songbirds of urban islands exemplifies, the population structure of island species is of special importance. Genetic diversity is demonstrably lower in small populations, including island populations (Ellsttrand and Elam 1993; Frankham 1996, 1997; Eldridge et al. 1999). These small population effects can result in a reduced evolutionary flexibility within these populations (Frankham 1996, 1997; Lacy 1997; Reed et al. 2003), as shown in studies of brown bears (Paetkau et al. 1998), pygmy rabbits (Estes-Zumpf et al. 2010), and turtles (Avise et al. 1992). For example, in an island population of black-footed rock wallaby (Petrogale lateralis), reduced genetic diversity, due to high levels of inbreeding, contributed to reduced fitness while not driving the populations to extinction (Eldridge et al. 1999).
Contrary to the potential genetic costs experienced by island-living species, several wildlife species such as raccoons, deer, and coyotes in the US (Tigas et al. 2002; Dharmarajan et al. 2009), badgers in the UK (Byrne et al. 2012), and rats globally (Clark 1981), thrive in anthropogenic landscapes, utilizing the landscape in a way that challenges traditional thinking about urban areas as wildlife barriers (Blanchong et al. 2013). For example, recent work on raccoons (Procyon lotor) found that raccoons living in urban environments readily disperse through the landscape and found no evidence of either isolation by barrier or isolation by distance, despite finding significant population structure at the neighborhood level, suggesting fine scale fragmentation is important in determining dispersal patterns in this landscape (Dharmarajan et al. 2009). Work demonstrating the role of the urban landscape as significant in shaping the population structure of wildlife species—either by creating a barrier or corridor for gene flow—has also been shown in bobcats, black bears, frogs (Hitchings and Beebee 1997; Dixon et al. 2006; Ruell et al. 2012). This suggests that some wildlife populations are capable of living alongside human populations, using the landscape in previously unexpected ways, which results in subsequent and unanticipated effects to the genetic structure of these wildlife populations.
Long-tailed macaques (Macaca fascicularis), found throughout Southeast Asia, exhibit strong female philopatry and male dispersal (Fooden 1995; Fa and Lindburg 1996; Lane et al. 2010; Gumert 2011). Macaques thrive in areas of high human density and disturbances by exploiting human land use changes to maintain gene flow between subpopulations (Fuentes et al. 2005; Lane et al. 2010). On the island of Bali, Indonesia, 42 populations of long-tailed macaque have been previously identified (Southern 2002; Fuentes et al. 2005; see Fig. 1). These populations exist in a complex, anthropogenic landscape, dominated by riparian corridors, terraced rice agricultural land, and community temples (Lansing 1991; Wheatley 1999). Each population consisted of approximately 22–300+ individuals in 1–5 groups who utilize a consistent range associated with specific locations and habitats most often including a temple and/or shrines and associated forest patches (Fuentes et al. 2005; Lane et al. 2010). Interactions with nearby human communities include feeding, both direct feedings and indirect provisioning via offerings, cooperative efforts between macaques and local community members to entertain tourists, and can include crop raiding, hunting of macaques, and bites and scratches from macaques (Lane-deGraaf et al. 2013a). Increases in urbanization in and around the island’s core in the last 100 years may have resulted in shifting macaque population dynamics, including altered patterns of dispersal and gene flow (Lane et al. 2011). The close association of these non-human primates with human populations can intensify effects on population structure. This occurs as humans create potential barriers to gene flow, through land use alterations (Radaspiel et al. 2008; Liu et al. 2009), or facilitate opportunities for gene flow, through the direct movement of animals or the preservation of natural corridors (Luckett et al. 2004; Nasi et al. 2008; Shepherd 2010).

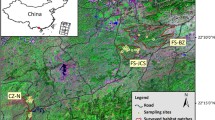
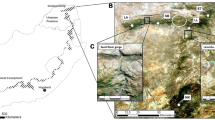
Map of Bali, including elevation, forest cover, and streams shown. Focal macaque populations in this study are marked in black
Long-term residential macaque populations, combined with recent landscape changes in the form of increased terracing for rice agriculture and growing urban areas potentially important in creating boundaries to gene flow, provide a unique system for examining the genetic structure of a cosmopolitan species across several populations on a small island as well as for evaluating the effect of recent landscape alterations on gene flow. Here, we characterized the genetic variation at 15 populations of long-tailed macaques across the island of Bali, Indonesia, using measures of heterozygosity (HE), allelic richness (AR), and genetic distance (FST), with the intent of determining the extent of population structure that exists on this relatively small island. We further used individual level landscape genetics analyses, including assignment and exclusion tests to identify first generation migrants and Bayesian cluster analyses and kriging analyses to identify significant genetic barriers at the individual level, providing a dual picture of historic and more contemporary gene flow between populations. Our goals, then, were twofold. First, we aimed to identify the level of genetic structuring among macaque populations across a relatively small island. Second, we sought to understand how this genetic structure is shaped by the landscape, including anthropogenic features of that landscape. We hypothesized that if historical gene flow has been occurring to nearest neighbor populations, then we expect a pattern of isolation-by-distance (IBD) over time to dominate the structure of macaque populations in Bali. Alternatively, if recent anthropogenic developments are influencing macaque movement, we expect human barriers to dominate island-wide macaque population structure.
Methods
Study design
Island-wide sampling was performed in order to best evaluate multiple hypotheses and to fully develop an overall picture of the population structure of the macaques across landscapes. The use of power analysis in genetic sampling is often challenging due to the frequency of rare alleles, the use and analysis of multiple loci, and the determination of F-statistics involving an additional layer of replication (Ryman et al. 2006; Landguth et al. 2010). However, given the cost and effort involved with sampling, a power analysis was performed to ensure our sampling was appropriate for producing interpretable results. Determination of sampling effort was based on censused population sizes and an estimation of 12 loci used for microsatellite genotyping, according to the following equation: x = 1/√(nm), where n is the number of individuals estimated in the population and m is the number of molecular markers (Patterson et al. 2006). This lower number of loci than actually sampled was chosen in an effort to accurately determine an appropriate number of samples to collect per population while assuming a likelihood of the occurrence of problematic, and thus uninformative, loci. We found that large populations (<300 individuals; U, PU, PL, AK, and S) required sampling from only 20 % of the total population size in order to make statistically and biologically relevant inferences, while smaller populations required sampling from as much as 45 % of the total population in order to make the same inferences. We also determined that one population—BT—required approximately sampling 130 % of the total population (n = 22) due to its small population size. While this population was not eliminated from the overall analysis, it was excluded from analyses that required additional replication steps beyond the level of F-statistics, such as the determination of effective population size and the assignment of first generation migrants. Finally, Landguth et al. (2010) determined that landscape genetics methods, including several employed here, were especially sensitive to analyses with extremely large sample sizes. Thus, our findings of sampling 20 % from the largest populations minimizes the potential for any method-introduced bias.
Sample collection
Blood and fecal samples (n = 345) were collected at 15 populations across the island of Bali, Indonesia, which is near the southern range of M. fascicularis (Fig. 1). Fecal samples (n = 226) were collected during single day collections from each population, following a census to determine population size. Only fresh samples were collected, and when necessary, monkeys were fed in order to facilitate fresh sampling. Upon collection, feces were immediately placed on ice. Samples were shipped to the US on dry ice and remained frozen at −85 °C until DNA extraction. Blood samples (n = 119) were collected at five of the fifteen populations by darting during the summer of 2007 (n = 57) or during the summer of 2001 (n = 62; by Lisa Jones-Engel and collaborators). Darting protocols have been previously described (Jones-Engel et al. 2006).
DNA was extracted using QIAGEN DNA Mini Kit and Stool Mini Kit (Valencia, CA), according to the manufacturer’s protocol, with an extended 30 min incubation time as the only modification to the protocol. Microsatellite loci were chosen for their known variability, ease of amplification, and locations across the macaque genome (Hadfield et al. 2001; Evans et al. 2001; Penedo et al. 2005). Loci span the genome, including locations on chromosomes 1, 6, 7, 8, 11, 13, 14, 17, and 22. Between 3 and 5 μl of extraction product was used for subsequent amplification.
PCR occurred in 30 μl final volumes, containing 20 pmol of each primer pair (ABI Human Mappairs), 0.25 mM of each dNTP (Thermo Scientific), 1× incubation buffer with 1.6 mM MgCl2 (Invitrogen), 1 U Taq DNA polymerase, and 1× BSA (bovine serum albumin, New England BioLabs) to minimize inhibition, in a (BioRad iCycler). Samples were amplified under one of two sets of cycler conditions (Hadfield et al. 2001; Penedo et al. 2005). Amplification conditions A (D1S550, D7S503, D8S272, D11S925, D13S317, D14S306, D17S1290, D22S345) began with an initial denaturation at 94 °C for 1 min, followed by 5 cycles consisting of a denaturing step at 94 °C for 1 min, an annealing step at 52 °C for 90 s, and an extension step at 72 °C for 90 s, followed by 45 cycles consisting of a denaturing step at 94 °C for 1 min, an annealing step at 57 °C for 90 s, and an extension step at 72 °C for 1 min, and a final extension step at 72 °C for 5 min. Amplification conditions B (D6S276, D6S2876, D6S2741, D6S291, MICA) began with an initial denaturation at 90 °C for 5 min, followed by 4 cycles consisting of a denaturing step at 94 °C for 1 min, an annealing step at 58 °C for 30 s, and an extension step at 72 °C for 30 s, followed by 30 cycles consisting of a denaturing step at 94 °C for 25 s, an annealing step at 58 °C for 30 s, and an extension step at 72 °C for 30 s, and a final extension step at 72 °C for 10 min. Amplification was verified by electrophoresis and visualization in 1 % agarose gels stained with ethidium bromide (0.5 μg/ml) in UV light.
Purified samples were genotyped at each of 13 loci on an ABI 3730x (Applied Biosystems) at the Yale DNA Analysis Facility on Science Hill, according to their protocols, using 1 μl of template and 0.5 μl of Liz 600 size standard (Applied Biosystems). Allele sizes were determined in GeneMapper v3.7 (Applied Biosystems). Linear regressions were performed to compare the genetic diversity of MHC specific loci used (n = 5) to neutral loci (n = 8) to control for the hypervariability of the MHC region in our analyses. No significant patterns were found (NA: r2 = 0.171, p = 0.284; HE: r2 = 0.094, p = 0.349), and so all loci were included in subsequent analyses.
Data validation
Given the suboptimal quality of DNA extracted from feces, which includes an increase in inhibition factors and sources of non-target DNA from food and gut microbiota, as well as the potential for cross contamination associated with using human-targeted primers, data confirmation and validation were important in ensuring the quality of our data, and ultimately, our findings (Paetkau 2003). Both positive (known blood and fecal samples) and negative control samples were used throughout the analyses to both confirm amplification and to eliminate the possibility of researcher-driven cross contamination. In addition to the above described sampling scheme designed to minimize repeated sampling of individuals, internal validation of allele calls and testing for null alleles within loci was performed. While recommendations for data checking include triple checking (from amplification through allele calling) homozygotes and sevenfold checking for all heterozygotes (Taberlet et al. 1996), the cost efficacy and feasibility of this are not conducive to large scale studies, such as this. However, more recent work with fecal DNA (Hoffman and Amos 2005; Pompanon et al. 2005) suggests that a replication of only 10 % of the dataset, in conjunction with conservative bounds for the detection of null alleles and large allele dropout, are sufficient to ensure confidence in datasets of DNA extracted from fecal samples. Thus, approximately 75 % of samples were amplified in duplicate. Allele calls for each amplification were made in duplicate, by two separate researchers, and compared for consistency in calling. When discrepancies occurred between calls, PCRs were performed a third time, allele calls were again performed in duplicate, and final assignments made. Samples that did not fit into the published repeat motif were reanalyzed. If a discrepancy existed, samples were analyzed a third time. Alleles were officially called when the most recent calls matched reanalyzed calls. When a discrepancy remained due to amplification difficulty or allele call standardization issues, samples were eliminated from the analysis. After completing this process, approximately 20 % of the dataset was reanalyzed and compared to the original allele size calls for internal consistency.
The dataset was initially screened for outliers and data entry errors by the creation of an allele frequency table generated in Convert 1.31 (Glaubitz 2004). A secondary examination of the dataset was performed using Micro-Checker 2.2.3 (Van Oosterhour et al. 2004). Presence of putatively null alleles, large allele dropout, and scoring error due to stuttering was checked by individual loci. Two loci (D6S276 and D6S2876) were determined to have a high likelihood of null alleles based on a determination of excess homozygotes for most allele size classes, with 95 % CI, and were subsequently eliminated from the analysis. The adjusted data set was then re-checked, and no additional loci were found to have a high a likelihood of null alleles, large allele dropout, or stuttering, and thus, were used for all subsequent analyses.
Genetic analyses
Deviations from Hardy–Weinberg equilibrium (HWE) were tested for at each locus within each population in GENEPOP v.4.1 (Raymond and Rousset 1995; Rousset 2008), using exact tests. Observed and expected heterozygosities, pairwise FST values, and allelic richness were determined using Arlequin 3.11 (Excoffier et al. 2005). Within locus tests of heterozygote excess or deficiency, significant in determining evidence of past bottlenecks, were calculated in Bottleneck v.1.2 (Cornuet and Luikart 1996), under the Infinite Alleles Model, a biologically appropriate model that captures the evolution of microsatellites. Both the test for mode shifts in allelic distributions and the test of heterozygosity excess or deficiencies (DH/sd) were used to determine the existence of past bottlenecks. Differentiation between populations, measured by F-statistics computed according to Weir and Cockerham, and Nei’s standard genetic distances were analyzed using FSTAT 2.9.3.2 (Goudet 1995). Linkage disequilibrium values (r2) and effective population sizes (NEF) for individual populations were calculated in LDNe (Waples and Do 1997), using a model of random mating individuals and the standard temporal method for determination of effective population sizes. LDNe calculates r2 based on Weir’s (1979) unbiased estimator, which controls for sample size limitations and can be standardized to accommodate variability in allele frequencies and generating pairwise values for linkage disequilibrium (Waples and Do 2008). Estimates of 95 % confidence intervals (CI) for NEF were also calculated in LDNe. LDNe utilizes a jackknife estimate for computing 95 % CI for estimates of effective population sizes, which outperformed traditional parametric estimates in capturing NEF especially in situations of non-independent loci. Thus, when estimates of NEF fall within 95 % CI, especially in an analysis including unique and rare alleles, these estimates are more likely to be accurate (Waples and Do 1997).
Assignment tests and the detection of first generation migrants were performed in GeneClass 2 v.2.0.h (Piry et al. 2004), with the assignment value set conservatively at 0.001 and using the Bayesian approach laid out in Rannala and Mountain (1997) and with the determination of first generation migrants using the MCMC resampling method of Paetkau et al. (2004) to better control for Type 1 errors, or the mis-assignment of individuals from their reference populations. Assignment tests were performed on all samples; while male macaques are known dispersers, less frequently, female macaques have been known to begin new populations via fission–fusion dynamics (Seuer et al. 2010). Detection of first generation migrants was performed with the criterion estimates computed as the ratio of the likelihood of the individual genotype within the population where the individual was sampled to the highest likelihood value of all available sampled populations, including the sampled population, which has been shown to provide the most power in the determination of first generation migrants (Paetkau et al. 2004; Piry et al. 2004). To prevent any significant levels of linkage disequilibrium from artificially inflating our findings, we used two additional tests to reduce the inclusion of linked loci in our analysis. First, we determined the total number of pairwise comparisons between loci with significant LD and eliminated any locus involved in a high number of pairings. With high levels of LD remaining, we then determined the total number of pairwise comparisons between loci within each population and eliminated any population from the analysis which exhibited significant LD in greater than 5 % of those comparisons before running the assignment and determination of first generation migrant tests. This allowed us to use assignment tests—which use linkage disequilibrium as a critical component in the analysis—without violating the assumptions of the tests through the inclusion of a population with a high number of significant pairwise linkages. Only one population—S—was removed from this analysis as a result of these more constrained conditions.
To test for isolation by distance effects, we performed an island wide Mantel test between matrices of genetic and geographic distances using R (R Core Development Team 2009) and the ‘ade4’ package, performed as a Monte Carlo analysis (Thioulouse and Dray 2007). The measure of genetic distance used was Rousset’s (1997) FST/(1 − FST), and geographic distance (km) was log transformed, as recommended by de Campos Telles et al. (2005).
Landscape characterization
Bayesian cluster analyses and kriging are two methods for detecting fine-scale variation in spatial genetic structure (Piertney et al. 1998; Manel et al. 2003; Cushman et al. 2006; Storfer et al. 2007; Grivet et al. 2008; Guillot et al. 2008). Bayesian cluster analyses identifies significant barriers in spatially explicit genetic data while kriging interpolates values from known values in spatially sampled data, creating spatially-explicit maps of these relationships (Manel et al. 2003; Storfer et al. 2007; Segelbacher et al. 2008). The inclusion of anthopogenic landscape variables in these analyses allows for the measurement of the effects of the physical and anthropogenic landscape on population structure.
Identifying barriers
Bayesian cluster analyses was performed on genotype data and latitude and longitude for each individual macaque using R (R Core Development Team 2009) and the ‘Geneland’ package (Guillot et al. 2008). Individual macaque locations were estimated through the use of a random number generator for both latitude and longitude, with maximum limits set to within a 100 ha buffer zone of the population from which the individual was sampled. Analyses in Geneland were performed with a maximum number of loci of 305, with 100,000 iterations and a thinning value of 200, with a subsequent burnin value of 200. Mantel tests were performed on any groups of population clusters identified in the process of cluster analysis to test for patterns of isolation by distance within population clusters.
Visualizing the landscape
Kriging interpolates the relatedness of nearby individuals/populations based on the genotypes and geospatial information of known individuals/populations (Holderegger et al. 2010). Because kriging interpolates the values of a single factor of interest against spatial coordinates, principal components analysis (PCA) was performed in R (R Core Development Team 2009) of macaque genotype data in the ‘stats’ package. PCA was used to condense the dataset and to identify specific components that capture a significant amount of variation within the genetic data. In doing this analysis, we identified four significant components of the genetic landscape that were used in subsequent analyses. PCA was previously performed on the environmental parameters, and found representative landscape components: the anthropogenic landscape (PC1), includes tourism rate, provisioning rate, and macaque population size; the mixed environment landscape (PC2), includes rice agriculture and urban area as well as water availability; and the non-anthropogenic environment (PC3), includes forest area and elevation (Lane et al. 2011).
Kriging was then run on each of the significant environmental and genetic landscape components in R using the ‘gstat’ package (Pebesma 2004) as determined by each component’s loading scores. To determine the statistical significance of concordant and discordant patterns between environment and genetic landscape, pairwise t tests were performed on the results of the kriging variograms.
Results
Descriptive statistics
Eleven putatively neutral loci were amplified successfully in a total of 345 individuals, spanning 15 populations. Population level pairwise FST values ranged from 0.015 (LM–BG) to 0.218 (MK–AN) and were all significantly different from zero, except for one pairing (AS–AN: FST = 0.015, p = 0.297; Table 1), demonstrating a significant level of genetic structure between all 15 macaque populations. Further, three populations were identified as having evidence to support recent genetic bottlenecks—BG, CK, and PL—as determined by a mode shift in allele frequency and by evidence of significant heterozygosity excess across loci in these three populations. Effective population sizes were significantly lower than censused population sizes in all populations (Z = −3.408, p = 0.001; Table 2). To test for a historic pattern of nearest neighbor migration, a Mantel test compared geographic distance to genetic distance and found no evidence of isolation by distance (obs. = 0.00952, p = 0.394).
After the elimination of S due to significant LD in greater than 5 % of its potential pairwise relationships, we determined that 16 of 304 individuals were found to be likely first generation migrants. This number excluded 11 individuals sampled at AN and nine individuals sampled at AS (Table 3); assignment tests showed that these individuals were equally as likely to have their origins in either population, further supported by a non-significant FST value between the populations. Dispersal patterns between more diverged populations showed biased dispersal with PU receiving half of all migrants from across the island. Specifically, the origins of immigrants detected in PU were as follows: BD (1), BP (1), LM (1), and U (5). However, PU had only one emigrant, who migrated to U. First generation migrants, originating from PL, were also detected in AS. One migrant was detected in CK, also emigrating from PL, its nearest neighbor. BP had one migrant from AK. LM also received one migrant, originating from BG. No first generation migrants were detected in AK, BG, BD, MK, PL, or TK, and no first generation migrants were detected from CK, MK, or TK.
Bayesian cluster analyses identified no evidence of significant barriers between populations within the island’s stretches of contiguous forest and those outside of the forest patch, showing no support for the hypothesis that the forest acts as a dispersal corridor. However, cluster analysis identified significant barriers in genotypic distributions, separating the seven core populations (PU, AN, AS, AK, S, BP, and U) from the remaining populations (CK, PL, MK, BD, TK, LM, and BG; See Fig. 1). This suggests the presence of a core-periphery population partition. A Mantel test comparing genetic and spatial distance between these seven core populations found no evidence of isolation by distance (obs. = −0.30713, p = 0.871). In contrast, cluster analysis identified significant barriers between each peripheral population (Fig. 1), less the two southernmost populations (U and BP), which clustered tightly with the core group. The inclusion of the two southernmost populations suggests that Denpasar, the island’s largest metropolitan area with a human population density of over 6,500 individuals/km2 (CITATION), does not act as a barrier for macaque dispersal. Further, the identification of genetic barriers between periphery populations indicates significant genetic differentiation across the island, with a series of small barriers resulting in an overall east–west running gradient between these populations. An additional Mantel test comparing the genetic and spatial distance between these peripheral populations also found no evidence of isolation by distance (obs. = 0.0489, p = 0.631).
PCA of genotypic data found four significant components, explaining 57.8 % of the variation in the genetic data. Using the PC findings to krige, we determined that the largest component of the genetic landscape (PC1GENETIC), at 25.5 %, is significantly concordant with the anthropogenic components of the landscape—tourism rate, offering weight, and macaque population size. Moreover, offering weight and water availability, the major components of environmental PC2ENVIRONMENT, were significantly concordant with genetic principal component 4, which explains only 8.1 % of the genetic variation. Finally, environmental principal component 3—the ‘natural’ landscape comprised of forest area and elevation predominantly—was not significantly concordant with any component of the genetic landscape (Table 2).
Discussion
Landscape complexity on Bali has left a footprint of two levels of macaque population structure, allowing both historic and contemporary patterns to emerge. FST values and low effective population sizes reflect historical genetic structure, resulting from relative isolation across the island via human activities like rice agriculture and community temple development that have been ongoing for more than 1,000 years (Wheatley 1999). Contemporary patterns of gene flow—described by assignment analyses, cluster analysis, and kriging results—reflect the connectivity between macaque populations influenced by human activities occurring within the last 100 years, such as expanding urban areas and road building.
The movement of even the occasional immigrant between populations can have long-term effects on population structure. In island populations, this effect can be more pronounced, leading to reductions in overall genetic diversity as populations lose structure via gene flow (Frankham 1997). In long-tailed macaque populations, female philopatry and male dispersal results in a model of long-term residence. Strong matrilineal patterns develop while male macaques disperse from their natal group, typically before reaching sexual maturity (Melnick and Hoelzer 1992). However, our results suggest significant genetic structure exists across populations of long-tailed macaques on the small island of Bali, indicating male movement has not been as pronounced in this anthropogenic context as previously found. Significant pairwise FST values between all but two populations suggest long-term genetic isolation across the island. We found no evidence of isolation-by-distance between populations, suggesting that macaque dispersal, when occurring, is not following a ‘nearest neighbor’ pattern, as has been previously suggested for macaques (Thierry 2007). Three populations show evidence of recent bottlenecks and occur in the farthest east and farthest west stretches of the island. These bottlenecks further support low levels of historic gene flow, suggesting that these populations may have undergone a period of extreme isolation resulting in founder effects, evidence of which is still discernible today.
Low effective population sizes and the identification of numerous first generation migrants suggest that the patterns of macaque movement through the landscape are not explained by FST values alone. Our findings indicate contemporary migration between populations may be shifting due to changes in human land use practices and human-macaque interactions. These shifts in migration and focal temples are likely the result of rapid growth in population sizes, combined with a skew in reproductive success due to the hierarchical social structure of macaques, and the increase in males putatively remaining in their natal populations beyond reaching sexual maturity. Assignment tests identified 16 first generation migrants across 14 populations. However, the pattern of migration is one of a magnet, with 50 % of all migrants dispersing into one population (PU). This population is the largest population on the island, with approximately 400 individuals and comprised of at least four matrilines at the time of this study, although recent census data estimates as many as 600 individuals (Brotcorne et al. 2011). Further, it is the best understood in terms of human interactions, with regular large-scale and supplementary smaller-scale feedings, regulated tourism, and an on-site management team. Despite the high number of immigrants, PU had only one successful emigrant, which suggests that anthropogenic impacts are changing the dynamics of macaque population social structure, enticing male macaques to remain in residence at this population far longer than previously found in macaque populations (Fuentes et al. 2011). In long-tailed macaques, male rank, which correlates in general with mating success, can be temporally highly variable (Thierry 2007). Recent work in gorillas (Gorilla beringi beringi) demonstrate that kin-based behavior can result in males remaining in their natal groups long-term, benefitting from these kin associations (Bradley et al. 2004). While this research focused on between group male–male patterns of relatedness, the implication for male behavior benefitting from kin associations, when applied to a primate with a matrifocal hierarchy system, can suggest that males may remain in their native populations as long as the group social dynamics allow it. Anecdotal behavioral evidence suggests that males are remaining long-term in their natal population in at least the largest of populations in Bali (Fuentes et al. 2011), and evidence of strong genetic structure presented here suggest that reproductive success may be higher for males remaining in residence.
The results of our Bayesian cluster analyses analysis identified significant barriers separating the core of the island from all remaining populations, supporting our hypothesis of a relatively homogenous genetic landscape in the core facilitated by riverine connections and long-term co-existence to human populations that derive an economic benefit from macaques while remaining genetically distinct from peripheral populations. This core—periphery demarcation suggests that the combination of physical, or the volcanic range along the northern edge of the island, and anthropogenic effects, specifically the close proximity to human populations that do not support a tourism industry and therefore highly provision macaque populations in the island core, serve as barriers to dispersal between these two sets of populations. The significant barriers partitioning individual peripheral populations from each other, however, are also likely due to anthropogenic effects (Lane-deGraaf et al. 2013b). Outside of the island core, the lack of tourism associated with macaque populations provides no incentive for large-scale provisioning of these populations. As a result, crop raiding in these areas occurs at a much higher frequency. Negative human-macaque interactions are frequent, including bites and scratches, and hunting macaques for food occurs (Lane-deGraaf et al. 2013a). This anthropogenic context, combined with lack of corridors of suitable habitat between populations, facilitates increased fragmentation between the peripheral populations. As land use patterns continue to shift and human population density increases, it is likely that barriers to dispersal and gene flow will increase among peripheral populations, further partitioning these populations from each other, while core populations will become increasingly linked due to the relative protection of macaques and coexistence with local communities which benefit from the economic advantage of international tourism.
Kriging allowed us to more methodically identify subtleties in genetic variation. The significant concordance of the genetic landscape with the anthropogenic components of the landscape, of tourism rate, offering weight, and macaque population size, and the mixed landscape components, of rice agriculture, urban area and water availability, demonstrate that human land use patterns, specifically tourism industry patterns and associated large-scale provisioning efforts, act as a significant driver of macaque population structure, shaping dispersal patterns across the island. Further, the finding of significant discordance between environmental principal component 3—the ‘natural’ landscape comprised of forest area and elevation predominantly—and the genetic landscape suggests that long-term anthropogenic effects play a greater role in shaping macaque population structure across the island than ‘natural’ landscapes, including protected habitats.
Much work has been done establishing the need for wildlife corridors to maintain genetic diversity in the face of urban communities encroaching on wildlife habitat (Ruell et al. 2012; Magle et al. 2010; Munshi-South 2012). These corridors are designed to allow dispersal, and subsequent gene flow, between populations that would consider human communities to be a barrier. Urban wildlife species, however, often use the anthropogenic landscape in unexpected ways—using unforeseen corridors and finding barriers where we would not predict. For example, a generalist omnivore species known to thrive in human communities—raccoons (Procyon lotor)—displayed significant population structure at the neighborhood level, despite no evidence for population barriers at the population level. Moreover, the evidence suggests that social interactions and behavior were significant in shaping the genetic structure of raccoons between neighborhoods and is likely important in other fine-scale analyses (Dharmarajan et al. 2009). Thus, changes to the urban landscape may shift the importance of factors which influence genetic structure from dispersal distance alone to more extrinsic factors, including social dynamics and behavior. Alternatively, black-tailed prairie dog populations, which require long distance dispersal to maintain gene flow between relatively isolated populations, were able to maintain connectivity, albeit at a slightly reduced rate, between distant populations in urban landscapes. This finding led the authors to suggest that despite urban development, prairie dogs had reached a possible endpoint for the genetic consequences associated with habitat loss and were likely to persist long-term (Magle et al. 2010). More broadly, then, our results suggest that barriers for some wildlife species are not barriers for all wildlife species, especially in an anthropogenic context.
These results suggest that the macaques of Bali are heavily influenced both by physical features of the Balinese environment and by human land use patterns, including provisioning, tourism, and urban development. While it is clear that human land use has had effects on macaque population structure, it is important to note that the effects have not been ubiquitous across the island. Large scale urban development, specifically Denpasar and the surrounding human communities, acts as a barrier to gene flow, while human communities in the core of the island act to homogenize population structure. The presence of large populations of macaques, which act as a tourism draw and thus provide an economic incentive to the surrounding human communities, benefit from routine, and often large-scale, provisioning and relative resource protection while remaining in the core of the island. Riverine corridors, rice agricultural land, and family gardens further facilitate dispersal within the island’s core by providing corridors for dispersal among these populations.
The combination of population-level analyses, such as pairwise FST comparisons and measurements of allelic richness and effective population sizes, and individual-level analyses, such as assignment tests, the determination of first generation migrants, Bayesian cluster analyses, and kriging, has more fully informed our understanding of the population structure of macaques than using either set of analyses independently. In this and other systems with significant landscape complexity, the inclusion of more individual-level analyses can capture the landscape genetics of populations. Doing so will allow us to understand how such a ubiquitous and widespread species exhibits such strong patterns of genetic structure on a relatively small geographic scale, and how these patterns can be used to develop our understanding of the relationship between population and social structure of macaque populations more widely.
References
Abernathy K, Peignot P, McCann C, Teffler P (2002) Solving the problem of illegal captive primates in Gabon. XIXth Congress of the International Primatological Society
Altmann J, Alberts SC (2003) Variability in reproductive success viewed from a life-history perspective in baboons. Am J Hum Biol 15:401–409
Avise JC, Bown BW, Lamb T, Meylan AB, Bermingham E (1992) Mitochondrial DNA evolution at a turtle’s pace: evidence for low genetic variability and reduced microevolutionary rate in the Testudines. Mol Biol Evol 9:457–473
Bercovitch FB (1991) Mate selection, consortship formation, and reproductive tactics in adult female savanna baboons. Primates 32:437–452
Blanchong JA, Sorin AB, Scribner KT (2013) Genetic diversity and population structure in urban white-tailed deer. J Wildl Manag 77:855
Bradley BJ, Doran-Sheehy DM, Lukas D, Boesch C, Vigilant L (2004) Dispersed male networks in western gorillas. Curr Biol 14:510–513
Brotcorne F, Wandia IN, Rompis ALT et al (2011) Recent demographic and behavioral data of Macaca fascicularis at Padangtegal, Bali, Indonesia. In: Gumert MD, Fuentes A, Jones-Engel L (eds) Monkeys on the edge: ecology and management of long-tailed macaques and their interface with humans. Cambridge University Press, Cambridge, pp 180–182
Byrne A, Paddy Sleeman D, O’Keeffe J, Davenport J (2012) The ecology of the European badger (Meles meles) in Ireland: a review. Biol Environ Proc Roy Irish Acad 112:105–132
Clark DA (1981) Foraging patterns of black rats across a desert-montane forest gradient in the Galapagos Islands. Biotropica 13:182–194
Cornuet JM, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144:2001–2014
Cushman SA, McKelvey KS, Hayden J, Schwartz MK (2006) Gene flow in complex landscapes: testing mutliple hypotheses with causal modeling. Am Nat 168(4):468–499
de Campos Telles MP, Coelho ASG, Chaves LJ, Diniz-Filho JAF, d’Ayala Valva F (2005) Genetic diversity and population sturcutre of Eugenia dysenterica DC. (“cagaiteira”-Myrtaceae) in Central Brazil: spatial analysis and implications for conservation and management. Conserv Genet 4:685–695
Dharmarajan G, Beasley JC, Fike JA, Rhodes OE (2009) Population genetic structure of raccoons (Procyon lotor) inhabiting a highly fragmented landscape. Can J Zool 87:814
Dixon JD, Oli MK, Wooten MC, Eason TH, McCown JW, Paetkau J (2006) Effectiveness of a regional corridor in connecting two Florida black bear populations. Conserv Biol 20:155–162
Eldridge MDB, King JM, Loupis AK, Spencer PBS, Taylor AC, Pope LC, Hall GP (1999) Unprecedented low levels of genetic variation and inbreeding depression in an island population of the black-footed rock-wallaby. Conserv Biol 13:531–541
Ellsttrand NC, Elam DR (1993) Population genetic consequences of small population size: implications for plant conservation. Annu Rev Ecol Syst 24:217–242
Estes-Zumpf WA, Rachlow JL, Waits LP, Warheit KI (2010) Dispersal, gene flow, and population genetic structure in the pygmy rabbit (Brachylagus idahoensis). J Mammal 91(1):208–219
Evans BJ, Supriatna J, Melnick DJ (2001) Hybridization and population genetics of two macaque species in Sulawesi, Indonesia. Evolution 55:1686–1702
Excoffier L, Laval G, Schneider S (2005) Arlequin (v 3.0): an integrated software package for population genetics data analysis. Evol Bioinform 1:47–50
Fa JE, Lindburg DG (1996) Evolution and ecology of macaque societies. Cambridge University Press, Cambridge
Fitch-Snyder HM (2002) History of captive conservation of tarsiers. Past, Present, and Future. Rutgers University Press, Tarsiers
Fooden J (1995) Systematic review of Southeast Asian long-tailed macaque, Macaca fascicularis (Raffles, 1821). Fieldiana Zool 81:1–205
Frankham R (1996) Relationship of genetic variation to population size in wildlife. Conserv Biol 10:1500–1508
Frankham R (1997) Do island populations have less genetic variation than mainland populations? Heredity 78:311–327
Fuentes A, Southern M, Suaryana KG (2005) Monkey forests and human landscapes: is extensive sympatry sustainable for Homo sapiens and Macaca fascicularis in Bali? In: Patterson J, Wallised J (eds) Commensalism and conflict: the primate-human interface. American Society of Primatology Publications, Norman, pp 168–195
Fuentes A, Rompis ALT, Arta Putra IGA, Watiniasih NL, Suartha IN, Soma IG, Wandia IN, Harya Putra IDK, Stephenson R, Selamat W (2011) Macaque behavior at the human-monkey interface: the activity and demography of semi-free ranging Macaca fascicularis at Padangtegal, Bali, Indonesia. In: Monkeys on the Edge, pp 159–182. Cambridge University Press, Cambridge
Glaubitz JC (2004) Convert: a user-friendly program to reformat diploid genotypic data for commonly used population genetic software packages. Mol Ecol Notes 4:309–310
Goudet J (1995) FSTAT: a computer program to calculate F-statistics. J Hered 86:485–486
Grivet D, Sork VL, Westfall RD, Davis FW (2008) Conserving the evolutionary potential of valley oak (Quercus lobata Née) in California. Mol Ecol 17:139–156
Guillot G, Santos F, Estoup A (2008) Analysing georeferenced population genetics data with Geneland: a new algorithm to deal with null alleles and a friendly graphical user interface. Bioinformatics 24(11):1406–1407
Gumert MD (2011) The common monkey of Southeast Asia: long-tailed macaque populations, ethnophoresy, and their occurrence in human environments. In: Gumert MD, Fuentes A, Jones-Engel L (eds) Monkeys on the edge: ecology and management of long-tailed macaques and their interface with humans. Cambridge University Press, Cambridge
Hadfield RM, Pullen JG, Davies KF, Wolfensohn SE, Kemnitz JW, Weeks DE, Bennett ST, Kennedy SH (2001) Toward developing a genome-wide microsatellite marker set for linkage analysis in the rhesus macaque (Macaca mulatta): identification of 76 polymorphic markers. Am J Primatol 54:223–231
Hitchings SP, Beebee TJC (1997) Genetic substructuring as a result of barriers to gene flow in urban Rana temporaria (common frog) populations: implications for biodiversity conservation. Heredity 79:117–127
Hoffman JI, Amos W (2005) Microsatellite genotyping errors: detection approaches, common sources and consequences for paternal exclusion. Mol Ecol 14:599–612
Holderegger R, Buehler D, Gugerli F, Manel S (2010) Landscape genetics of plants. Trends Plant Sci 15:675–682
James RA, Leberg PL, Quattro JM, Vrijenhoek RC (1997) Genetic diversity in black howler monkeys (Alouatta pigra) from Belize. Am J Phys Anthropol 102:329–336
Jones-Engel L, Engel GA, Schillaci MA, Froelich J, Paputungan U, Kyes RC (2004) Prevalence of enteric parasites in pet macaques in Sulawesi, Indonesia. Am J Primatol 62(2):71–82
Jones-Engel L, Engel GA, Schillaci MA, Lee B, Heidrick J, Chalise M, Kyes RC (2006) Considering human-primate transmission of measles virus through the prism of risk analysis. Am J Primatol 68:868–879
Lacy RC (1997) Importance of genetic variation to the viability of mammalian populations. J Mammal 78:320–335
Landguth EL, Cushman SA, Schwartz MK, McKelvey KS, Murphy M, Luikart G (2010) Quantifying the lag time to detect barriers in landscape genetics. Mol Ecol 19:4179–4191
Lane KE, Lute M, Arta Putra IGA, Rompis A, Wandia IN, Hollocher H, Fuentes A (2010) Pests, Pestilence, and People: The Long-tailed macaque and its role in the cultural complexities of Bali. Indonesian Primates, Springer, Berlin
Lane KE, Holley C, Hollocher H, Fuentes A (2011) The anthropogenic environment lessens the intensity and prevalence of gastrointestinal parasites in Balinese long-tailed macaques (Macaca fascicularis). Primates 52:117–128
Lane-deGraaf KE, Hollocher H, Fuentes A (2013a) Balinese perceptions of pathogen exposure in human communities surrounding long-tailed macaque populations. Am J Primatol. doi:10.1002/ajp.22218
Lane-deGraaf KE, Kennedy RC, Madey GR, Fuentes A, Hollocher H (2013b) Exploring the use of agent-based models in understanding patterns of pathogen transmission. BMC Ecol 13:36. doi:10.1186/1472-6785-13-35
Lansing JS (1991) Priests and programmers: technologies of power in the engineered landscape of Bali. Princeton University Press, Princeton
Liu Z, Ren B, Wu R, Zhao L, Hao Y, Wang B, Wei FW, Long Y, Li M (2009) The effect of landscape features on population genetic structure in Yunnan snub-nosed monkeys (Rhinopithecus bieti) implies an anthropogenic genetic discontinuity. Mol Ecol 18:3831–3846
Luckett J, Danforth E, Linsenbardt K, Pruetz J (2004) Planted trees as corridors for primates at El Zota Biological Field Station, Cost Rica. Neotrop Primates 12:143–146
MacDougall-Shackleton EA, Clinchy M, Zanette L, Neff BD (2011) Songbird genetic diversity is lower in anthropogenically versus naturally fragmented landscapes. Conserv Genet 12:1195–1203
Magle SB, Ruell EW, Antolin MF, Crooks KR (2010) Population genetic structure of black-tailed prairie dogs, a highly interactive species, in fragmented urban habitat. J Mammol 91(2):326–335
Manel S, Schwartz MK, Luikart G, Taberlet P (2003) Landscape genetics: combining landscape ecology and population genetics. TREE 18(4):189–197
Melnick DJ, Hoelzer GA (1992) Differences in male and female macaque dispersal lead to contrasting distributions of nuclear and mitochondrial DNA variation. Int J Primatol 53:379–393
Munshi-South J (2012) Urban landscape genetics: canopy cover predicts gene flow between white-footed mouse (Peromyscus leucopus) populations in New York City. Mol Ecol 21(6):1360–1378
Nasi R, Koponen P, Poulsen JG, Buitenzorgy M, Rusmantoro W (2008) Impact of landscape and corridor design on primates in a large-scale industrial tropical plantation landscape. Biodivers Conserv 17:1105–1126
Paetkau D (2003) An empirical exploration of data quality in DNA-based population inventories. Mol Ecol 12:1375–1387
Paetkau D, Waits LP, Clarkson PL, Craighead L, Vyse E, Ward R, Storbeck C (1998) Variation in genetic diversity across the range of North American brown bears. Conserv Biol 12:418–429
Paetkau D, Slade R, Burden M, Estoup A (2004) Genetic assignment methods for the direct, real-time estimation of migration rate: a simulation-based exploration of accuracy and power. Mol Ecol 13:55–65
Patterson N, Price AL, Reich D (2006) Population structure and eigen analysis. PLoS Genet 2:2074–2093
Pebesma EJ (2004) Multivariable geostatistics in S: the gstat package. Comput Geosci 30(7):683–691
Penedo MCT, Bontrop RE, Heijmans CMC, Otting N, Noort R, Rouweler AJM, de Groot N, de Groot NG, Ward T, Doxiadis GGM (2005) Microsatellite typing of the rhesus macaque MHC region. Immunogenetics 57:198–2009
Piertney SB, MacColl ADC, Bacon PJ, Dallas JF (1998) Local genetic structure in red grouse (Lagopus lagopus scotius): evidence from microsatellite DNA markers. Mol Ecol 7:1645–1495
Piry S, Alapetite A, Cornuet J-M, Paetkau D, Baudoin L, Estoup A (2004) GENECLASS2: a software for genetic assignment and first-generation migrant detection. J Hered 95:536–539
Pompanon F, Bonin A, Bellemain E, Taberlet P (2005) Genotyping errors: causes, consequences and solutions. Nature Reveiws Genetics 6:1–13
Proctor MF, McLellan BN, Strobeck C, Barclay RMR (2005) Genetic analysis reveals demographic fragmentation of grizzly bears yielding vulnerably small populations. Proc R Soc B Ser 272:2409–2416
Radaspiel U, Rakotondravony R, Chikhi L (2008) Natural and anthropogenic determinants of genetic structure in the largest remaining population of the endangered golden-brown mouse lemur, Microcebus ravelobensis. Am J Primatol 70:860–870
Rannala B, Mountain JL (1997) Detecting immigration by using multilocus genotypes. Proc Natl Acad Sci 94:9197–9201
Raymond M, Rousset F (2007) GENEPOP (v. 1.2): population genetics software for exact tests and ecumenicism. Heredity 86:248–249
Reed DH, Lowe EH, Briscoe DA, Frankham R (2003) Inbreeding and extinction: effects of rate of inbreeding. Conserv Genet 4:405–410
Rousset F (1997) Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145:1219–1228
Rousset F (2008) Genepop’007: a complete re-implementation of the genepop software for Windows and Linux. Mol Ecol Resour 8:103–106
Ruell E, Riley S, Douglas M, Antolin M, Pollinger J, Tracey J, Lyren L, Boydston E, Fisher R, Crooks K (2012) Urban habitat fragmentation and genetic population structure of bobcats in coastal southern California. Am Midl Nat 168:265–280
Ryman N, Palm S, Andre C, Carvalho GR, Dahlgren TG, Jorde PE, Laikre L, Larsson LC, Palme A, Ruzzante DE (2006) Power for detecting genetic divergence: differences between statistical methods and marker loci. Mol Ecol 15:231–2045
Segelbacher G, Manel S, Tomiuk J (2008) Temporal and spatial analyses disclose consequences of habitat fragmentation on the genetic diversity in capercaillie (Tetrao urogallus). Mol Ecol 17:2356–2367
Seuer C, King AJ, Conradt L, Kerth G, Lusseau D, Mettke-Hofmann C, Schaffner CM, Williams L, Zinner D, Aureli F (2010) Collective decision-making and fission-fusion dynamics: a conceptual framework. Oikos 120(11):1608–1617
Shepherd CR (2010) Illegal primate trade in Indonesia exemplified by surveys carried out over a decade in North Sumatra. Endangered Species Res 11:201–205
Southern MW (2002) An assessment of potential habitat corridors and landscape ecology for long-tailed macaques (Macaca fascicularis) on Bali, Indonesia. Dissertation, Central Washington University
Storfer A, Murphy MA, Evans JS, Goldberg CS, Robinson S, Spear SF, Dezzani R, Delmelle E, Vierling L, Waits LP (2007) Putting the ‘landscape’ in landscape geentics. Heredity 98:128–142
Taberlet P, Griffin S, Goossens B, Questiau S, Manceau V, Escaravage N, Waits LP, Bouvet J (1996) Reliable genotyping of samples with very low DNA quantities using PCR. Nucleic Acids Res 24(16):3189–3194
Thierry B (2007) The macaques. In: Campbell C, Fuentes A, MacKinnon K, Bearder S, Stumpf R (eds) Primates in Perspective. Oxford University Press, Oxford
Thioulouse J, Dray S (2007) Interactive multivariate data analysis in R with the ‘ade4’ and ‘ade4TkGUI’ packages. J Stat Softw 22:1–14
Tigas LA, Van Vuren DH, Sauvajot RM (2002) Behavioral responses of bobcats and coyotes to habitat fragmentation and corridors in an urban environment. Biol Conserv 108:299–306
Van Oosterhour C, Hutchinson WF, Wills DPM, Shipley P (2004) Microchecker: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538
Waples RS, Do C (1997) LDNE: a program for estimating effective population size from data on linkage disequilibrium. Mol Ecol Res 8:753–756
Waples RS, Do C (2008) LDNe: a program for estimating effective population size from data on linkage disequilibrium. Mol Ecol Res 8(4):735–756
Weir BS (1979) Inferences about linkage disequilibrium. Biometrics 35:235–254
Wheatley BP (1999) The sacred monkeys of Bali. Wave Island Press, Wave
Acknowledgments
The authors would like to thank Arta Putra and I Nengah Wandia for assistance in the field and Malachi McHenry and Michelle Lute for their helpful comments. This work was funded by the Leakey Foundation and the National Science Foundation (BSC 0629787).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Lane-deGraaf, K.E., Fuentes, A. & Hollocher, H. Landscape genetics reveal fine-scale boundaries in island populations of Indonesian long-tailed macaques. Landscape Ecol 29, 1505–1519 (2014). https://doi.org/10.1007/s10980-014-0069-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10980-014-0069-0