
Abstract
Habitat fragmentation may strongly impact population genetic structure and reduce the genetic diversity and viability of small and isolated populations. The white-headed langur (Trachypithecus leucocephalus) is a critically endangered primate species living in a highly fragmented and human-modified habitat in southern China. We examined the population genetic structure and genetic diversity of the species and investigated the environmental and anthropogenic factors that may have shaped its population structure. We used 214 unique multi-locus genotypes from 41 social groups across the main distribution area of T. leucocephalus, and found strong genetic structure and significant genetic differentiation among local populations. Our landscape genetic analyses using a causal modelling framework suggest that a large habitat gap and geographical distance represent the primary landscape elements shaping genetic structure, yet high levels of genetic differentiation also exist between patches separated by a small habitat gap or road. This is the first comprehensive study that has evaluated the population genetic structure and diversity of T. leucocephalus using nuclear markers. Our results indicate strong negative impacts of anthropogenic land modifications and habitat fragmentation on primate genetic connectivity between forest patches. Our analyses suggest that two management units of the species could be defined, and indicate that habitat continuity should be enforced and restored to reduce genetic isolation and enhance population viability.
Similar content being viewed by others
Introduction
It is widely recognised that continued population isolation caused by natural or anthropogenic habitat fragmentation can lead to modified population genetic structure, reduced genetic variation within subpopulations and a decline of the total effective population size (Keyghobadi, 2007; Radespiel and Bruford, 2014). These consequences are particularly detrimental for species with small populations, as they are more vulnerable to inbreeding depression, which can lower individual fitness and viability and potentially result in local extinctions (Keller and Waller, 2002; Frankham, 2005). Therefore, functional connectivity among habitat patches that ensures gene flow is essential for long-term population persistence. Identifying the environmental factors that impede or facilitate dispersal across landscapes has thus become a priority in conservation research of endangered species with fragmented distribution patterns (Lindenmayer et al., 2008; Luque et al., 2012). Early detection of gene flow discontinuity and genetic differentiation can reveal landscape–organism interactions, identify key barriers to dispersal, determine conservation priorities and facilitate the design of appropriate strategies to counteract genetic isolation (Segelbacher et al., 2010; Leslie, 2013).
Primates are underrepresented in landscape genetic investigations, in part due to the fact that most of these species exhibit high mobility and thus are expected to be less restricted by landscape barriers to dispersal (Quéméré et al., 2010; Salmona et al., 2015). In addition, the genetic consequences of dispersal barriers may not manifest until long after the establishment of the associated landscape features owing to long generation times in most primate species, which prohibits timely evaluation of the impact of recent landscape changes. However, a number of studies have shown strong impacts of habitat discontinuity caused by human land modification on the genetic structure of primate populations (Grativol et al., 2001; Oklander et al., 2010; Quéméré et al., 2010; Blair and Melnick, 2012), indicating significant influences of landscape anthropogenic factors on primate genetic patterns.
Traditional dispersal rate measurement in a fragmented habitat by direct observation, mark-recapture or radio-tracking methods is often difficult, costly and inaccurate (Koenig et al., 1996). Molecular genetic analysis, coupled with non-invasive sampling, has provided an alternative approach for evaluation of effective gene flow among populations (Griesser et al., 2014). This approach is particularly favourable for the study of migration patterns in endangered primates living in challenging habitats. For instance, genetic analyses have shown the impacts of habitat fragmentation on dispersal behaviour and population genetic structure in the black-and-white howler (Alouatta caraya; Oklander et al., 2010) and black-handed spider monkey (Adeles geoffroyi; Hagell et al., 2013). The recent development of landscape genetic statistical tools allowed further assessment of the influence of environmental features on genetic patterns in endangered primates (Quéméré et al., 2010; Blair et al., 2012). In addition, evaluation of genetic diversity, population structure and demographic history is increasingly incorporated into conservation studies, and has provided essential knowledge for assessing long-term population viability, defining appropriate conservation units and designing management strategies (Bastos et al., 2010; Chaves et al., 2011).
Asian colobine monkeys face a severe threat of extinction as a result of habitat destruction and fragmentation and human exploitation. Among the 55 currently recognised species of Asian colobines, 41 (75%) are classified as vulnerable, endangered or critically endangered on the International Union for Conservation of Nature (IUCN) Red List (Roos et al., 2014). Asian colobines are also among the most disproportionally understudied groups of primates concerning their population genetic data. Except for a few species (Rhinopithecus roxellana, Pan et al., 2009; R. bieti, Liu et al., 2009; R. brelichi, Yang et al., 2012 and Kolleck et al., 2013; Trachypithecus delacouri, Ebenau et al., 2011), very little information is available on genetic diversity and population structure for most Asian colobines. Hence, there is an urgent need to understand the population genetic characteristics of Asian colobines to support science-based conservation design. The white-headed langur (Trachypithecus leucocephalus) is an Asian colobine endemic to a narrow range of karst forests in Guangxi Province, South China. With ~1000 individuals remaining in the wild, T. leucocephalus is classified as critically endangered by the IUCN (IUCN, 2016). Although the species was first described in the 1950s (Tan, 1957), it remains one of the least known primates; its historical range and population status before the 1980s is unclear. Population surveys of T. leucocephalus conducted during the last 30 years indicated a contraction of nearly 80% in the total range of the species (from 360 to 80 km2), extirpations of several local populations and population size reduction of ~60% in one of the largest populations, Fusui (FS), from 1987 to 1997 (Hu et al., 1998; Ran, 2003). The main threats to the species included habitat loss and deterioration due to land exploitation for crop cultivation, firewood cutting, limestone mining and poaching (Burton et al., 1995; Huang et al., 2002). Poaching has been largely eliminated in recent years, but habitat destruction and fragmentation have not improved and remain imminent threats to the survival of the species. There are three main extant populations: ~550 langurs in FS, 300 langurs in Chongzuo (CZ) and 80 langurs in Nonggang (NG; Chongzuo Nature Reserve, 2010). Habitat conditions vary considerably across the range of T. leucocephalus. In stark contrast to an ideal continuous habitat in NG with primitive vegetation and minimal human accessibility, habitats in FS and CZ are highly disrupted by human land use; the limestone hills inhabited by the langur are separated into patches by flat land that has been cleared and exploited as farmland and residences (Li, 1993; Huang et al., 2002). Native vegetation cover generally remains only on steep limestone hills unsuitable for crop cultivation. These habitat patches vary in size, distance from other patches and degree of functional connectivity. It is unclear how habitat fragmentation and recent population decline have influenced individual dispersal and genetic structure within and among the habitat patches. Thus, T. leucocephalus provides an ideal system for studying the scale and extent of the effects of anthropogenic land modifications on population genetic patterns of Asian colobine monkeys, which suffer from a persistent threat of human disturbance.
Previous population genetic study of T. leucocephalus indicates very low genetic diversity and significant divergence between the populations (ΦST=0.375, P<0.001; Wang et al., 2015). However, these results were based on a 350 bp fragment of mitochondrial DNA (mtDNA) hypervariable region I (HVRI) from only 77 individuals. To gain a comprehensive understanding of the genetic diversity and population structure of T. leucocephalus at a finer scale and identify contributing habitat features, we genotyped 15 microsatellite loci, mtDNA control region sequences and sex-specific markers of faecal samples collected non-invasively from social groups across its main distribution areas. To assess population genetic structure and genetic differentiation among inferred subpopulations, we used individual-based, spatially explicit and nonspatial Bayesian clustering algorithms, neighbour-net tree analysis and F-statistics. To identify landscape factors that influence gene flow, we applied a causal modelling approach to test whether geographical distance and anthropogenic habitat fragmentation (large and small habitat gaps, and roads) are correlated with genetic distance among individuals of T. leucocephalus. The causal modelling framework can evaluate the effect of each landscape factor independent of other factors, hence disentangling the influences of multiple factors on gene flow. To gain an understanding of genetic diversity of the species, we evaluated the population genetic diversity parameters of the total population and inferred genetic subpopulations, and compared these with genetic diversity data of other endangered primates. Finally we made recommendations for conservation of population viability on the basis of our results.
Materials and methods
Study area and faecal sample collection
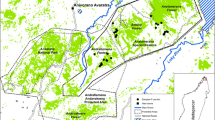
We conducted faecal sample collection in the Chongzuo White-Headed Langur National Reserve in Guangxi Province, China. The reserve comprises an area of 256 km2 and includes the distribution area of two main populations, FS and CZ, which are inhabited by more than 90% of the extant T. leucocephalus population. Nearly all flatlands in the region are exploited as agricultural land, villages and road networks. The remaining natural vegetation, composed of evergreen and semi-deciduous forests, only covers the steep karst limestone hills of the reserve. FS and CZ are further divided into several habitat patches, the largest of which are FS-Jiuchongshan (FS-JCS), FS-Buzun (FS-BZ), CZ-north patch (CZ-N) and CZ-south patch (CZ-S) (Figure 1). FS and CZ are separated by 40–50 km of flat landscape of farmland, roads and human settlements; FS-JCS and FS-BZ are separated by ~3 km of farmland and a village; CZ-N and CZ-S are divided by a national road (Figure 1).

Satellite map (Landsat 7 EMT+, Path126, Row 044/045, RGB=742, data source: http://landsat.usgs.gov/) of the sampling areas in Guangxi Province, China. The inset map shows the location of the study areas (red dot) in China. Natural forests are shown in dark green, agricultural lands in pink and water bodies in navy. Dots represent the sampled sleeping sites of social groups included in the study. Surveyed habitat patches are indicated: FS-JCS, Fusui-Jiuchongshan; FS-BZ, Fusui-Buzun; CZ-N, Chongzuo-North; CZ-S, Chongzuo-South.
T. leucocephalus is highly folivorous, inhabits karst limestone hills and occurs mainly in social groups of 3–30 individuals (mean=11.7) (Jin, 2008; Jin et al., 2009). A total of 403 faecal samples from 41 social groups were collected non-invasively from the limestone caves that langurs used as sleeping sites across the FS (N=245) and CZ (N=158) areas in dry winter months (mostly in January) between 2012 and 2014. The sampled social groups were relatively evenly distributed in the sampling areas (Figure 1). T. leucocephalus is the only colobine species occurring at the sampling sites. The only other primate in the reserve is the macaque (Macaca mulatta), but it generally does not co-occur with T. leucocephalus; moreover, the faeces of the two species are readily distinguishable. Caution was taken to select fresh (<3 days old) and separate faecal pellets to ensure DNA quality and avoid cross-individual contamination.
Faecal samples were submerged in 95–100% ethanol and stored at −20 °C until DNA was extracted.
DNA extraction, sex identification, mtDNA sequencing, microsatellite genotyping and individual identification
Faecal DNA extraction, purification and concentration determination were described in detail previously (Wang et al., 2015). Molecular sex identification was carried out by PCR amplification of the sex chromosome-linked DEAD-box gene that shows sexual dimorphism in size in a range of primate species (Villesen and Fredsted, 2006), using conditions described previously (Wang et al., 2015). We determined the sex of the originating animal for 367 (91%) of the collected samples. Samples that failed to show an unambiguous result in molecular sex identification were considered as sex unknown. Sex information was accounted for during individual identification. Samples with identified and unknown sex were all included in the analyses.
We sequenced a fragment of 350 bp of the mtDNA hypervariable region I (HVRI) of the 403 collected samples that contained all known variable sites of the entire control region in our study population (Wang et al., 2015). Primer sequences, PCR conditions, sequencing information and procedures designed to exclude the possibility of contamination by nuclear insertion of mtDNA fragments (numts; Thalmann et al., 2004) followed established protocols (Wang et al., 2015). The mtDNA haplotype information was accounted for in individual identification, but not used in other data analyses of this study.
All faecal samples were genotyped at 15 variable microsatellite loci (WT-58, WT-97, WT-111, WT-168, WT-173, WT-181, WT-223, WHL-60, WHL-121, WHL-122, WHL-138, WHL-206, WHL-220, WHL-287 and WHL-293) with fluorescent-labelled primers by following the protocols specifically developed for non-invasive samples from T. leucocephalus (Wang et al., 2016). Because DNA extracted from faecal samples is usually degraded and low in quantity, allelic dropout (ADO), false alleles (FA) and null alleles are likely to be common in PCR using such templates and lead to genotyping errors (Taberlet et al., 1999). We followed the multiple-tube approach suggested by Taberlet et al. (1996) and modified by Frantz et al. (2003), and determined the consensus genotype for each locus after 2–7 repetitions using independently extracted DNA. A heterozygous genotype was determined when each allele was amplified in at least two independent PCRs, and a homozygous genotype was determined based on at least three independent homozygote PCR results. We discarded samples that failed to produce a consensus genotype following the above criteria after seven PCR repetitions. We estimated that the rate of ADO varied from 2.4 to 17.3%, FA ranged from 0 to 5.1%, and genotyping accuracy was in the range of 90–99% across the 15 assessed loci using 86 samples randomly drawn from the collected faecal samples and our genotyping procedures (Wang et al., 2016).
MICRO-CHECKER v 2.2.3 (van Oosterhout et al., 2004) was used to check for possible null alleles, ADO and scoring errors due to stuttering. The significance of deviations from Hardy–Weinberg equilibrium (HWE) and the fixation index (FIS) for each locus, as well as linkage disequilibrium (LD) among loci, were assessed with GENEPOP v 4.0 (Rousset, 2008), followed by the sequential Bonferroni test (Rice, 1989) to correct for multiple comparisons by bootstrapping with 1000 replicates across loci to generate 95% confidence intervals.
The probabilities of identity for unrelated individuals (PID) and siblings (PID-sib) across loci were calculated using GENALEX v 6.5 (Peakall and Smouse, 2006, 2012). Samples showing identical genotypes at all microsatellite loci, mtDNA haplotype and sex were considered to be samples from a single individual. Samples with identical genotypes at all loci, but with differing sex or mtDNA haplotypes, were regarded as originating from distinct individuals. Samples that had identical genotypes at all but one locus were reamplified to ensure unique genotypes.
Population genetic structure inferred from microsatellite data
For microsatellite data, population differentiation was evaluated first using a classic Bayesian clustering approach implemented in STRUCTURE v 2.3.4 (Pritchard et al., 2000) without a priori designation of sample populations. This approach assigns individuals to putative populations such that HWE is maximised within each population. We used the admixture model and correlated allele frequencies within populations (Falush et al., 2003). Simulations were run using 1 000 000 Markov chain Monte Carlo (MCMC) iterations, a burn-in of 100 000 and 10 independent runs for each value of K (assumed number of populations) to check for consistency in the results. We set the value of K from 1 to 8, the maximum number being the number of putative populations (FS-JCS, FS-BZ, CZ-N and CZ-S) plus 4 (Evanno et al., 2005). The most likely value of K was determined by examining the log probability of the data [ln Pr (X|K)] and ΔK (Evanno et al., 2005) over multiple runs using STRUCTURE HARVESTER (Earl and vonHoldt, 2012). The run with the highest posterior probability of the 10 STRUCTURE runs was exported.
Next, a neighbour-net tree was constructed based on Nei’s standard genetic distance (DS; Nei, 1972) between individuals. The neighbour-net algorithm constructs highly resolved networks and represent these in a splits graph (Bryant and Moulton, 2004), hence providing a visualisation of genetic variation among individuals. The neighbour-net analysis was conducted using POPULATION v 1.2.32 (http://bioinformatics.org/populations/) and visualised in SPLITSTREE v 4.13.1 (Huson, 1998).
Finally, the spatially explicit Bayesian clustering method implemented in the R package GENELAND v 4.0.4 (Guillot et al., 2005b) was used to detect genetic discontinuities between populations or individuals using microsatellite data. This method assigns individuals to putative populations by maximising HWE and gametic phase equilibrium within genetic clusters, and uses the spatial coordinates as a prior in the model. We used the uncorrelated allele frequency model (Dirichlet-model) for microsatellites, as the correlated model can be more prone to algorithm instabilities and sensitive to violation of model assumptions (Wei et al., 2015). The presence of null alleles was allowed (Guillot et al., 2008). We first ran five independent MCMC simulations with 1 000 000 iterations for the number of clusters and K varying from 1 to 10, of which every 100th iteration was saved. To assign the individuals to each of the genetic clusters, the model was run 20 more times, treating the number of clusters as known, based on the value of K inferred from the previous runs. Correlated and uncorrelated allele frequency models were each used 10 times in the following runs. The posterior probability of population membership was calculated using a burn-in period length of 1000 iterations (Guillot et al., 2005a, 2009).
Population genetic diversity, genetic differentiation and gene flow inferred from microsatellite data
To assess genetic diversity of three genetic subpopulations inferred by GENELAND, we used CERVUS v 3.0.6 (Kalinowski et al., 2007) to calculate the observed (A) number of alleles, polymorphism information content (PIC), and observed (HO) and expected (HE) heterozygosities at each microsatellite locus and for each population. Allelic richness (Ar) was calculated using HP-RARE (Kalinowski, 2005). The effective number of alleles (Na) was calculated using POPGENE v 1.32 (Yeh et al., 1997).
To estimate genetic differences among inferred genetic subpopulations, we calculated pairwise FST (Weir and Cockerham, 1984) and RST (Slatkin, 1995) statistics between the populations using microsatellite data and ARLEQUIN v 3.5.1.2 (Excoffier and Lischer, 2010). The significance of pairwise comparisons was tested with 10 000 permutations.
We estimated the effective number of migrants per generation (Nem, where Ne is the effective population size and m is the migration rate) between inferred genetic subpopulations using a maximum likelihood approach in MIGRATE-N v 3.6.4 (Beerli and Felsenstein, 2001; Beerli, 2006). We used FST-based estimates as the starting parameter values. The sampling increment was set to 20, and the burn-in was set to 10 000. We performed three independent runs, each of 10 short chains with 10 000 steps and three long chains with 1 000 000 steps. All other settings were left at default values. The results of the three independent long chain search runs were checked for convergence and combined to produce the final results.
Causal modelling analyses of landscape factors
On the basis of field observations, we proposed four potential driving factors of genetic structure in T. leucocephalus: geographical distance (factor D), the large habitat gap between FS and CZ (factor L), the small habitat gap between FS-JCS and FS-BZ (factor S) and the national roads between CZ-N and CZ-S and between FS and CZ (factor R). We did not test the effects of other environmental factors such as forest cover, habitat connectivity within patches and levels of human disturbance, because these landscape parameters are similar and relatively consistent among patches. A matrix of genetic distance (G) among individuals was estimated by Rousset’s ar (Rousset, 2000) and calculated using SPAGEDI v 1.5 (Hardy and Vekemans, 2002). We tested one isolation-by-distance hypothesis modelled with genetic distances between pairs of individuals predicted to increase linearly with Euclidian distances between their sampling locations. In addition, we tested three isolation-by-barrier hypotheses (isolation-by-large habitat gap, isolation-by-small habitat gap and isolation-by-road) with categorical model matrices in which 0s and 1s represented pairs of individuals on the same or opposite side of the barrier, respectively.
As simple correlational analyses between genetic distance and individual environmental variables can produce spurious correlations (Cushman and Landguth, 2010b), we applied a causal modelling approach, in which alternative hypotheses are compared based on relative statistical support to identify landscape factors influencing gene flow within and across habitat patches. Causal modelling provides estimates of statistical significance of a series of partial Mantel tests that can be used to find explanations that are most consistent with the expectations of the causal model (Cushman et al., 2006). This approach has been shown to be highly effective at reducing the risk of spurious correlations between landscape factors and genetic patterns and identifying the correct driving process (Cushman and Landguth, 2010b). We calculated four simple Mantel tests and 12 partial Mantel tests to evaluate the support for 15 organisational hypotheses representing all possible combinations of causality by the four landscape factors described above (D, L, S and R). We first computed the correlation coefficient (r) and associated P-value for each simple test between the genetic distance matrix and matrices corresponding to each of the four landscape factors. We then performed partial Mantel tests on each factor while controlling for one other factor, and compared the significant and non-significant results to the expectations under the 15 organisational hypotheses to identify those that were best supported. The relative support for the hypotheses was measured by the support rate of each hypothesis, which was calculated as the ratio of the number of tests consistent with the expectations to the total number of relevant tests. To obtain the confidence interval for each hypothesis, we randomly resampled 75% of the 214 individuals without replacement for 1000 iterations (Quéméré et al., 2010). After identifying factor L as a major landscape feature that shaped genetic variation, we further analysed the correlations between genetic distance (ar) and the geographical distances between individuals within FS and CZ by simple Mantel tests.
All Mantel tests were conducted in ARLEQUIN. Statistical significance was calculated by 10 000 iterations at a 95% confidence interval (CI).
Results
Microsatellite genotyping and individual identification
Of 403 field-collected faecal samples (FS-JCS: 216; FS-BZ: 29; CZ-N: 133; CZ-S: 25), 20 were discarded due to their low (<50%) PCR success rate or missing data at >5 loci. The remaining 383 (95%) samples yielded genotype data at 10–15 microsatellite loci and were included in the subsequent analyses. The probability of identity by chance based on the 15 loci was computed as cumulative PID (5 × 10−10) and PID-Sib (5.9 × 10−5), suggesting that the panel of loci had high power to distinguish individuals in the population. We identified 187 unique genotypes based on the microsatellite data, and distinguished 27 additional individuals based on sex (N=25) or mtDNA sequences (N=2), resulting in a total of 214 unique individuals (FS-JCS: 119; FS-BZ: 12; CZ-N: 67; CZ-S: 16). All unique individuals with identical microsatellite genotype, but different sexes or haplotypes, were found in the same social group. The number of times that each individual was sampled varied from 1 to 10 (mean±s.d.: 1.8±1.3). The reoccurring individuals were always from the same social group, substantiating the accuracy of individual identification in our genetic analysis.
Population genetic structure inferred from microsatellite loci
For microsatellite data, we first conducted STRUCTURE analyses, which showed consistent results over repeated runs for each tested K-value (1–8). The rate of change (ΔK) reached a maximum value when K=2, indicating the likely presence of two genetic clusters (Figure 2). The genetic partitioning of the FS and CZ samples explicitly corresponded with their geographical origins, with high (all q>0.84; mean=0.99) population assignment probability for all individuals (Figure 2). No further divisions of genetic clusters were detected when K was increased to 5 (Supplementary Figure S1; Supplementary Information). Excluding identical microsatellite genotypes had no effect on genetic clustering of the FS and CZ samples (data not shown). To exclude interference caused by samples from other genetic groups in the analysis of subpopulation structure, we also ran separate STRUCTURE analyses on the FS and CZ samples (that is, a hierarchical STRUCTURE analysis; Evanno et al., 2005), but failed to observe a clear subdivision in either cluster (data not shown). The partitioning of samples into two genetic clusters that were strongly correlated with their geographical populations was also confirmed by a neighbour-net tree based on Nei’s standard genetic distance of the microsatellite loci between individuals (Figure 3).

Results of STRUCTURE analysis based on microsatellite data. (a) Estimation of the number of genetic clusters (K) from 50 independent runs for K=1–8. The left panel shows the mean and s.d. of ln probability of K, and the right panel shows the rate of change (ΔK). K=2 is shown as the best fit of the data. (b) The two genetic clusters were based on genotypes at 15 microsatellite loci from 214 unique T. leucocephalus individuals. The colours indicate the proportion of each individual assigned to each of the inferred clusters. The assigned genetic clusters correspond with the two main distribution areas, Fusui and Chongzuo. The sampling areas of the individuals are indicated at the bottom. CZ-N, Chongzuo-North; CZ-S, Chongzuo-South; FS-BZ, Fusui-Buzun; FS-JCS, Fusui-Jiuchongshan.

Neighbour-net tree of 214T. leucocephalus individuals constructed based on 15 microsatellite loci. The colours of the samples indicate their geographical origin. FS-JCS, Fusui-Jiuchongshan; FS-BZ, Fusui-Buzun; CZ-N, Chongzuo-North; CZ-S, Chongzuo-South.
Assuming uncorrelated allelic frequencies between sampling sites, GENELAND inferred the presence of three genetic groups based on the highest average posterior probability, with clear spatial discontinuity between the CZ, FS-JCS and FS-BZ patches and individuals from each of these sampling sites forming a group (Figure 4). No division was detected within the CZ samples. The genetically distinct groups assigned by the GENELAND analyses approximately corresponded to those inferred by STRUCTURE and by neighbour-net tree clustering, with the exception of the FS cluster, which was further divided into the FS-JCS and FS-BZ groups by GENELAND. FS-BZ has the smallest population size of the four analysed patches. The twelve sampled individuals from the FS-BZ patch were found to show assignment probabilities similar to those from the FS-JCS patch (using STRUCTURE, Figure 2); these FS-BZ individuals were considered together with the FS-JCS individuals in the neighbour-net tree analysis (Figure 3). Taken together, the individual-based Bayesian statistical analyses and neighbour-net tree analysis of microsatellite data indicate that the T. leucocephalus population is genetically structured into two or three subpopulations.

Posterior probabilities of spatial population membership for 214T. leucocephalus individuals inferred by GENELAND based on 15 microsatellite loci. The posterior probability distribution suggests K=3 as the best fit for the data. The contour lines in the maps indicate the spatial positions of genetic discontinuities. Lighter shading indicates a higher probability of belonging to the genetic subpopulation. FS-JCS, Fusui-Jiuchongshan; FS-BZ, Fusui-Buzun; CZ, Chongzuo.
Genetic diversity, genetic differentiation and gene flow analyses of microsatellite loci
The 15 microsatellite loci had a number of alleles ranging from 2 to 8 per locus across all samples. For the inferred genetic subpopulations (FS-JCS, FS-BZ and CZ), mean allelic richness across the loci was low (2.5–3.6). The mean observed heterozygosity and expected heterozygosity of the three subpopulations were in the range of 0.51–0.53 and 0.43–0.53, respectively (Table 1). The private allele frequency of the CZ population was 0.83, whereas that of the FS population was 1.04. HWE tests showed significant deviation from HWE for locus WHL-121 in the CZ population, WT-111 in the FS-JCS population and WT-97 in the FS-BZ population, whereas no significant deviation from HWE was detected in any subpopulation across all loci. The MICROCHECKER results showed a high frequency of null alleles for two loci: WHL-206 in CZ (0.094) and WHL-293 in FS-JCS (0.101). No significant linkage disequilibrium between loci was detected in any subpopulation, suggesting no physical linkage between the loci. Overall inbreeding coefficient (FIS) values were negative, but not significantly different from zero in each genetic subpopulation (Table 1), suggesting no apparent signature of inbreeding.
We assessed genetic differentiation among inferred subpopulations by pairwise FST using the microsatellite data. Statistical tests showed significant pairwise FST among CZ, FS-JCS and FS-BZ samples (Table 2). Values of pairwise FST ranged from 0.104 to 0.223, suggesting strong genetic divergence and low gene flow between the subpopulations. Calculation of the FST value between CZ-N and CZ-S samples revealed significant genetic differentiation (FST=0.066, P<0.001), although no substructuring within the CZ population was detected in the population structure analyses. Measurement of genetic differentiation by RST showed similar patterns to those revealed by the FST analysis (Table 2).
MIGRATE analyses estimated bidirectional migration rates ranging from 0.27 to 0.92 individuals per generation between the three inferred genetic subpopulations, with the highest migration rates between the FS-JCS and FS-BZ patches and lowest migration rates between the FS-BZ and CZ patches (Table 3).
Causal modelling analyses of landscape factors
Coincidences of genetic boundaries and landscape features (large and small habitat gaps) suggest that anthropogenic land use may generate dispersal barriers for T. leucocephalus. However, other environmental factors, in particular geographical distance, have likely shaped the observed population genetic structure independent of the effects of barriers on gene flow. To identify the landscape factors structuring the genetic patterns across habitat patches, we first applied simple Mantel tests of four factors, geographical distance (D), roads (R), large habitat gaps (L) and small habitat gaps (S), for the degree of association with genetic distances (G) among individuals. The results of the simple Mantel tests showed that all factors except small habitat gaps were significantly correlated with genetic distance (following a Bonferroni correction; P<0.001) with high correlation values (0.47–0.54; Table 4). We then compared the alternative hypotheses of each and all combinations of the four factors with partial Mantel tests in a causal modelling framework. Table 5 shows the expectations of the 15 organisational hypotheses (in columns) of landscape factors driving population genetic structure and the results of 12 partial Mantel tests (in rows) for causal modelling analysis; the highlighted boxes indicate where the result of a partial Mantel test was consistent with the expectation of a hypothesis. The support rate for each hypothesis (that is, the ratio of the number of tests consistent with the expectations to the total number of relevant tests) varied from 0.10 to 0.84, with hypothesis 3 (L, that is, genetic structure is influenced by large habitat gaps only) being the most highly supported hypothesis. With a support rate of 0.83, Hypothesis 6 (D+L, that is, genetic structure is influenced by both geographical distance and large habitat gaps) was the next most highly supported hypothesis. The support rates of hypothesis 1 (D) and hypothesis 11 (D+R+L) were 0.80 and 0.66, respectively, whereas the support rates for the other eight hypotheses were <0.60. Three of the four most highly supported hypotheses included the influence of geographical distance (D) or large habitat gaps (L), one included that of roads (R) and none included that of small habitat gaps (S), suggesting strong impacts of the former two factors on genetic patterns.
To control for the effect of large habitat gaps (the most highly supported factor in causal modelling across the FS and CZ populations) and elucidate the effect of geographical distance at a finer spatial scale, we evaluated correlations between genetic distance and geographical distance for individuals within the FS and CZ populations separately using simple Mantel tests. A significant pattern of isolation by distance (IBD) was revealed in the FS population (r=0.095, P=0.013), but not in the CZ population (r=0.058, P=0.128) (Supplementary Figure S2; Supplementary Information).
Discussion
Spatial genetic structure and potential barriers to gene flow
Accurate assessment of the spatial genetic patterns of species living in heterogeneous habitats is an effective means of identifying landscape features and processes that influence genetic structure. As applying different analytical methods to the same data often yields inconsistent conclusions concerning patterns of genetic structure, the results of various methods should be compared to ensure accurate and comprehensive estimation of population genetic parameters (Balkenhol et al., 2009; Landguth et al., 2010). We found an overall consistent delineation of two genetic subpopulations (FS and CZ) using the nonspatial Bayesian clustering approach STRUCTURE and genetic distance-based neighbour-net networks based on microsatellite data. The results of the spatial Bayesian clustering method GENELAND are in agreement with the genetic divergence between the FS and CZ populations, yet further divide the FS samples into JCS and BZ groups. GENELAND is one of the most powerful statistical methods of inferring genetic discontinuities and has shown high detection success rates within fewer generations following the establishment of barriers (Blair et al., 2012; Ruiz-Gonzalez et al., 2015). High levels of genetic differentiation among the three subpopulations are supported by the large and significant pairwise FST values (0.10–0.22) obtained using microsatellite data.
The boundaries between the inferred genetic clusters coincided precisely with the spatial distribution of anthropogenic habitat gaps (for example, agricultural lands, human residential areas and so on), confirming the barrier effects of human land modification to dispersal of T. leucocephalus. Furthermore, the absence of apparent admixed individuals between the FS and CZ populations revealed by the STRUCTURE analysis suggests a very low level of gene flow between populations and strong isolation caused by large non-habitat areas separating forest patches. Causal modelling analysis identified large habitat gaps and geographical distance as the primary landscape factors contributing to spatial genetic patterns across the distribution range of T. leucocephalus, in agreement with the strong genetic differentiation between the FS and CZ populations.
Within the two main habitat areas, the FS population showed a significant genetic pattern of isolation-by-distance, whereas the CZ population did not. The signature of isolation-by-distance is critically dependent on the relative spatial scale of the study area and the dispersal distance of the species under analysis. Sampling within the dispersal extent may obscure the effects of geographical distance on the genetic structure of the organism (Cushman and Landguth, 2010a). We do not have direct observational data on the dispersal distance of T. leucocephalus. On the basis of the first-generation migrants detected using microsatellite data, the geographical distances between migrant individuals and their inferred natal groups ranged from 0.2 to 5.3 km in the FS population (Wang and Yao, 2017). The habitat size of the FS population is larger than that of the CZ population (the largest distance between sampled groups was 13.6 km in FS and 8.3 km in CZ). Therefore, the size of the CZ patch may be closer to the maximum dispersal distance of the langur, which may account for the less pronounced pattern of isolation-by-distance of the CZ population in comparison with that of the FS population.
The genetic discontinuity between the FS-JCS and FS-BZ populations detected by the GENELAND analysis of microsatellite data coincided with a small habitat gap (shortest distance was approximately 3 km) consisting of farmland and human residences, indicating that even small non-habitat zones can pose dispersal barriers for T. leucocephalus. Genetic differentiation (measured by pairwise FST) and migration rates revealed by microsatellite data suggest that the FS-JCS and FS-BZ populations are moderately differentiated, but still functionally connected by low levels of gene flow; however, the two populations share no common mtDNA haplotypes despite their geographical proximity, indicating complete genetic isolation of the two patches (Wang et al., 2015). The discrepancy between the genetic patterns shown by the two types of molecular markers may be best explained by male-biased dispersal in T. leucocephalus. Observational studies have shown that the male offspring of a reproductive group often disperse before sexual maturity, whereas females mostly remain and reproduce in their natal groups (Jin, 2008; Zhao et al., 2011). Sexual differences in dispersal tendency and capacity likely lead to a stronger genetic structure in the more philopatric sex (female in this case), which thus show greater genetic differentiation as revealed by sex-specific markers (mtDNA) in comparison with that revealed by bi-parentally inherited markers (microsatellites) (for example, Prugnolle and de Meeus, 2002).
Although none of the genetic structure analyses detected substructuring within the CZ population, genetic differentiation measured by pairwise FST analysis of microsatellite data reached a significant and relatively high value (0.066) between samples from the CZ-N and CZ-S patches, suggesting that genetic differentiation is occurring between these populations, which are separated by a national road. Roads have been shown to impede gene flow and promote genetic isolation in many mammalian species (for example, Epps et al., 2005; Perez-Espona et al., 2008). However, previous studies indicate that the effect of roads on primate genetic structure is insignificant or ambiguous (Radespiel et al., 2008; Quéméré et al., 2010). Many characteristics determine the impacts of roads on wild animals, including their year of construction, size, traffic density, and the presence of fences (Balkenhol and Waits, 2009; Holderegger and Di Giulio, 2010). In our case, the national road in question was likely first paved in the 1970s, but it may have existed in a primitive form for a much longer time. Although the road is currently of moderate size and traffic load, the residences of a village along the road where it cuts across the CZ area may present an additional barrier to the langurs as a result of their behavioural avoidance of humans. Furthermore, the small population size of the CZ populations, particularly CZ-S, may have enhanced the road-induced genetic isolation and differentiation due to strong genetic drift.
Genetic diversity and population demographic history
The levels of genetic diversity assessed by microsatellite heterozygosities and number of alleles were noticeably lower than those reported for several other endangered primate species with similar total population sizes (Table 6). In addition, the CZ population displayed microsatellite diversity comparable to that of the FS population (Table 1), yet it was entirely devoid of mtDNA variation (Wang et al., 2015). It is worth noting that cross-species comparisons of genetic diversity parameters based on different microsatellite loci are prone to ascertainment bias in that the genetic diversity level in a species can be high due to the inclusion of highly polymorphic markers. However, the panel of microsatellite loci in our study was developed specifically for T. leucocephalus, and we selected the most polymorphic loci of isolated microsatellites from the genome (Wang et al., 2016). Therefore, we think that the lower microsatellite diversity in this species compared with the other endangered primate species is mostly likely caused by the overall low genetic diversity of the genome rather than marker selection. In addition, the sample size of our study was larger than those of studies of other endangered primate species, and therefore the difference in genetic variation was not attributed to limited sampling. The phylogenetic relationship revealed by mtDNA sequences shows that T. leucocephalus constitutes a monophyletic clade nested within the François’ langur (Trachypithecus francoisi), from which it diverged 0.46–0.27 Myr ago (Liu et al., 2013). Thus, possible explanations for low genetic variation in the species may include its relatively short evolutionary history, restricted historical population size and recent population declines due to habitat deterioration and hunting by humans. The stark contrast between the genetic diversity levels of the CZ population measured by microsatellite and mtDNA data might be a result of the establishment of this population by a small number of female founders, giving rise to low mtDNA diversity, followed by genetic exchange with other populations (such as the FS population) primarily mediated by male dispersal, resulting in relatively high nuclear marker (microsatellite) variation.
Implications for conservation management
Little historical data are available about human activity in the study area. The earliest record of the region is the establishment of Xiangjun County during the Qin Dynasty nearly 2200 years ago. Agricultural activity and land modifications may have increased subsequently, but the timescale and spatial extent of habitat destruction are unclear. Despite the relatively rapid mutation rate of microsatellites, the short history of human activity precludes meaningful calculations of isolation time between the habitat areas. Furthermore, population genetic processes such as genetic drift, gene flow and bottleneck effects can obscure the estimation of population divergence times based on genetic distance and a constant mutation rate (Relethford, 1996; Arbogast et al., 2002). Therefore, the question still remains as to when anthropogenic factors began to impact the population genetics of the langur.
Although no genetic signature of inbreeding was detected in this study, nor signs of genetic erosion, such as reduced birth rates or increased disease-associated mortality, have been reported in wild T. leucocephalus, the low genetic diversity level and strong population structure of the species increase the risk of inbreeding depression and further loss of genetic viability in remnant populations. Tracking genetic patterns and population dynamics will allow detection and alleviation of anthropogenic dispersal barriers before their influences on genetic connectivity fully manifest. On the basis of the microsatellite and mtDNA data (Wang et al., 2015), the T. leucocephalus population can be regarded as two management units (MUs; Moritz, 1994) corresponding to FS and CZ, due to their demographic independency as assessed by strong genetic differentiation and very low between-area migration rates. Genetic divergence between habitat patches within each area (FS-JCS and FS-BZ; CZ-N and CZ-S) is less pronounced than that between the habitat areas (FS and CZ), most likely attributed to much shorter geographical distances and weaker barrier effects of landscape features between patches, which result in less severe genetic isolation. However, as human disturbance and land modification continue to intensify as a result of population growth and economic development, genetic separation among habitat patches may be rapidly aggravated. Given the already low genetic variation in the species, reproductive isolation will greatly accelerate the process of genetic erosion by drift and inbreeding depression, particularly in small populations (Keller and Waller, 2002; Keyghobadi, 2007). Conservation efforts should focus on enforcement of habitat connectivity among adjacent patches that are divided by narrow habitat gaps or roads. Genetic isolation can be prevented by regulating human activity and restoring vegetation corridors for dispersal routes between small populations. Considering the close proximity between adjacent patches and current migration patterns, corridor building would be easy to implement and more practical than human-assisted dispersal or ex situ conservation. Gene flow and inbreeding status should be closely monitored by non-invasive genetic methods to evaluate the effectiveness of conservation efforts and assist in future management planning.
Data accessibility
Sampling locations and microsatellite genotypes will be deposited in the Dryad digital repository and a doi will be provided. Data for mitochondrial HVRI sequences are accessible in GenBank under accession numbers KP772243–KP772251.
Data archiving
Genotype and landscape data for this study are available from the Dryad Digital Repository: doi:10.5061/dryad.746k3.
References
Arbogast BS, Edwards SV, Wakeley J, Beerli P, Slowinski JB . (2002). Estimating divergence times from molecular data on phylogenetic and population genetic timescales. Annu Rev Ecol Syst 33: 707–740.
Balkenhol N, Waits LP . (2009). Molecular road ecology: exploring the potential of genetics for investigating transportation impacts on wildlife. Mol Ecol 18: 4151–4164.
Balkenhol N, Waits LP, Dezzani RJ . (2009). Statistical approaches in landscape genetics: an evaluation of methods for linking landscape and genetic data. Ecography 32: 818–830.
Bastos HB, Gonçalves EC, Ferrari SF, Silva A, Schneider MPC . (2010). Genetic structure of red-handed howler monkey populations in the fragmented landscape of Eastern Brazilian Amazonia. Genet Mol Biol 33: 774–780.
Beerli P . (2006). Comparison of Bayesian and maximum likelihood inference of population genetic parameters. Bioinformatics 22: 341–345.
Beerli P, Felsenstein J . (2001). Maximum likelihood estimation of a migration matrix and effective population sizes in n subpopulations by using a coalescent approach. PNAS 98: 4563–4568.
Blair C, Weigel DE, Balazik M, Keeley ATH, Walker FM, Landguth E et al. (2012). A simulation-based evaluation of methods for inferring linear barriers to gene flow. Mol Ecol Resour 12: 822–833.
Blair ME, Melnick DJ . (2012). Scale-dependent effects of a heterogeneous landscape on genetic differentiation in the Central American squirrel monkey (Saimiri oerstedii. PLoS One 7: e43027.
Bryant D, Moulton V . (2004). Neighbor-net: an agglomerative method for the construction of phylogenetic networks. Mol Biol Evol 21: 255–265.
Burton FD, Snarr KA, Harrison SE . (1995). Preliminary report on Presbytis francoisi leucocephalus. Int J Primatol 10: 311–327.
Chaves PB, Alvarenga CS, CdB Possamai, Dias LG, Boubli JP, Strier KB et al. (2011). Genetic diversity and population history of a critically endangered primate, the northern muriqui (Brachyteles hypoxanthus. PLoS ONE 6: e20722.
Chongzuo Nature Reserve. (2010) Population Census of the White-headed Langur. Forestry Bureau of Guangxi Province.
Cushman SA, Landguth EL . (2010a). Scale dependent inference in landscape genetics. Landsc Ecol 25: 967–979.
Cushman SA, Landguth EL . (2010b). Spurious correlations and inference in landscape genetics. Mol Ecol 19: 3592–3602.
Cushman SA, McKelvey KS, Hayden J, Schwartz MK . (2006). Gene flow in complex landscape: testing multiple hypotheses with causal modeling. Am Nat 168: 486–499.
Earl DA, vonHoldt BM . (2012). STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Res 4: 359–361.
Ebenau A, Nadler T, Zinner D, Roos C . (2011). Genetic population structure of the critically endangered Delacour’s langur (Trachypithecus delacouri in Van Long Nature Reserve, Vietnam. Vietnam J Primatol 5: 1–15.
Epps CW, Palsboll PJ, Wehausen JD, Roderick GK, Ramey RR, McCullough DR . (2005). Highways block gene flow and cause a rapid decline in genetic diversity of desert bighorn sheep. Ecol Lett 8: 1029–1038.
Evanno G, Regnaut S, Goudet J . (2005). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14: 2611–2620.
Excoffier L, Lischer HL . (2010). Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10: 564–567.
Falush D, Stephens M, Pritchard JK . (2003). Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164: 1567–1587.
Frankham R . (2005). Genetics and extinction. Biol Conserv 126: 131–140.
Frantz AC, Pope LC, Carpenter PJ . (2003). Reliable microsatellite genotyping of the Eurasian badger (Meles meles using faecal DNA. Mol Ecol 12: 1649–1661.
Grativol AD, Ballou JD, Fleischer RC . (2001). Microsatellite variation within and among recently fragmented populations of the golden lion tamarin (Leontopithecus rosalia. Conserv Genet 2: 1–9.
Griesser M, Halvarsson P, Sahlman T, Ekman J . (2014). What are the strengths and limitations of direct and indirect assessment of dispersal? Insights from a long-term field study in a group-living bird species. Behav Ecol Sociobiol 68: 485–497.
Guillot G, Estoup A, Mortier F, Cosson JF . (2005a). A spatial statistical model for landscape genetics. Genetics 170: 1261–1280.
Guillot G, Leblois R, Coulon A, Frantz AC . (2009). Statistical methods in spatial genetics. Mol Ecol 18: 4734–4756.
Guillot G, Mortier F, Estoup A . (2005b). GENELAND: a computer package for landscape genetics. Mol Ecol Notes 5: 712–715.
Guillot G, Santos F, Estoup A . (2008). Analysing georeferenced population genetics data with GENELAND: a new algorithm to deal with null alleles and a friendly graphical user interface. Bioinformatics 24: 1406–1407.
Hagell S, Whipple AV, Chambers CL . (2013). Population genetic patterns among social groups of the endangered Central American spider monkey (Ateles geoffroyi in a human-dominated landscape. Ecol Evol 3: 1388–1399.
Hardy OJ, Vekemans X . (2002). SPAGEDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes 2: 618–620.
Holderegger R, Di Giulio M . (2010). The genetic effects of roads: a review of empirical evidence. Basic Appl Ecol 11: 522–531.
Hu G, Wei Y, Li Z . (1998). The population dynamics of the white-headed langur Presbytis leucocephalus in Bapen. J Guangxi Normal Univ 16: 64–67.
Huang CM, Wei FW, Li M, Quan GQ, Li HH . (2002). Current status and conservation of white-headed langur (Trachypithecus leucocephalus in China. Biol Conserv 104: 221–225.
Huson DH . (1998). SplitsTree: analyzing and visualizing evolutionary data. Bioinformatics 14: 68–73.
IUCN. (2016). The IUCN red list of threatened species. Version 2016-3. Available at: www.iucnredlist.org. Accessed on 20 June 2016.
Jin T . (2008). Female reproductive strategy of wild white-headed langurs (Trachypithecus leucocephalus: social relationship, reproduction and allomothering behavior. PhD thesis, Peking University, Beijing, China.
Jin T, Wang DZ, Zhao Q, Yin LJ, Qin DG, Ran WZ et al. (2009). Social organization of white-headed langurs (Trachypithecus leucocephalus in the Nongguan Karst Hills, Guangxi, China. Am J Primatol 71: 206–213.
Kalinowski ST . (2005). HP-RARE 1.0: a computer program for performing rarefaction on measures of allelic richness. Mol Ecol Notes 5: 187–189.
Kalinowski ST, Taper ML, Marshall TC . (2007). Revising how the computer program CERVUS accommodates genotyping error increases success in paternity assignment. Mol Ecol 16: 1099–1106.
Keller LF, Waller DM . (2002). Inbreeding effects in wild populations. Trends Ecol Evol 17: 230–241.
Keyghobadi N . (2007). The genetic implications of habitat fragmentation for animals. Can J Zool 85: 1049–1064.
Koenig WD, VanVuren D, Hooge PN . (1996). Detectability, philopatry, and the distribution of dispersal distances in vertebrates. Trends Ecol Evol 11: 514–517.
Kolleck J, Yang M, Zinner D, Roos C . (2013). Genetic diversity in endangered Guizhou snub-nosed monkeys (Rhinopithecus brelichi: contrasting results from microsatellite and mitochondrial DNA data. PLoS One 8: e73647.
Landguth EL, Cushman SA, Schwartz MK, McKelvey KS, Murphy M, Luikart G . (2010). Quantifying the lag time to detect barriers in landscape genetics. Mol Ecol 19: 4179–4191.
Leslie KA (2013). Molecular genetic tools for evaluating the consequences of habitat fragmentation. In: Marsh LK, Chapman CA (eds). Primates in Fragments: Complexity and Resilience. Springer:Berlin, Germany, pp 389–398.
Li ZY . (1993). Preliminary investigation of the habitats of Presbytis francoisi and Presbytis leucocephalus, with notes on the activity pattern of Presbytis leucocephalus. Folia Primatol 60: 83–93.
Lindenmayer DB, Hobbs RJ, Montague-Drake R . (2008). A checklist for ecological management of landscapes for conservation. Ecol Lett 11: 78–91.
Liu Z, Ren B, Wu R, Zhao L, Hao Y, Wang B et al. (2009). The effect of landscape features on population genetic structure in Yunnan snub-nosed monkeys (Rhinopithecus bieti implies an anthropogenic genetic discontinuity. Mol Ecol 18: 3831–3846.
Liu Z, Wang B, Nadler T, Liu G, Sun T, Huang C et al. (2013). Relatively recent evolution of pelage coloration in Colobinae: phylogeny and phylogeography of three closely related langur species. PLoS One 8: e61659.
Luque S, Saura S, Fortin M-J . (2012). Landscape connectivity analysis for conservation: insights from combining new methods with ecological and genetic data. Landsc Ecol 27: 153–157.
Moritz C . (1994). Applications of mitochondrial DNA analysis in conservation: a critical review. Mol Ecol 3: 401–411.
Nei M . (1972). Genetic distance between populations. Am Nat 106: 283–292.
Oklander LI, Kowalewski MM, Corach D . (2010). Genetic consequences of habitat fragmentation in black-and-gold howler (Alouatta caraya populations from Northern Argentina. Int J Primatol 31: 813–832.
Pan D, Hu HX, Meng SJ, Men ZM, Fu YX, Zhang YP . (2009). A high polymorphism level in Rhinopithecus roxellana. Int J Primatol 30: 337–351.
Peakall R, Smouse P . (2006). GENAlEX 6.5: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Resour 6: 288–295.
Peakall R, Smouse P . (2012). GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research- an update. Bioinformatics 28: 2537–2539.
Perez-Espona S, Perez-Barberia FJ, McLeod JE, Jiggins CD, Gordon IJ, Pemberton JM . (2008). Landscape features affect gene flow of Scottish Highland red deer (Cervus elaphus. Mol Ecol 17: 981–996.
Pritchard JK, Stephens M, Donnelly P . (2000). Inference of population structure using multilocus genotype data. Genetics 155: 945–959.
Prugnolle F, de Meeus T . (2002). Inferring sex-biased dispersal from population genetic tools: a review. Heredity 88: 161–165.
Quéméré E, Crouau-Roy B, Rabarivola C, Louis EE, Chikhi L . (2010). Landscape genetics of an endangered lemur (Propithecus tattersalli within its entire fragmented range. Mol Ecol 19: 1606–1621.
Radespiel U, Bruford MW . (2014). Fragmentation genetics of rainforest animals: insights from recent studies. Conserv Genet 15: 245–260.
Radespiel U, Rakotondravony R, Chikhi L . (2008). Natural and anthropogenic determinants of genetic structure in the largest remaining population of the endangered golden-brown mouse lemur, Microcebus ravelobensis. Am J Primatol 70: 860–870.
Ran WZ . (2003). The current status of wild-ranged white-headed langur: distribution, number, habitat use, and population viability analysis. PhD thesis, Peking University, Beijing, China.
Relethford JH . (1996). Genetic drift can obscure population history: Problem and solution. Hum Biol 68: 29–44.
Rice WR . (1989). Analyzing tables of statistical tests. Evolution 43: 223–225.
Roos C, Boonratana R, Supriatna J, Fellowes JR, Groves CP, Nash SD et al. (2014). An updated taxonomy and conservation status review of Asian primates. Asian Primates J 4: 2–38.
Rousset F . (2000). Genetic differentiation between individuals. J Evol Biol 13: 58–62.
Rousset F . (2008). GENEPOP’ 007: a complete re-implementation of the GENEPOP software for Windows and Linux. Mol Ecol Resour 8: 103–106.
Ruiz-Gonzalez A, Cushman SA, Madeira MJ, Randi E, Gómez-Moliner BJ . (2015). Isolation by distance, resistance and/or clusters? Lessons learned from a forest-dwelling carnivore inhabiting a heterogeneous landscape. Mol Ecol 24: 5110–5129.
Salmona J, Teixeira H, Rasolondraibe E, Aleixo-Pais I, Kun-Rodrigues C, Rakotonanahary AN et al. (2015). Genetic diversity, population size, and conservation of the Critically Endangered Perrier’s sifaka (Propithecus perrieri. Int J Primatol 36: 1132–1153.
Segelbacher G, Cushman SA, Epperson BK, Fortin M-J, Francois O, Hardy OJ et al. (2010). Applications of landscape genetics in conservation biology: concepts and challenges. Conserv Genet 11: 375–385.
Slatkin M . (1995). A measure of population subdivision based on microsatellite allele frequencies. Genetics 139: 457–462.
Taberlet P, Griffin S, Goossens B, Questiau S, Manceau V, Escaravage N et al. (1996). Reliable genotyping of samples with very low DNA quantities using PCR. Nucleic Acids Res 24: 3189–3194.
Taberlet P, Waits LP, Luikart G . (1999). Noninvasive genetic sampling: look before you leap. Trends Ecol Evol 14: 323–327.
Tan PC . (1957). Rare catches by Chinese animal collectors. Zoo Life 12: 61–63.
Thalmann O, Hebler J, Poinar HN, Pääbo S, Vigilant L . (2004). Unreliable mtDNA data due to nuclear insertions: a cautionary tale from analysis of humans and other great apes. Mol Ecol 13: 321–335.
van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P . (2004). MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4: 535–538.
Villesen P, Fredsted T . (2006). A new sex identification tool: One primer pair can reliably sex ape and monkey DNA samples. Conserv Genet 7: 455–459.
Wang W, Qiao Y, Pan W, Yao M . (2015). Low genetic diversity and strong geographical structure of the Critically Endangered white-headed langur (Trachypithecus leucocephalus inferred from mitochondrial DNA control region sequences. PLoS One 10: e0129782.
Wang W, Qiao Y, Zheng Y, Yao M . (2016). Isolation of microsatellite loci and reliable genotyping using noninvasive samples for a critically endangered primate, Trachypithecus leucocephalus. Integr Zool 11: 250–262.
Wang W, Yao M : Fine-scale genetic structure analyses reveal dispersal patterns in a critically endangered primate, Trachypithecus leucocephalus. Am J Primatol,. (2017) e-pub ahead of print 18 January 2017 doi:10.1002/ajp.22635.
Wei S-J, Cao L-J, Gong Y-J, Shi B-C, Wang S, Zhang F et al. (2015). Population genetic structure and approximate Bayesian computation analyses reveal the southern origin and northward dispersal of the oriental fruit moth Grapholita molesta (Lepidoptera:Tortricidae) in its native range. Mol Ecol 24: 4094–4111.
Weir BS, Cockerham CC . (1984). Estimating F-statistics for the analysis of population structure. Evolution 38: 1358–1370.
Yang M, Yang Y, Cui D, Fickenscher G, Zinner D, Roos C et al. (2012). Population genetic structure of Guizhou snub-nosed monkeys (Rhinopithecus brelichi as inferred from mitochondrial control region sequences, and comparison with R. roxellana and R. bieti. Am J Phys Anthropol 147: 1–10.
Yeh FC, Yang R-C, Boyle TBJ, Ye Z-H, Mao JX . (1997) POPGENE, The User-friendly Shareware for Population Genetic Analysis. Molecular Biology and Biotechnology Centre, University of Alberta: Edmonton, AB, Canada.
Zhao Q, Borries C, Pan WS . (2011). Male takeover, infanticide, and female countertactics in white-headed leaf monkeys (Trachypithecus leucocephalus. Behav Ecol Sociobiol 65: 1535–1547.
Acknowledgements
We thank Barbara Mable and three anonymous reviewers for their valuable comments on earlier versions of the manuscript. We thank Qihai Zhou, Jintong Lu, Xinyang Li, Yuning Meng and the crew at Bapen Conservation Station for their assistance with sample collection, and Zuhong Liang, Meiying Huang and Yanping Yang for their help with field work. We also thank Haoran Xue for his help with data analysis. Financial support for this study was provided by the National Natural Science Foundation of China (31572283 and 31272329) to MY. We thank SCINET for language editing of this manuscript.
Author contributions
MY and WW conceived and designed the research and wrote the manuscript. WW performed the experiments and acquired data. WW and MY analysed data. SL assisted with data analysis. WW, YQ, WP and MY collected the samples. All authors read and approved the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on Heredity website
Supplementary information
Rights and permissions
About this article

{kind=link}
{kind=link}
Cite this article
Wang, W., Qiao, Y., Li, S. et al. Low genetic diversity and strong population structure shaped by anthropogenic habitat fragmentation in a critically endangered primate, Trachypithecus leucocephalus. Heredity 118, 542–553 (2017). https://doi.org/10.1038/hdy.2017.2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/hdy.2017.2
- Springer Nature Switzerland AG
This article is cited by
-
Looking Ahead: Predicting the Possible Ecological and Physiological Response of Galago Moholi to Environmental Change
International Journal of Primatology (2023)
-
The reference genome and transcriptome of the limestone langur, Trachypithecus leucocephalus, reveal expansion of genes related to alkali tolerance
BMC Biology (2021)
-
The complete mitochondrial genome of Indian gaur, Bos gaurus and its phylogenetic implications
Scientific Reports (2020)
-
Low genetic diversity in a critically endangered primate: shallow evolutionary history or recent population bottleneck?
BMC Evolutionary Biology (2019)
-
Considering landscape connectivity and gene flow in the Anthropocene using complementary landscape genetics and habitat modelling approaches
Landscape Ecology (2019)