
Abstract
Forests affected by fragmentation are at risk of losing their primate populations over the long term. The impact of fragmentation on primate populations has been studied in several places in Africa, Asia and South America; however, there has been no discernible pattern of how primates react to forest disturbance and fragmentation. In fragmented habitats, the local extinction probability of a species increases due to a decrease in patch area and an increase in genetic isolation. Here we used microsatellite markers and mitochondrial DNA sequences to investigate how habitat fragmentation impacts on the genetic diversity and structure of a samango monkey population inhabiting forest patches in the Soutpansberg mountain range of northern South Africa. We sampled four local populations across the length of the mountain range and an additional outlying population from the Great Escarpment to the south. Our results indicate that local populations along the mountain range were historically more connected and less distinct than at present. In more recent times, a lack of contemporary gene flow is leading to a more pronounced genetic structure, causing population subdivision across the mountain and likely isolating the Soutpansberg population from the escarpment population to the south. Based on our results, we suggest that natural and anthropogenic fragmentation are driving population genetic differentiation, and that the matrix surrounding forests and their suitability for samango monkey utilisation play a role at the local scale. The degree of genetic isolation found for samango monkey populations in our study raises concerns about the long-term viability of populations across the mountain range.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Forest habitat loss and fragmentation and the resulting isolation of animal populations is a global issue of our time (FAO 2018). Changes in forest environments can lead to a reduction of specific habitat structures and niches, which has been reported to threaten the survival of forest-dependent species such as koala (Phascolarctos cinereus), crested Guinea-fowl (Guttera edouardi) and numerous other species of forest birds (Bennun et al. 1996; McAlpine et al. 2006; Maseko et al. 2017). If habitat fragmentation leads to reduced gene flow between populations, it could have profound effects on the genetic viability of populations, due to two main genetic problems (Lacy 1997; Dudash and Fenster 2000). Firstly, small populations are more vulnerable to genetic drift causing random changes in allele frequencies, resulting in a loss of genetic variability (Lacy 1997). Secondly, isolated populations experience greater inbreeding, as immigration and emigration are impeded. Inbreeding, especially over several generations, increases the chance of homozygosity of deleterious recessive alleles and thus their expression. At the same time, heterozygosity is lost and this can lead to reduced adaptability (Lacy 2000). The combination of drift and inbreeding can increase the level of genetic structure observed between populations. Thus, monitoring and managing changes in species population size, connectivity and genetic diversity in fragmented forest habitat should be considered a priority in order to protect biodiversity.
African forests are characterised by a diverse mammalian fauna, including primates, which have been shown to play vital roles in forest ecosystem functioning, structure and resilience (Estrada et al. 2017). Primates disperse seeds, play integral roles in food webs as consumers and prey, and participate in a diverse array of coevolved relationships with other species (Marsh 2003; Seufert et al. 2009; Linden et al. 2015). Forests affected by outside disturbance and/or isolation are at risk of losing their primate populations over time. Although the impact of fragmentation on primate populations has been studied in many places in Africa, Asia and South America, there is no discernible pattern of how primates react to forest disturbance and fragmentation (Marsh 2003, 2013). This is because the ability of primate populations to sustain themselves in disturbed and fragmented forests is very species- and circumstance-specific, and as a result, so are conservation and management recommendations (Gibbons and Harcourt 2009). In South Africa, indigenous, high-canopy, evergreen forests are the most restricted and naturally fragmented biome (Eeley et al. 1999; Mucina and Geldenhuys 2006), covering only 0.4% of the country’s land surface area (Berliner 2005). Given that forests have the highest biodiversity per unit area of any biome in South Africa (Berliner 2005), the extent to which habitat fragmentation impacts on forest animal species at the population genetic level (on a landscape scale) is generally understudied and varies by species and geographic area. For example, Eggert et al. (2008) found that for a small population of elephants confined largely to Afromonate forest in the southern Cape of South Africa, genetic diversity has thus far not been affected by isolation, as these elephants are likely remnants of once widespread populations of South Africa. Moir et al. (2021) surveyed forest-utilizing bats and reported that vulnerability to fragmentation varies among bat species. Lastly, Madisha et al. (2017) suggested that samango monkey populations inhabiting two forest fragments in the Eastern Cape Province in South Africa did not display negative genetic factors associated with isolation due to male-mediated migration.
Our study focuses on a population of South Africa’s only diurnal, forest-dwelling primate species, the samango monkey (Cercopithecus albogularis), whose distribution pattern mirrors that of South African forests. The species is nationally listed as Vulnerable and the subspecies C. a. schwarzi found in the study area as Endangered (Linden et al. 2016). In this study, we investigate how historical and recent forest habitat fragmentation may impact on the genetic diversity and structure of a samango monkey population in a longitudinal (running form east to west) mountain range, the Soutpansberg, in far northern South Africa. Forest fragmentation in the Soutpansberg is a result of natural processes as well as anthropogenic activities (Scott 1987; Munyati and Kabanda 2009), and the matrix (portion of unsuitable habitat in the landscape) surrounding forest fragments is very diverse. This makes it an ideal landscape to investigate how samango monkeys are genetically impacted under various fragmentation and surrounding matrix scenarios. Being a mountain range, the study area can further be considered a biogeographic island characterised by a certain degree of isolation from its surroundings. Given the landscape characteristics, we expect that the samango monkey population is genetically subdivided within the mountain range and that gene flow between the mountain and the closest samango monkey populations further south has become very restricted. We propose that population subdivision is driven by two main processes: (1) natural habitat fragmentation driven by paleoclimatic changes or geographic barriers such as distance and topographic features, and (2) anthropogenic habitat fragmentation caused by land transformation. To determine whether natural and anthropogenic barriers are leading to population subdivision, we analysed microsatellite markers and mitochondrial DNA (mtDNA) to estimate the extent of genetic variation and gene flow among populations. Results from this study will inform conservation planning for samango monkey populations inhabiting this mountain range.
Materials and methods
Study species
Samango monkeys live in multifemale groups led by a single adult male, with females being philopatric as is common in forest guenons (Cords 2001). Males emigrate from their natal group roughly a year before reaching sexual maturity at about 6–7 years of age (Henzi and Lawes 1987; Ekernas and Cords 2007). Extra-group males are described to range widely and interact with more than one group of females (Swart and Lawes 1996). Samango monkey group sizes vary between 16 and 60 individuals across study sites in South Africa (Lawes 1990, 1992; Coleman and Hill 2014; Novak et al. 2014; Linden et al. 2015; Wimberger et al. 2017), as do home range sizes, from 15 to 54 ha (Lawes et al. 1990; Coleman and Hill 2014; Wimberger et al. 2017). Neighbouring groups’ home ranges often overlap (Lawes 1992; Lawes and Henzi 1995; Novak et al. 2014), and extra-group male ranges may overlap considerably with group home ranges (Swart et al. 1993). Neighbouring groups can be “sister-groups” because of group fission, for example due to group size (Swart and Lawes 1996; Linden pers. obs.). Using the average age of first breeding for females (Oklander et al. 2017), the generation time of samango monkeys is ~ 7 years (Cords 2012).
Study area and study population
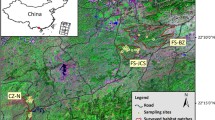
The Soutpansberg is situated in South Africa’s far northern Limpopo Province (Fig. 1a). It is a ~ 210 km-long and ~ 60 km-wide (widest point), east–west-orientated mountain range with an altitudinal range between 200 m in the far east and 1748 m (Lajuma) in the far west (Hahn 2017a) (Fig. 1a, b). High-canopy, evergreen forests suitable for samango monkeys are only found on the southern ridges of the mountain (Fig. 1b), with forest patches in the more arid western Soutpansberg being smaller and naturally more confined compared to forests in the east (Linden et al. in prep.).

Geographic setting. a Map of South Africa detailing the Great Escarpment, Limpopo Province with the Soutpansberg, and Hogsback. b Satellite image showing the five study sites across the Soutpansberg. LA Lajuma, SB Schoemansdal/Buzzard Mountain, LL Levuvhu/Luonde, ET Entabeni/Thathe Vondo and northern escarpment (MK Magoebaskloof). Geographic barriers are detailed in the two enlargements; the Sand River gorge (inset c) and the gap between the Soutpansberg and escarpment, the Levuvhu River area (inset d). The area circled in white shows the ~ 30 km samango monkey distribution gap in the middle Soutpansberg (Linden et al. in prep. a). The southern portion of the Sand River gorge is between 700 m and 1.6 km wide from cliff top to cliff top, and the river bed is between 56 and 140 m. Satellite image: European Space Agency (ESA), Sentinel 2 (2019)
This study focused on five samango monkey populations, of which four were from the Soutpansberg sensu stricto (Lajuma [LA], Schoemansdal/Buzzard Mountain [SB], Levuvhu/Luonde [LL], Entabeni/Thathe Vondo [ET]) and one from the northern parts of the Great Escarpment (Magoebaskloof [MK]) south of the Soutpansberg (Fig. 1a, b). We chose populations across the Soutpansberg according to geographic distance from each other, matrix surrounding forests and position in relation to main barriers.
The LA population is located in the far west of the mountain range, west of a main topographic feature, the north–south-running Sand River gorge (Fig. 1b, c). The SB population is located east of the Sand River gorge, also in the western part of the Soutpansberg (Fig. 1b). The surrounding matrix of both western populations is composed of natural vegetation (Fig. 2).

Land cover map showing the four sampling areas in the Soutpansberg (LA Lajuma, SB Schoemansdal/Buzzard Mountain, ET Entabeni/Thathe Vondo, LL Luonde/Levuvhu, MK Magoebaskloof) in the context of land use/transformation. Enlarged maps for each sampling area show the matrix surrounding indigenous forests (the category “natural vegetation” includes the Soutpansberg Mountain Bushveld vegetation type). Map source: South African National Land Cover (NLC) (2014)
The ET population is the easternmost population sampled and the LL population is closest to the escarpment, south of the mountain (Fig. 1b). The surrounding matrix of both the LL and ET populations is characterised largely by silvicultural and agricultural areas (Fig. 2). The eastern and western populations are further separated by a ~ 30 km-wide distribution gap found in the middle Soutpansberg (Linden et al. in prep.) (Fig. 1b).
It is important to mention that the SB study area received about six individuals, including at least one male, from the ET study area as part of a Department of Water Affairs and Forestry (DWAF) translocation programme in the 1980s (Greaves J pers. comm.). Samango monkeys were found to cause damage to pine trees in timber plantations in the ET area, and the problem was managed by trapping and translocating individuals (Droomer 1985; von dem Bussche and van der Zee 1985). We included the SB population, as the survival and successful integration of translocated individuals into existing groups was not monitored and thus their fate is unknown. Samples from the MK population were included to determine the degree of differentiation and gene flow between the Soutpansberg and escarpment populations, as we consider this the most likely historical migration route into the Soutpansberg. Here, a population is not considered equal to a group of samango monkeys. A certain geographic area may contain several groups of samango monkeys making up a local population in a particular forest patch, and a study by Madisha et al. (2017) in Hogsback (South Africa) showed that gene flow between neighbouring samango groups was high. To avoid re-sampling the same individuals of a local population, different groups were sampled in each area. Through a concurrent distribution survey (Linden et al. in prep.) knowledge was available on groups occurring in forest patches in the SB and LL areas. For MK, Dalton et al. (2015) collected the five samples from one known group and the additional sample added in our study originated from a group resident on a private property ~ 6 km away from the latter.
Sample collection and DNA extraction
Faecal samples
We collected 42 faecal samples between 2012 and 2015 from the four geographic areas across the Soutpansberg: LA, SB, LL and ET (Table 1). We sampled two samango monkey groups per geographic locality (forest patch) (Table 1). When collecting the samples in the field, strict precautions were used to avoid contamination (Goossens et al. 2003). When possible, faecal samples were collected immediately after defecation to obtain high-quantity and high-quality DNA and avoid degradation (Wasser et al. 1997). However, this was not always possible in the field, as all but one population (LA) sampled (consisting of two habituated groups) were completely unhabituated. Sample collection on the two habituated groups in LA was achieved by following the samango monkeys, observing for any individual defecating and collection of samples within 5 min. For all other groups sampled, following distances were greater, making observations of individuals defecating and detecting faeces on the forest floor challenging. Thus, this resulted in samples being collected between 5 to 60 min after defecation. As most shed cells are found at the “front end” and the outside of faeces, as much of the outer layer as possible was collected to maximise DNA yield (Goossens et al. 2003). Faecal samples were directly transferred into absolute ethanol (sample/ethanol 1:3) (Gerloff et al.1999) and then stored in a refrigerator no warmer than 4°C (Goossens et al. 2003). Samples were processed in the lab within 4 weeks of collection. DNA from faecal samples was extracted using the ZR Faecal DNA MiniPrep™ (Zymo Research) extraction kit according to the manufacturer’s recommendations.
Tissue samples
Eleven tissue samples were collected opportunistically from 2012 to 2015 in one locality (LL population) where samango monkeys are regularly killed through car collisions (Linden et al. 2020) (Table 1). The total number of groups sampled through roadkill could not be established, as only single individuals were found. As the particular stretch of road was driven several times a week, road kills were detected relatively quickly after they occurred. Additionally, one samango carcass was sampled from an individual in Magoebaskloof (MK) which was reportedly found dead (possibly killed by an eagle), and one tissue sample was available from one of the Lajuma groups as part of a trapping exercise to place ear tags on animals for another project (using the same methodology and trapping permit as detailed in Dalton et al. 2015). Tissue samples from all individuals were taken from the muscle and ear (skin and cartilage) and immediately stored in absolute ethanol in a refrigerator (4 °C). Samples were processed in the lab within 4 weeks of collection. DNA extraction from tissue samples was conducted using the ZR Genomic DNA™ Tissue MiniPrep kit (Zymo Research) following the extraction protocol as outlined by the manufacturer. Another 19 tissue samples from Dalton et al. (2015) were included in the analysis of this study: 14 for LA and five for MK (Table 1).
Microsatellite genotyping
All samples were initially genotyped for polymorphism at the 21 microsatellite loci used for samango monkey tissue samples in Dalton et al. (2015) following methods described therein. Faecal DNA amplification was carried out using a 15 micro litre (µl) reaction volume and polymerase chain reaction (PCR) was conducted with 2X Platinum Multiplex PCR Master Mix (Life Technologies™). The final reaction conditions were as follows: 1 X Master Mix, 0.5 μM of each of the forward and reverse primers, 10–20 ng genomic DNA template. The conditions for PCR amplification were as follows: 5 min at 95 °C initial denaturation, 30 cycles for 30 s at 95 °C, 30 s at 50 °C and 1 min at 72 °C, followed by extension at 72 °C for 20 min. The PCR reaction was carried out in a T100™ Thermal Cycler (Bio-Rad Laboratories, Inc.). PCR products were pooled together and run against Genescan™ 500 LIZ® internal size standard on an ABI 3500 Genetic Analyzer (Applied Biosystems, Inc.). Samples were genotyped using GeneMapper software version 4.0 (Applied Biosystems, Inc.). Because of lower DNA quantity and quality obtained from faecal samples, and to control for allelic dropout, each PCR amplification was repeated three times (Goossens et al. 2003). Samples were scored as heterozygotes at a locus if both alleles appeared clearly distinguishable twice among the three replicates. Homozygotes were scored if at least two replicates showed identical homozygote profiles. Of the 42 faecal samples collected, 13 had more than 50% missing data and were excluded from further analysis. GenAlEx version 6.5 (Peakall and Smouse 2012) was used to identify matching genotypes to ensure that only unique individuals were included. Thus, the final microsatellite data set included 61 samples (32 tissue and 29 faecal) from five different samango populations (Table 1). Of the 21 markers initially genotyped for polymorphism, 13 (D4S243, D12S67, D9S922, D3S1768, D8S1106, D15S108, D1S518, D18S536, D10S1432, D11S925, D13S765, D5S1475, D1S207) were polymorphic and gave consistent results for both tissue and faecal samples.
Genetic diversity and relatedness
MICRO-CHECKER was used (Van Oosterhout et al. 2004) to detect potential genotyping errors, allelic dropout and null alleles for each microsatellite locus within our data set. Linkage disequilibrium (LD) between pairs of microsatellite loci within each population, deviations from Hardy–Weinberg equilibrium (HWE) and the fixation index (FIS) were calculated using ARLEQUIN version 3.5.1.2 (Excoffier and Lischer 2010).
To determine the most appropriate relatedness estimator to use for this data set, we used the package related v1.0 (Pew et al. 2015) in R v3.6.2 (R Core Team 2019) to simulate, from the allele frequencies, 100 pairs of each of the following relatedness categories: parent-offspring (PO), full-siblings (FS), half-siblings (HS) and unrelated (UR). The performance of six relatedness estimators was tested by estimating relatedness of these simulated pairs and determining which estimator correlated best with the simulated values by calculating Pearson’s correlation coefficient (cor.test function in R). The estimators tested were as follows: dyadic maximum likelihood “DyadML” (Milligan 2003), Lynch-Li (1993), Lynch and Ritland (1999), Queller and Goodnight (1989), triadic maximum likelihood “TrioML” (Wang 2007) and Wang (2002). Based on the above simulation and the STRUCTURE results, and considering sample size, pairwise relatedness was estimated within the following subgroups, in order to use the most appropriate background allele frequencies, LA, SB-ET and LL-MK, using the triadic likelihood estimator (TrioML) in the R package related (note: the setting “allow.inbreeding” was set to “TRUE”, as currently in v1.0 of the package this option is inverted, so in order to not account for inbreeding, this setting must be set to “TRUE”; Frasier T pers. comm).
Information about the sex and group provenance of samples from LA (two groups were sampled at this locality) were used to test whether individuals were more related within sexes or within groups in this population than is expected by chance. This was done using the grouprel function of the related package in R. This function calculates the average relatedness within each of the specified groups (groups and sex, in this case), then “generates a distribution of ‘expected’ relatedness values by randomly shuffling individuals between groups, while keeping each group size constant, and calculating the average relatedness within each group for each randomization step” (related tutorial). Here, we used 1000 randomisation steps. The function then determines whether the observed relatedness is significantly different from the random distribution of relatedness values generated. A 0.05 level of significance was applied.
To estimate genetic diversity within populations, the mean number of alleles per locus (NA), observed heterozygosities (HO), expected heterozygosities (HE) and unbiased expected heterozygosity (uHE) were determined using GenAlEx version 6.5 (Peakall and Smouse 2012). Allelic richness (Ar) was estimated correcting for sample size through rarefaction using HP-RARE v. June-6-2006 (Kalinowski 2005).
Genetic structure
To assess overall population structure, two approaches were used: a model-based Bayesian clustering algorithm implemented in STRUCTURE version 2.3.4 (Pritchard et al. 2000), and a non-model-based discriminant analysis of principal components (DAPC) (Jombart et al. 2010). STRUCTURE was used to determine the most probable number of populations and to assign individuals to their most likely population of origin. STRUCTURE was run with a model assuming admixture, without any prior population information and with correlated allele frequencies. We used a burn-in period of 100,000 followed by 700,000 repetitions of Markov chain Monte Carlo (MCMC). All the runs were replicated ten times for K = 1–10. The optimum K was identified using the Evanno method (ΔK) (Evanno et al. 2005) as implemented in STRUCTURE HARVESTER (Earl 2012) and by evaluating the log likelihood of the K (Ln Pr(X|K)) curve. The DAPC was performed using the adegenet version 3.1.9, an R package dedicated to the multivariate analysis of genetic markers (Jombart 2008). Here, the most likely number of clusters (between 1 and 30) associated with the lowest Bayesian information criterion (BIC) was determined using the find.cluster function in adegenet 3.19. Optimisation α-score analysis determined that seven principal components should be retained for assignment (Fig. Supplementary 1); thus the DAPC was performed using the dapc function in adegenet retaining seven principal components. We determined population differentiation by calculating hierarchical F-statistics across a range of population grouping scenarios in an analysis of molecular variance (AMOVA) framework (Excoffier et al. 1992). The scenario with the highest among-population variation (FST) thus describes the most likely pattern of population differentiation. We also explicitly tested Wright’s (1943) model of isolation by distance (IBD) to determine the role played by geographic distance in shaping the observed genetic structure. This was achieved by a Mantel test (Mantel 1967) between matrices of pairwise FST and geographic distance in ARLEQUIN version 3.5.1.2 (Excoffier and Lischer 2010). Euclidean (straight-line) distances between populations were determined in ArcGIS version 10.5 (Esri®), taking the centre of each sampling site.
Fine-scale genetic structure
Inference of recent migration rates
To estimate the reaction of samango monkeys to recent anthropogenic activity, contemporary levels of gene flow between each population were inferred using BIMr (Bayesian inference of migration rates; Faubet and Gaggiotti 2008). This analysis assumes that sampling took place before migration, permitting migration rates from 0 to 1, and the migration rate matrix generated reflects gene flow over the last generation. For this analysis, MCMC sampling was implemented to determine a posterior estimate of the pairwise migration matrix. Posterior estimates consisted of posterior mean and mode and 95% highest posterior density intervals (HPDIs). BIMr was run with a MCMC of 10 million, a burn-in of 2 million, thinning interval of 10,000 and a sample size of 1000. We performed the analysis using the F-model, as we assume population admixture before the last generation of migration and as it has been shown to improve MCMC convergence when population differentiation is weak. We performed five independent runs with eight repeats each, resulting in a total of 40 migration rate estimates for each pair.
mtDNA sequencing and analysis
To gain insight into the phylogeny of the Soutpansberg population, we sequenced two mitochondrial gene regions (Cyt B and 16S) for four LL tissue samples using primers and protocols from Meyer et al. (2011) and methods presented in Dalton et al. (2015). In addition, we included sequences available from Dalton et al. (2015) for LA (4 samples), Hogsback (7 samples) and MK (5 samples) in our analysis (Fig. 1a, b). Sexes of individuals sampled are detailed in a table in Supplementary 2. Generated chromatograms were viewed and edited in the Chromas program embedded in MEGA6 (Tamura et al. 2013). We placed Cyt B and 16S sequences in a concatenated alignment of 910 nucleotides. MEGA version 6 (Tamura et al. 2011) was used for the construction of the phylogenetic trees. Sequence alignment was conducted using ClustalW, which is incorporated in MEGA. Phylogeny reconstruction was completed using a maximum likelihood (ML) statistical method with a bootstrap of 1000 replications. The ML phylogenetic tree was constructed with inclusion of Cercopithecus a. moloneyi samples from Zambia (JQ256962), Tanzania (JQ256964) and Malawi (JQ256971), C. a. monoides (JQ256963), C. m. bourtourlinii (JQ256959), C. a. albogularis (JQ256956), C. a. albotorquatus_(JQ256969), C. m. opisthosticus (JQ256973), C. m. mitus (JQ256974), C. n. nictitans (JQ256951), C. m. doggetti (JQ256965, JQ256958, JQ256953), C. a. kolbi (JQ256955), C. a. francescae (JQ256970), C. m. heymansi (JQ256967), C. m. kandti (JQ256972, JQ256968) and C. m. stuhlmanni (JQ256957), to the Cyt B/16S concatenated analysis, with A. nigroviridis (NC023965), M. ogouensis (JQ256995), P. papio (NC020009), M. m. lasiotus (KF830702), M. mulatta (AY612638), M. thibetana (EU294187), M. sylvanus (AJ309865), L. albigena (JQ256999), C. atys (JQ256998), M. leucophaeus (JQ257001), T. gelada (JQ257000, FJ785426), P. hamadryas (Y18001), L. aterrimus (KJ434960), P. badius (DQ355301, EU004482) and C. guereza (EU004483) as outgroups.
A recent study on samango monkeys assessed the hypervariable 1 region (HVR1) of the mitochondrial genome and microsatellite genotypes of samples collected from two localities within the Hogsback area in the Eastern Cape Province, South Africa (Madisha et al. 2017). The authors identified population structure with the mitochondrial data, but not with the microsatellite data, indicating male-mediated gene flow between the two localities. Interestingly, three male immigrants were identified based on the mismatch of their mitochondrial lineage with their sampling location (Madisha et al. 2017). To determine whether any male immigrants (i.e., dispersers) were sampled in our study that could indicate potential male-mediated gene flow, concatenated Cyt B and 16S mitochondrial sequences were analysed for three populations, namely LA, MK and LL, using DnaSP V. 6.12.01 (Librado and Rozas 2009) to identify haplotypes and PopART version 1.7 (Leigh and Bryant 2015) to generate a minimum spanning network.
Results
Genetic diversity and relatedness
MICRO-CHECKER detected null alleles for one locus (D4S243) in the LA population, one locus (D13S765) in the SB population, two loci (D12S67, D10S1432) in the LL population and one locus (D1S207) in the MK population (Table Supplementary 3). These markers additionally deviated from HWE in the respective populations, most likely due to the presence of null alleles. These markers were retained in the data set, because they did not show null alleles across multiple populations. Evidence of linkage disequilibrium following Bonferroni correction was detected between three marker pairs in the LA population (D9S922 and D3S1768, D9S922 and D10S1432, D3S1768 and D10S1432), and between one marker pair in the ET population (D1S518 and D10S1432). However, there were no consistent patterns of LD between any loci across populations, and thus the loci were retained in the data set (Table Supplementary 4). The probability of identity per population across 15 markers ranged from 1.2E−09 to 3.8E−07, and the genotype accumulation curve plateaued at eight loci, indicating sufficient discriminatory power for individualisation of samango monkey samples. We found no evidence of re-sampling of the same individual, as each individual had a unique profile.
The TrioML estimator had the highest correlation (R = 0.78, p < 2.2E−16) with simulated relatedness values in the data set (Fig. Supplementary 5) and was thus used for all further relatedness analyses. The highest mean relatedness (r) was observed in the SB population (r = 0.135), followed by MK (r = 0.126), ET (r = 0.093), LA (r = 0.084) and LL (r = 0.078) (Fig. Supplementary 6). Two separate groups were sampled in LA, named group B and group H. Thus, group provenance was used to determine whether fine-scale structure existed within this forest, i.e. are individuals within groups more related than expected by chance within the forest patch. We found that individuals were not more related within groups than is expected by chance (group B: p value < 0.417, group H: p value < 0.273). The same analysis was conducted for each sex in the LA population. We found that neither females (p value < 0.137) nor males (p value < 0.848) were more related than expected by chance.
For the five sampling areas, genetic diversity estimates were similar, with the mean number of alleles (NA) and the expected heterozygosity (HE) across loci and populations being 3.37 and 0.53, respectively (Table 2). NA was highest in the LL population (3.54) and lowest in both the SB and ET populations (3.15). Expected heterozygosity was highest in the MK (0.56) population and lowest in the LA (0.48) population. Variability was similar in SB (HE = 0.53) and LL (HE = 0.54) and was second lowest in ET (HE = 0.51). Unbiased expected heterozygosity (uHE) ranged between 0.49 and 0.62. Results show that observed heterozygosity (HO) was lower than HE in all populations sampled (Table 2). Further analysis using a two-tailed pairwise t test (α = 0.025) showed this difference to be significant in the ET population but not in LA, SD, LL or MK. Private alleles were observed in all five populations: eight for ET, six for LL, five for MK, four for LA and one for SB. FIS was highest in the ET population (0.35, P = 0.007), followed by LL (0.16, P = 0.054) and LA (0.13, P = 0.057). The other two populations (SB, MK) showed slightly negative FIS values (Table 2, P = 0.699 and 0.815, respectively).
Genetic structure
Our analyses of population structure using model-based (STRUCTURE) and non-model-based (DAPC) algorithms indicated the existence of genetically distinct units of samango monkeys. The most likely number of populations was identified as K = 3, based on the log likelihood of the data (the curve plateaued at K = 3, which also has the lowest standard deviation) and the deltaK plot, with the grouping of individuals as (1) LA, (2) SB and ET, (3) LL and MK (Fig. 3), although we show results for K = 2–5. However, as the K value reported by STRUCTURE represents the uppermost genetic hierarchical level, it may not be a perfect reflection of true population structure (Waples and Gaggiotti 2006). Thus, we compared STRUCTURE results for K = 2 to K = 5 to the DAPC and AMOVA results.

Top panels a and b show the mean log likelihood Ln P(X|K) and DeltaK as a function of the number of genetic clusters (K) averaged over 10 consecutive STRUCTURE runs for each K (error bars indicate one standard deviation) with (a) probability (−LnPr) of K = 1–10 and b delta K values for real population structures of K = 1–10. Bottom panel c shows Bayesian assignment probabilities for K = 2 to K = 5 of microsatellite genotypes. Each individual is represented by a single vertical bar, with lengths proportional to the estimated membership in each cluster. LA Lajuma, SB Schoemansdal/Buzzard Mountain, ET Entabeni/Thathe Vondo, LL Luonde/Levuvhu, MK Magoebaskloof
The number of clusters can be determined using the Bayesian information criterion (BIC). Ideally, the optimal number of clusters corresponds to the lowest BIC. Here, the BIC value decreased to its lowest at three and increased after five; thus the hierarchical population structure inferred using DAPC supports three to five genetic clusters (Fig. 4a and b; Fig. Supplementary 1 and 7). The two main axes of the DAPC analysis explained 99.29% of the total variability among clusters. In both cluster scenarios (STRUCTURE and DAPC), there were a few outlier individuals that were collected in one locality but were placed in a different genetic cluster based on genetic data. Results from the AMOVA analysis across the samango monkey populations demonstrated the highest proportion of variation being observed when populations were separated into five groups, namely LA, SB, LL, ET and MK (Table 3).

a Discriminant analysis of principal components (DAPC) of samango monkeys grouped into five clusters. Each point represents individual; colours and ellipses indicate their assignment to one of the five genetic clusters inferred by DAPC. The bottom left graph inset displays the variance explained by the principal component axes used for the DAPC (in dark grey). The bottom right inset displays in relative magnitude the variance explained by the two discriminant axes plotted (in dark grey). b Population structure of samango monkeys provided by DAPC. Colours represent different assigned clusters, and each individual is represented by a single vertical bar. LA Lajuma, SB Schoemansdal/Buzzard Mountain, ET Entabeni/Thathe Vondo, LL Luonde/Levuvhu, MK Magoebaskloof
Microsatellite-based pairwise FST values and associated P values are indicated in Table 4. As FST is highly sensitive to diversity (alleles per locus), it can potentially pose a problem when comparing populations of varying diversities. Hence, we additionally calculated Jost’s D to estimate genetic differentiation (Table Supplementary 8 and 9). Here, FST and Jost’s D values showed the same patterns for our sampled populations, with moderate to high genetic differentiation observed between all populations (FST = 0.048–0.252), with an average of 0.18642 (P < 0.001). In general, populations that were geographically closer displayed lower pairwise FST values. Exceptions are the ET and LL groups that are geographically close (27.35 km), with a higher FST value (0.134), and the SB and ET groups that are moderately geographically distant (59.36 km) but show a very low FST value (0.074). The straight-line distance from the furthest east to the furthest west population sampled was 90.45 km (LL to ET) and the shortest distance between Soutpansberg and escarpment populations was 91.83 km (LL to MK) (Table 4). The Mantel test showed a significant correlation between genetic distance and geographic distance (r = 0.6744, Z = 106.84, P = 0.002), with 45% (r2 = 0.4548) of genetic difference being explained by distance (Fig. Supplementary 10).
Fine-scale genetic structure
Inference of recent migration rates
MCMC trace plots of each pairwise estimate were checked, and runs with poor MCMC convergence were excluded (Faubet and Gaggiotti 2008). We used pairwise migration estimates of the five best runs to calculate the average migration rate (Robin et al. 2015). Our results show that overall recent migration rates between populations were so low (2.4E−11–5.8E−11) that they can be considered 0 (Table Supplementary 11).
mtDNA phylogeny and haplotypes
The phylogenetic tree we constructed using the maximum likelihood analysis (Fig. Supplementary 12) shows the monophyletic separation of the highly polytypic Cercopithecus nictitans group (including the species C. albogularis and C. mitis) from the various outgroups (A. nigroviridis, M. ogouensis, P. papio, M. m. lasiotus, M. mulatta, M. thibetana, M. sylvanus, L. albigena, C. atys, M. leucophaeus, T. gelada, P. hamadryas, L. aterrimus, P. badius and C. guereza). For the populations in our study area, branching patterns place the MK population as sister group to the Soutpansberg populations (56% bootstrap). The separation of the LL and LA populations within the Soutpansberg is not well supported (24% bootstrap), but individuals are grouping according to their geographic locality. In the haplotype alignment including both sexes, 10 variable positions were identified, and seven haplotypes were detected. The overall haplotype diversity was h = 0.87. A distinct correlation was found between locations and haplotypes. Haplotypes 1 and 2 were detected only in LA, haplotypes 3 and 4 were detected only in MK, and haplotypes 5, 6 and 7 were identified only in LL (Fig. Supplementary 13).
Discussion
Genetic diversity of populations
Genetic diversity across all five sampled populations varied in terms of allelic richness (Ar between 2.32 and 2.02) and heterozygosity (HE between 0.48 and 0.56). For the ET population, we found that the HO was significantly lower than the HE, with a significantly positive inbreeding coefficient of (0.35, P = 0.007). In the wild, a deficiency of observed heterozygotes could be caused by inbreeding between closely related individuals, decreasing population size (genetic drift) or the Wahlund effect (Wahlund 1928), when sampling two structured subpopulations with spatial overlap. Although positive FIS values may indicate inbreeding, they can also be caused by null alleles (Brookfield 1996), unrecognised fine-scale structure (Hartl and Clark 1987) or small sample size. Null alleles were absent in the ET population. Furthermore, ET’s high allelic richness and He values suggest a relatively recent loss of genetic diversity through population bottlenecking (Cornuet and Luikart 1996). The lowest expected heterozygosity was observed in the LA population (HE 0.48) in the far western part of the mountain range and reflects the lower number of alleles in this population compared to others. LA’s observed number of heterozygotes was also low, similar to that of LL, and both with inbreeding coefficients that are approaching significance (FIS LA = 0.13, P = > 0.057; FIS LL = 0.16, P = 0.054). In contrast to the isolation and lower diversity observed in the Soutpansberg, the highest genetic diversity and lowest inbreeding values were observed in the MK population (HE 0.56) from the escarpment south of the Soutpansberg, despite this population having the smallest sample size (n = 6, Table 1). The MK population is situated in the Woodbush-De Hoek forest which is, with 6626 ha, the largest indigenous forest in Limpopo Province and the second largest in all of South Africa (Scheepers 1978; Cooper 1985), likely supporting a much larger, genetically interconnected samango monkey population.
The mean relatedness (r = 0.078–0.135) within each population was low to moderate (Fig. Supplementary 6), indicating that most of the individuals in a population were unrelated. In populations that are stable, with low rates of immigration, high rates of recruitment via births, and transfer of reproductive status between related females, high relatedness is expected. For example, a study on wild common marmosets (Callithrix jacchus) reported within-population relatedness as r = 0.73 to 0.471 (Nievergelt et al. 2000). Low mean relatedness identified in this study may be due to the sampling of two groups per geographic location. However, within most locations, there were several outliers that correspond to first- and second-order relatedness levels (r = 0.25–0.75), such as half-siblings, full-siblings and parent-offspring. This would be expected from a female philopatric, group-living species.
In the LA population, individuals were not more related within groups than could be expected by chance. Given female philopatry in this species, this may be an unexpected result. However, in samango monkeys and closely related blue monkeys (C. mitis) from Kenya, behavioural observations show that neighbouring groups may be “sister-groups” due to group fission (e.g. when the initial group grew too large and subsequently split) (Cords and Rowell 1986; Swart and Lawes 1996). For some female-bonded primate species, genetic studies indicate that relatedness within groups may increase if groups split along genetic lines (e.g. matrilineal lines) (e.g. Olivier et al. 1981; van Horn et al. 2007). However, for blue monkeys it was observed that groups do not always fission cleanly along matrilineal lines (Cords 2012), which may result in less pronounced within-group relatedness (Silk and Kappeler 2017). The two neighbouring groups sampled in LA were a result of a past fission event (Linden pers. obs.) that may not have occurred cleanly along matrilineal lines, given the observed relatedness patterns. If the fission event occurred along matrilineal lines, or if two independent groups (i.e. not sister-groups) were sampled, one would expect a higher likelihood that individuals within groups would be more related than expected by chance. Additionally, if a larger proportion of each group were sampled in the LA population, it may reveal the expected pattern of higher within-group relatedness.
Analysis of two mitochondrial gene regions (Cyt B and 16S) for the LA, LL and MK populations using tissue samples identified separation of individuals according to their geographic locality. However, minimal genetic differences were observed within groups (Fig. Supplementary 12). Shared haplotypes between populations were not identified, not supporting the existence of male immigration between LA, LL and MK, with the latter population separated from others by five mutational changes (Fig. Supplementary 13). This contrasts with the results of Madisha et al. (2017), who identified high levels of gene flow and male immigrants between groups. We suggest the reason for Madisha et al. (2017) finding evidence for male immigration is linked to the comparatively close geographic proximity (~ 1 km) between their study groups and a larger sample size. The three localities in our study were much further apart (> 60 km, Table 4), significantly decreasing the chance of males dispersing between populations and reducing the likelihood of sampling such individuals. It is possible that more comprehensive sampling could detect male immigrants between populations. However, given the structure observed in both the microsatellite and mitochondrial data, we suggest that we did not detect male immigrants due to true lack of dispersal and gene flow.
Non-invasive faecal samples could not be analysed in this study using mitochondrial markers due to co-amplification of prey remains and degradation of DNA due to exposure to environmental factors and/or due to the presence of inhibitors (Taberlet et al. 1999; Snyder-Mackler et al. 2016). A study published by Ang et al. (2020) described a shotgun sequencing method using faecal DNA to obtain mitogenomes, a promising new tool that could increase the possibility of genetic analysis from non-invasive samples. Future studies on samango monkeys in this region could focus on patterns of differentiation using maternally transmitted mtDNA such as HVR1 and paternally transmitted Y–chromosome markers to assess male- and female-mediated gene flow, primarily among neighbouring groups.
Natural fragmentation (paleoclimatic changes, geographic barriers)
Our results indicate that local samango monkey populations were historically more connected across the mountain range and with populations further south than what we see today. This is evident from the STRUCTURE analysis supporting the identification of historic gene flow between MK, LL and ET (K = 3; Fig. 3c). When studying the different ΔKs from the STRUCTURE analysis, it emerges that they follow a pattern of separating populations from east to west, with the far western populations (LA+SB) clustering together, and the eastern populations (LL+ET+MK) clustering together at K = 2. This pattern could be caused by long periods of reduced gene flow, or a more rapid cessation of gene flow at some time in the past. The Last Glacial Maximum (LGM) at 21,000 BP resulted in a substantial contraction of forest cover in southern Africa due to much drier climatic conditions (Deacon 1983). Pollen records and radiocarbon dating of peat deposits in the eastern Soutpansberg suggest that at around 12,000–10,000 years BP, forests were well developed in mountain ravines and under south-facing cliffs and that slopes were largely covered by open vegetation (grassland and fynbos elements) (Scott 1987). From between 10,000 and 6500 years BP the study found a reduction in forest elements and increase in savanna elements, and from 6500 years onwards, more swamp and mesic woodland elements suggest relatively moist conditions. These paleohistorical changes in forest extent and connectivity suggest that over time, gene flow was probably reduced to some extent between samango populations in the Soutpansberg.
Our results show that heterozygosity of populations within the Soutpansberg decreases with increasing distance to the escarpment population (MK) and from east to west (Table 2), with uHE being highest in the MK (0.62) and ET (0.58) populations, moderate in the SB and LL populations (0.56), and lowest in the LA population (0.49). A similar pattern was observed for pairwise fixation indices and geographic distances, with the highest differentiation observed between populations geographically furthest apart (> 100 km), namely between LA and MK (FST = 0.189) and ET and MK (FST = 0.252). Apart from the close relationship between ET and SB (see below under anthropogenic fragmentation), we detected geographic patterns of pairwise FST that were indicative of IBD across the mountain range.
The LA population was the only population sampled occurring west of a major topographic feature, the Sand River gorge (Fig. 1). We could however not find any evidence that this gorge necessarily poses or posed a barrier to samango monkeys, as the pairwise FST value between LA and its closest neighbour SB is low compared to other FST values and, rather, in line with what is expected from isolation by distance (Table 4). Interestingly, the differentiation between LL and ET is higher than expected (FST = 0.134) by geographic distance (27 km), suggesting the involvement of other landscape-related processes such as anthropogenic practices in the matrix surrounding forest fragments.
Anthropogenic fragmentation
Anthropogenic fragmentation may cause a far more rapid cessation of gene flow than natural processes. Samples from LL and MK were the last to be separated into distinct clusters, at K = 5 (Fig. 3c), indicating that these populations were likely more connected historically or until more recently. Regarding the connectivity between the escarpment and the Soutpansberg, old missionary records from this intervening area, the Levuvhu River area (Fig. 1d), show evidence of a once far more extensive lowland forest associated with the river and its tributaries draining out the southern slopes (Gründler 1897), offering a migration corridor for forest fauna. However, due to more recent anthropogenic landscape change this geographic gap between the Soutpansberg and escarpment (Fig. 2) likely poses much more of a barrier today than it did historically. Humans first settled in the mountain range from around 300 AD onwards (Eastwood and Eastwood 2006), and Scotts (1987) showed a sharp decline in tree pollen at around 1500 BP and suggested that this is linked to the burning and clearing of woody vegetation by the first people in the area. The arrival of people of European descent saw the beginnings of commercial silviculture in 1890s and the expansion and commercialisation of agriculture in the early 1900s (Scholes et al. 1995; Tempelhoff 1999). As indicated, anthropogenic land use and land transformation vary markedly between the eastern and western Soutpansberg.
Both the LA and SB populations from the west occur in areas where much of the matrix surrounding forests is composed of natural vegetation (Fig. 2), namely Soutpansberg Mountain Bushveld (Mucina and Rutherford 2006). This woodland/thicket vegetation type is considered novel, having been established (due to cumulative effects of anthropogenic activities) from the 1920s onwards in grassland areas not utilised for silvi- and agriculture (Hahn 2017b). As samango monkeys have been recorded to utilise this vegetation type in daily foraging sorties (Linden et al. 2015), we do not consider it to be a distribution barrier. Here we suggest that geographic distance between suitable forest patches continues to play a main role in the isolation of populations in the western Soutpansberg.
Contrary to this, the LL and ET populations in the eastern part of the Soutpansberg are surrounded by extensive commercial silvi- and agricultural areas (Fig. 2). These two populations showed a higher genetic differentiation than would be expected by geographic distance, and the ET population showed the highest inbreeding index of all studied populations. Although it has been shown that samango monkeys utilise timber plantations to some degree (Droomer 1985; von dem Bussche and van der Zee 1985; Linden et al. in prep), the distance of 27 km between them can apparently not be overcome. Further, when samangos enter the surrounding matrix, their exposure to threats linked to human proximity (e.g. roads, domestic dogs, power lines) rapidly increases, potentially hampering successful dispersal (Linden et al. 2016, 2020).
In addition to anthropogenically driven fragmentation of populations, our results also show evidence for human-mediated connectivity. Across all analysis, the SB population consistently showed results deviating from patterns found for all other populations, and the most plausible explanation for this is the translocation of individuals from ET to SB in the 1980s (John Greaves pers. comm.). STRUCTURE grouped SB and ET at K = 3, and the IBD analysis showed the weakest differentiation between ET and SB (FST = 0.074) despite geographic distance (59 km) and LL being an intervening population between them. The SB population further stood out among the Soutpansberg populations showing no signs of inbreeding (FIS −0.04). Although negative FIS values can be the result of random sampling errors, they may indicate recent outbreeding events such as the translocation of individuals from ET to SB. Given the longevity of samango monkeys (~ 30 years; Cords 2012), we consider it highly unlikely we randomly sampled original individuals translocated 30 years ago, suggesting that individuals successfully integrated into existing groups and reproduced.
Conclusion
This study provides the first analysis of the genetic diversity and structure of the Soutpansberg samango monkey populations and demonstrates the genetic consequences of increased habitat fragmentation and population isolation in a free-ranging African primate. Extensive anthropogenic changes to the landscape in the eastern parts of the Soutpansberg and distance between high-canopy forest patches in the western parts appear to have reduced the suitability of environmental conditions for samango monkeys to disperse between sampling areas. This was evident from analyses of population structure through the DAPC and AMOVA analysis, as overall the most likely current scenario was five distinct populations, corresponding to each sampling area. In addition, the lack of contemporary gene flow inferred from bidirectional migration rates among populations suggests that while historically the Soutpansberg samango monkey population might have been naturally divided into two or potentially three main genetic populations, recent anthropogenically driven fragmentation has resulted in greater isolation, thus driving the inflation of measures of population structure.
The degree of samango monkey population subdivision and the apparent lack of contemporary migration between populations found in our study raises concerns about the long-term viability of populations across the mountain, with eastern populations being of particular concern due to the immense anthropogenic pressure and western populations likely being entirely cut off from the eastern and south-eastern (escarpment) populations, leaving them without any rescue effect possibilities in the future.
References
Ang A, Roesma DI, Nijman V, Meier R, Srivathsan A (2020) Faecal DNA to the rescue: Shotgun sequencing of non-invasive samples reveals two subspecies of Southeast Asian primates to be Critically Endangered species. Sci Rep 10(1):1–16
Bennun L, Dranzoa C, Pomeroy D (1996) The forest birds of Kenya and Uganda. J East Afr Nat Hist 85:23–48
Berliner BB (2005) Systematic conservation planning for the forest biome of South Africa. Approach, methods and results of the selection of the priority forests for conservation action. Technical report prepared for the Water and Forestry Support Programme and the Department of Water Affairs and Forestry (DWAF), South Africa
Brookfield J (1996) A simple new method for estimating null allele frequency from heterozygote deficiency. Mol Ecol 5(3):453–455. https://doi.org/10.1046/j.1365-294X.1996.00098.x
Coleman BT, Hill RA (2014) Living in a landscape of fear: the impact of predation, resource availability and habitat structure on primate range use. Anim Behav 88:165–173. https://doi.org/10.1016/j.anbehav.2013.11.027
Cooper KH (1985) The conservation status of indigenous forests in Transvaal, Natal and O.F.S., South Africa. Report, Wildlife Society of Southern Africa, Durban
Cords M (2001) Friendship among adult female blue monkeys (Cercopithecus mitis). Behaviour 139:291–314
Cords M (2012) The 30-year blues: what we know and don’t know about life history, group size and group fission of blue monkeys in the Kakamega Forest, Kenya. In: Kappeler PM, Watts DP (eds) Long-term field studies of primates. Springer, Berlin, pp 289–312
Cords M, Rowell TE (1986) Group fission in blue monkeys of the Kakamega Forest. Kenya Folia Primatol 46(2):70–82
Cornuet J-M, Luikart G (1996) Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 144:2001–2014
Dalton DL, Linden B, Wimberger K, Nupen L, Tordiffe ASW, Taylor PJ, Madisha MT, Kotze A (2015) New insights into samango monkey speciation in South Africa. PLoS ONE 10(3):e0117003. https://doi.org/10.1371/journal.pone.0117003
Deacon HJ (1983) Another look at the Pleistocene climates of South Africa. S Afr J Sci 79:325–328
Droomer EAP (1985) Volume and value loss owing to samango monkey damage to pine stands in the Northern Transvaal. South Afr for J 134(1):47–51. https://doi.org/10.1080/00382167.1985.9629594
Dudash MR, Fenster CB (2000) Inbreeding and outbreeding depression in fragmented populations. In: Young AG, Clarke GM (eds) Genetics, demography, and viability of fragmented populations. Cambridge University Press, Cambridge, pp 35–53
Earl DA (2012) BM vonHoldt. STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Res 4:359–361. https://doi.org/10.1007/s12686-011-9548-7
Eastwood E, Eastwood C (2006) Capturing the Spoor. An exploration of southern African rock art. David Philip, New Africa Books (Pty) Ltd, Claremont
Eeley HAC, Lawes MJ, Piper SE (1999) The influence of climate change on the distribution of indigenous forest in KwaZulu-Natal. South Africa J Biogeogr 26(3):595–617. https://doi.org/10.1046/j.1365-2699.1999.00307.x
Eggert LS, Patterson G, Malsonado JE (2008) The Knysna elephants: a population study conducted using faecal DNA. Afr J Ecol 46:19–23. https://doi.org/10.1111/j.1365-2028.2007.00794.x
Ekernas LS, Cords M (2007) Social and environmental factors influencing natal dispersal in blue monkeys, Cercopithecus mitis stuhlmanni. Anim Behav 73:1009–1020. https://doi.org/10.1016/j.anbehav.2006.11.007
Estrada A, Garber PA, Rylands AB et al (2017) Impending extinction crisis of the world’s primates: why primates matter. Sci Adv 3(1):e1600946. https://doi.org/10.1126/sciadv.1600946
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620. https://doi.org/10.1111/j.1365-294X.2005.02553.x
Excoffier L, Lischer HEL (2010) Arlequin Suite ver. 3.5, a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567. https://doi.org/10.1111/j.1755-0998.2010.02847.x
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131(2):479–491
FAO (2018) The State of the World’s Forests 2018 - Forest pathways to sustainable development. Licence: CC BY-NC-SA 3.0 IGO, Rome
Faubet P, Gaggiotti OE (2008) A new Bayesian method to identify the environmental factors that influence recent migration. Genetics 178:1491–1504. https://doi.org/10.1534/genetics.107.082560
Gerloff U, Hartung B, Fruth B, Hohmann G, Tautz D (1999) Intracommunity relationships, dispersal pattern and paternity success in a wild living community of Bonobos (Pan paniscus) determined from DNA analysis of faecal samples. Proc Royal Soc B 266:1189–1195. https://doi.org/10.1098/rspb.1999.0762
Gibbons MA, Harcourt AH (2009) Biological correlates of extinction and persistence of primates in small forest fragments: a global analysis. Trop Conserv Sci 2(4):388–403. https://doi.org/10.1177/194008290900200402
Goossens B, Anthony N, Jeffery K, Johnson-Bawe M, Bruford MW (2003) Collection, storage and analysis of non-invasive genetic material in primate biology. In: Setchell JM, Curtis DC (eds) Field and laboratory methods in primatology. Cambridge University Press, Cambridge, pp 295–308
Gründler W (1897) Geschichte der Bawenda Mission in Nord-Transvaal. Berlin no. 43, Buchhandlung der Berliner evangelischen Missionsgesellschaft
Hahn N (2017a) Endemic flora of the Soutpansberg, Blouberg and Makgabeng. S Afr J Bot 113:324–335. https://doi.org/10.1016/j.sajb.2017.09.006
Hahn N (2017b) An historic account of the extinct high rainfall grasslands of the Soutpansberg, South Africa. Trans R Soc South Africa 73(1):20–32. https://doi.org/10.1080/0035919X.2017.1346528
Hartl D, Clark AG (1987) Principles of Population Genetics, 2nd edn. Sinauer Associates, Sunderland, Massachusetts
Henzi SP, Lawes M (1987) Breeding season influxes and the behaviour of adult male samango monkeys (Cercopithecus mitis albogularis). Folia Primatol 48:125–136. https://doi.org/10.1159/000156290
Jombart T (2008) Adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405. https://doi.org/10.1093/bioinformatics/btn129
Jombart T, Devillard S, Balloux F (2010) Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet 11:94. https://doi.org/10.1186/1471-2156-11-94
Kalinowski ST (2005) hp-rare 1.0: a computer program for performing rarefaction on measures of allelic richness. Mol Ecol Notes 5(1):187–189. https://doi.org/10.1111/j.1471-8286.2004.00845.x
Lacy RC (1997) Importance of genetic variation to the viability of mammalian populations. J Mammal 78:320–335. https://doi.org/10.2307/1382885
Lacy RC (2000) Structure of the VORTEX simulation model for population viability analysis. Ecol Bull 48:191–203. https://doi.org/10.2307/20113257
Lawes MJ (1990) The distribution of the Samango Monkey (Cercopithecus mitis erythrarchus Peters, 1852 and Cercopithecus mitis labiatus I. Geoffroy, 1843) and Forest History in Southern Africa. J Biogeogr 17(6):669–680. https://doi.org/10.2307/2845148
Lawes MJ (1992) Estimates of population density and correlates of the status of the samango monkey Cercopithecus mitis in Natal, South Africa. Biol Conserv 60:197–210. https://doi.org/10.1016/0006-3207(92)91252-N
Lawes MJ, Henzi SP (1995) Inter-group encounters in blue monkeys: how territorial must a territorial species be? Anim Behav 49:240–243. https://doi.org/10.1016/0003-3472(95)80173-1
Lawes MJ, Henzi SP, Perrin MR (1990) Diet and feeding behaviour in Samango Monkeys (Cercopithecus mitis labiatus) in Ngoye Forest. South Africa Folia Primatol 54:57–69. https://doi.org/10.1159/000156426
Leigh JW, Bryant D (2015) Popart: full-feature software for haplotype network construction. Methods Ecol Evol 6:1110–1116. https://doi.org/10.1111/2041-210X.12410
Li CC, Weeks DE, Chakravarti A (1993) Similarity of DNA fingerprints due to chance and relatedness. Hum Hered 43:45–52
Librado P, Rozas J (2009) DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25:1451–1452. https://doi.org/10.1093/bioinformatics/btp187
Linden B, Linden JD, Fischer F, Linsenmair KE (2015) Seed dispersal by South Africa’s only forest-dwelling guenon, the samango monkey (Cercopithecus mitis). Afr J Wildl Res 45(1):88–99. https://doi.org/10.3957/056.045.0110
Linden B, Foord S, Horta-Lacueva QJB, Taylor PJ (2020) Bridging the gap: how to design canopy bridges for arboreal guenons to mitigate road collisions. Biol Conserv 246:108560. https://doi.org/10.1016/j.biocon.2020.108560
Linden B, Wimberger K, Ehlers-Smith Y, Child MF (2016) A conservation assessment of Cercopithecus albogularis. In: Child MF, Roxburgh L, Do Linh San E, Raimondo D, Davies-Mostert HT (eds) The Red List of Mammals of South Africa, Swaziland and Lesotho. South African National Biodiversity Institute and Endangered Wildlife Trust, South Africa
Lynch M, Ritland K (1999) Estimation of pairwise relatedness with molecular markers. Genetics 152:1753–1766
Madisha MT, Dalton DL, Jasen R, Kotze A (2017) Genetic assessment of an isolated endemic samango monkey (Cercopithecus albogularis labiatus) population in the Amathole Mountains, Eastern Cape Province. South Africa Primates 59(2):197–207. https://doi.org/10.1007/s10329-017-0636-5
Marsh LK (2003) Primates in fragments. Ecology and conservation. Kluwer Academic/Plenum Publishers, New York
Marsh LK (2013) Primates in fragments. Complexity and resilience. Springer, New York, USA
Maseko MST, Ramesh T, Kalle R, Downs CT (2017) Response of crested Guinea-fowl (Guttera edouardi), a forest specialist, to spatial variation in land use in iSimangaliso Wetland Park, South Africa. J Ornithol 158:469–477
McAlpine CA, Rhodes JR, Callaghan JG, Bowen ME, Lunney D, Mitchell DL, Possingham HP (2006) The importance of forest area and configuration relative to local habitat factors for conserving forest mammals: a case study of koalas in Queensland. Australia Biol Conserv 132:153–165
Meyer D, Rinaldi ID, Ramlee H, Perwitasari-Farajallah D, Hodges JK, Roos C (2011) Mitochondrial phylogeny of leaf monkeys (genus Presbytis, Eschscholtz, 1821) with implications for taxonomy and conservation. Mol Phylogenet Evol 59(2):311–319. https://doi.org/10.1016/j.ympev.2011.02.015
Milligan BG (2003) Maximum-likelihood estimation of relatedness. Genetics 163:1153–1167
Moir M, Richards LR, Rambau RV, Cherry MI (2021) Functional diversity and trait filtering of insectivorous bats relate to forest biogeography and fragmentation in South Africa. J Biogeogr. https://doi.org/10.1111/jbi.14069
Mucina L, Geldenhuys CJ (2006) Afromontane, subtropical and azonal forests, Chapter 12. In: Mucina L, Rutherford MC (eds) The vegetation of South Africa, Lesotho and Swaziland. South African National Biodiversity Institute, Strelitzia 19, Pretoria, pp 584–615
Mucina L, Rutherford MC (Eds) (2006) The vegetation of South Africa, Lesotho and Swaziland. South African Biodiversity Institute, Pretoria
Munyati C, Kabanda TA (2009) Using multitemporal Landsat TM imagery to establish land use pressure induced trends in forest and woodland cover in sections of the Soutpansberg Mountains of Venda region, Limpopo Province. South Africa Reg Environ Change 9:41–56. https://doi.org/10.1007/s10113-008-0066-4
Nievergelt CM, Digby LJ, Ramakrishnan U, Woodruff DS (2000) Genetic analysis of group composition and breeding system in a wild common marmoset (Callithrix jacchus) population. Int J Primatol 21:1–20
Novak K, le Roux A, Richards SA, Scheijen CPJ, Hill RA (2014) Human observers impact habituated samango monkeys’ perceived landscape of fear. Behav Ecol 25(5):1199–1204. https://doi.org/10.1093/beheco/aru110
Oklander LI, Miño CI, Fernândez G, Caputo M, Corach D (2017) Genetic structure in the southernmost populations of black-and-gold howler monkeys (Alouatta caraya) and its conservation implications. PLoS ONE 12(10):e0185867. https://doi.org/10.1371/journal.pone.0185867
Olivier TJ, Ober C, Buettner–Janusch J, Sade DS (1981) Genetic differentiation among matrilines in social groups of rhesus monkeys. Behav Ecol Sociobiol 8:279–285
Peakall R, Smouse PE (2012) GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research - an update. Bioinformatics 28:2537–2539. https://doi.org/10.1093/bioinformatics/bts460
Pew J, Muir PH, Wang J, Frasier TR (2015) related: An R package for analysing pairwise relatedness from codominant molecular markers. Mol Ecol Resour 15:557–561. https://doi.org/10.1111/1755-0998.12323
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959. https://doi.org/10.1111/j.1471-8286.2007.01758.x
Queller DC, Goodnight KF (1989) Estimating relatedness using genetic markers. Evolution 43:258–275. https://doi.org/10.2307/2409206
Robin VV, Gupta P, Thatte P, Ramakrishnan U (2015) Islands within islands: two montane palaeo-endemic birds impacted by recent anthropogenic fragmentation. Mol Ecol 24(14):3572–3584. https://doi.org/10.1111/mec.13266
Scheepers JC (1978) The vegetation of Westfalia-Estate on the north-eastern Transvaal escarpment. Mem Bot Surv South Africa 42:1–230
Scholes RJ, Berns J, Everard DA, Scott D, Van Thienhoven M, Viljoen P (1995) The costs and benefits of forestry, case studies from Mpumalanga. Report FOR-DEA 939, Environmentek, CSIR, Pretoria
Scott L (1987) Late quaternary forest history in Venda, southern Africa. Rev Palaeobot Palynol 53:1–10. https://doi.org/10.1016/0034-6667(87)90008-X
Seufert V, Linden B, Fischer F (2009) Revealing secondary seed removers: results from camera trapping. Afr J Ecol 48:914–922. https://doi.org/10.1111/j.1365-2028.2009.01192.x
Silk JB, Kappeler PM (2017) Sociality in primates. In: Rubenstein DR, Abbot P (eds) Comparative social evolution. Cambridge University Press, Cambridge, pp 253–383
Snyder-Mackler N, Majoros WH, Yuan ML, Shaver AO, Gordon JB, Kopp GH, Schlebusch SA, Wall JD, Alberts SC, Mukherjee S, Zhou X (2016) Efficient genome-wide sequencing and low-coverage pedigree analysis from noninvasively collected samples. Genetics 203:699–714. https://doi.org/10.1534/genetics.116.187492
Swart J, Lawes MJ (1996) The effect of habitat patch connectivity on samango monkey (Cercopithecus mitis) metapopulation persistence. Ecol Model 93:57–74. https://doi.org/10.1016/0304-3800(95)00211-1
Swart J, Lawes MJ, Perrin MR (1993) A mathematical model to investigate the demographic viability of low-density samango monkey (Cercopithecus mitis) populations in Natal, South Africa. Ecol Model 70:289–303. https://doi.org/10.1016/0304-3800(93)90062-W
Taberlet P, Waits LP, Luikart G (1999) Noninvasive genetic sampling: look before you leap. Trends Ecol Evol 14:323–327. https://doi.org/10.1016/S0169-5347(99)01637-7
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28:2731–2739. https://doi.org/10.1093/molbev/msr121
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 60. Mol Biol Evol 30(12):2725–2729. https://doi.org/10.1093/molbev/mst197
Tempelhoff JWN (1999) Townspeople of the Soutpansberg. A centenary history (1899–1999). Greater Louis Trichardt Transitional Council, Louis Trichardt
Van Horn RC, Buchan JC, Altmann J, Alberts SC (2007) Divided destinies: group choice by female savannah baboons during social group fission. Behav Ecol Sociobiol 61(12):1823–1837. https://doi.org/10.1007/s00265-007-0415-1
Van Oosterhout C, Hutchinson WF, Wills DP, Shipley P (2004) MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4(3):535–538. https://doi.org/10.1111/j.1471-8286.2004.00684.x
von dem Bussche GH, Van der Zee D (1985) Damage by Samango monkeys, Cercopithecus (mitis) albogularis, to pine trees in the Northern Transvaal. South Afr for J 134:47–51. https://doi.org/10.1080/00382167.1985.9629573
Wahlund S (1928) Zusammensetzung von Populationen und Korelationserscheinungen vom Standpunkt der Vererbungslehre aus betrachtet. Hereditas 11:175–195
Wang J (2002) An estimator for pairwise relatedness using molecular markers. Genetics 160:1203–1215
Wang J (2007) Triadic IBD coefficients and applications to estimating pairwise relatedness. Genet Res 89:135–153. https://doi.org/10.1017/S0016672307008798
Waples RS, Gaggiotti O (2006) Invited review: What is a population? An empirical evaluation of some genetic methods for identifying the number of gene pools and their degree of connectivity. Mol Ecol 15:1419–1439
Wasser S, Houston C, Koehler G, Cadd G, Fain S (1997) Techniques for application of fecal DNA methods to field studies of Ursids. Mol Ecol 6:1091–1097. https://doi.org/10.1046/j.1365-294x.1997.00281.x
Wimberger K, Novak K, Hill RA (2017) Reliance on exotic plants by two groups of threatened samango monkeys, Cercopithecus albogularis labiatus, at their southern range limit. Int J Primatol 38(2):151–171. https://doi.org/10.1007/s10764-016-9949-2
Wright S (1943) Isolation by distance. Genetics 28(2):114–138
Acknowledgements
We would like to thank members of the public who reported samango carcasses (road kill or due to predation) allowing for tissue sample collection (Caswell Munyai, Koos Steyn, Irene Barnhoorn, Colin Schoeman, Stefan Foord, Mark Harman, Jacques Bouwer). We further thank private landowners, local communities and Komatiland for granting access to properties for faecal sample collection, Jabu Linden for field assistance and input on the manuscript and Norbert Hahn for valuable discussions and loan of historical publications.
Funding
Birthe Linden acknowledges the University of Venda for the PhD grant. Desiré L. Dalton acknowledges the South African National Biodiversity Institute for organisational core grant funding, and Peter J. Taylor acknowledges funding of the Department of Science and Technology (DST) and the National Research Foundation (NRF) through the South African Research Chairs Initiative (SARChI) Chair on Biodiversity Value and Change, hosted at University of Venda and co-hosted by the Centre for Invasion Biology at University of Stellenbosch (NRF Grant number 87311).
Author information
Authors and Affiliations
Contributions
Conceptualisation: BL, PJT, DLD; Methodology: BL, PJT, DLD; Formal analysis and investigation: BL, DLD, YM, DdJ, AVW; Writing—original draft preparation: BL; Writing—review and editing: PJT, DLD, YM, AVW, DdJ; Funding acquisition: BL, PJT, DLD; Resources: BL, PJT, DLD; Supervision: PJT, DLD.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflicts of interest to declare that are relevant to the content of this article.
Ethical approval
Relevant permits for non-invasive collection of faecal samples, opportunistic collection of tissue samples from roadkilled samango monkeys and sample storage were acquired from the Limpopo Provincial Government (Republic of South Africa), Department of Economic Development, Environment and Tourism (LEDET) (Permit Code 0089-MKT001-00002) and the Department of Environmental Affairs (DEA) (Republic of South Africa) (Standing Permit No. S07507). For samples collected on private or communal land, landowner permission was acquired. For samples collected in national forests relevant permits were sourced (Komatiland Forests, License Number: 19017 and License Number: 19023).
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
About this article

Cite this article
Linden, B., Dalton, D.L., Van Wyk, A. et al. Potential drivers of samango monkey (Cercopithecus albogularis) population subdivision in a highly fragmented mountain landscape in northern South Africa. Primates 63, 245–260 (2022). https://doi.org/10.1007/s10329-022-00981-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10329-022-00981-7