
Abstract
The microenvironment of a tumor has emerged recently as a critical contributor to the development of cancer. Within this environment, fibroblasts and immune cells are the cell lineages that seem to be active mediators of tumour development. The activated fibroblasts that are also present during wound healing and chronic inflammation have been studied extensively. Their activation leads to altered gene expression profiles that markedly increase growth factor and cytokine secretion, leading to major alterations in the immune cell microenvironment. To better understand normal tissue development, wound healing and the chronic inflammation that leads to cancer, we review here information available on the role of fibroblasts and immune cells in normal breast development and in cancer. We also discuss the immunogenicity of breast cancer compared to other cancers and the contribution of the immune microenvironment to the initiation, progression and metastasis of tumors. Also reviewed is the limited knowledge on the role of immune cells and fibroblasts in normal development and whether the risk of cancer increases when their control is not tightly regulated.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
While cancer research historically has focused on the intrinsic properties of tumor cells, more recently the microenvironment within which tumor cells are located has emerged as a critical component of cancer development and growth. This microenvironment includes fibroblasts and immune cells, components of the soil in which tumors develop. The six hallmarks of cancer, proposed by Hanahan and Weinberg in 2000 [1], provided an organizing principle with which to understand tumorigenesis. This was updated in 2011 to include these microenvironmental factors, highlighting their significance [2]. This review will discuss fibroblasts and immune cells in both normal development and in pathological situations such as wound healing, chronic tissue damage and cancer. We will highlight the inter-relationship between these cell types and discuss how an understanding of their unique roles may aid research focused on enhancing immune surveillance and inhibiting tumor development.
Involvement of Fibroblasts and Immune Cells in Normal Mammary Gland Development
Mammary Gland Development
At birth, the mammary gland consists of only rudimentary ducts running from the nipple into the fat pad. At 3-4 weeks of age, ovarian hormones begin to stimulate mammary gland growth via epithelial extension and branching into the fat pad [3]. Small bulb-like structures called terminal end buds (TEBs) form at the end of each duct and undergo high levels of proliferation, promoting invasion through the fat pad to give rise to new ducts [4]. During pregnancy, the mammary gland undergoes extensive structural and hormonal changes to allow subsequent secretion of milk to the newborn during lactation [3]. Upon the onset of pregnancy, an increase in progesterone and prolactin stimulates extensive proliferation of epithelial cells to form tertiary side-branches and immature alveoli. By mid-pregnancy, these developing alveoli have polarized, forming individual spherical alveoli connected to the ductal network, capable of synthesizing and secreting milk after parturition [5]. Involution of the mammary gland is a highly complex process, in which the lactating gland undergoes apoptosis and tissue remodeling to revert the gland back to its pre-pregnant, non-lactating state. Following weaning, the first 48 hours of involution are reversible (Stage 1), characterized by the induction of cell death and limited extracellular matrix (ECM) proteolysis. The next stage is irreversible, characterized by the collapse of alveolar structures, differentiation of adipocytes, extensive ECM proteolysis and loss of ability to produce milk [6,7].
Involvement of Fibroblasts in Development
Fibroblasts are thought to arise from a number of cellular and tissue origins, including from primary mesenchymal cells such as pericytes and smooth muscle cells, and from bone marrow derived precursors such as mesenchymal stem cells and fibrocytes [8,9]. They are the principal component of connective tissue and are responsible for the synthesis and removal of the ECM via the expression of collagens, fibronectin, laminins, elastins, proteoglycans, integrins, matrix metalloproteinases (MMPs), tissue inhibitors of matrix metalloproteinases (TIMPs) and a host of other ECM proteins. Fibroblasts are important for the regulation and maintenance of tissue homeostasis, synthesizing and remodeling the ECM, promoting maturation of epithelial cells and facilitating contraction of granulation tissue during wound healing [10,11]. Under normal conditions, fibroblasts exist in a relatively quiescent state, proliferating slowly and synthesizing only low levels of ECM proteins and MMPs to maintain ECM integrity. After tissue injury, mechanical tension at the wound site stimulates the differentiation of resident and recruited normal fibroblasts (NFs) into proto-myofibroblast intermediates, then active myofibroblasts.
During puberty, mammary fibroblasts surrounding the branching TEBs become activated in response to estrogen and growth hormones released by the ovary and pituitary gland, respectively. They secrete fibroblast growth factors (FGFs) that stimulate TEBs to promote luminal epithelial cell expansion and ductal branching and their differentiation into myoepithelial cells [12]. Other growth factors secreted by the microenvironment, such as epidermal growth factor (EGF) and growth hormone (GH), to which the fibroblasts respond, are critical for correct mammary gland morphogenesis and ductal branching during puberty [13,14]. While little is known about the role of fibroblasts in pregnancy, signaling through the fibroblast growth factor receptor (FGFR2-IIIb) is essential to stimulate normal lobuloalveolar development during pregnancy [15]. Fibroblasts in the involuting mammary gland contain elevated MMPs, fibronectin and laminins, as well as higher levels of fibrillar collagens [16,17], all of which contribute to ECM/basement membrane breakdown and tissue remodeling during involution.
Contribution of the Immune System to Mammary Gland Development
Immune cells have been documented to supply soluble growth and survival factors, matrix remodelling enzymes, reactive oxygen species, and other bioactive molecules that influence cancer cell proliferation, angiogenesis, invasion, and metastasis [18–20]. Immune cells respond to pathogens such as invading viruses or bacteria by producing chemokines that recruit specialized defender cells to the site of injury to phagocytose the intruders. This first line of defense is known as inflammation and is a protective response involving host cells, blood vessels and proteins that work to eliminate the initial cause of cell injury, remove the necrotic cells and initiate repair. Inflammation can be either acute or chronic in nature, distinguished by the duration or type of infiltrating inflammatory cells present. Acute inflammation is rapid and involves predominantly innate immune cells such as neutrophils, whereas chronic inflammation occurs over an extended time (days to years) and involves both innate and adaptive immune cells, mainly macrophages and lymphocytes. The immune infiltrate within the mammary gland during development and reproduction has not been described in detail, however it is clear that immune cells have an active role in the growth of the gland during puberty, pregnancy and lactation, and perhaps most importantly, during mammary gland involution, where controlled cell death and tissue remodeling are required to return the breast to its pre-pregnant state.
Macrophages are localized to the neck region of TEBs during puberty [21]. Their recruitment is a result of colony stimulating factor 1 (CSF-1) secretion by mammary myoepithelial cells. CSF-1 is a major regulator of macrophage survival, proliferation, differentiation and recruitment [22] and hence, CSF-1 deficient (Csf-1 op/op) mice exhibit a severe macrophage deficiency in many tissues. The Csf-1op/op mice revealed a role for macrophages in several stages of mammary development. These mice exhibit an atrophic mammary gland ductal system during puberty with a reduced number of TEBs and reduced ductal length and branching [23]. In pregnancy, they have reduced secondary and tertiary branching, with premature alveolar growth and milk protein expression. There are also lactation defects, including an accumulation within the alveolar cells of milk proteins that are neither secreted nor cleared from the ducts [24]. Macrophages are also important for mammary stem cell function, as Csf-1 op/op mice have a reduced stem cell frequency and activity, indicating that macrophages might also facilitate the maintenance and/or proliferation of mammary stem cells during puberty [25]. In addition to this, acute macrophage depletion using the CD11b–diphtheria toxin receptor (DTR) transgenic mice, showed that macrophages are involved in both alveolar bud formation as well as their breakdown [26]. Moreover, acute macrophage depletion in transgenic mice carrying a suicide gene driven by the macrophage specific CSF-1 receptor promoter, revealed that macrophages are also important for the involution process [27].
During puberty, the mammary gland has elevated levels of mast cells within the stroma surrounding TEBs. Functional studies of mammary gland mast cells indicate that they contribute to mammary gland development by releasing growth factors such as VEGF, and dipeptidyl peptidase I that assists in mast cell degranulation [28,29]. Mice deficient in mast cells have defective mammary gland branching during puberty [28]. Mast cells also regulate post-lactational involution through their binding of plasma kallikrein [30].
Eosinophil infiltration also occurs during puberty, due to the increased secretion of the chemokine eotaxin by TEB epithelial cells. Eosinophil knock out mouse studies have highlighted the role of infiltrating eosinophils in driving ductal elongation and branching during mammary gland development [31,32]. In addition, eosinophil knock out mice (Il-5 -/-) were unable to secrete sufficient milk for their pups, indicating a lactation defect [32].
The role of T and B lymphocytes in mammary development and pregnancy is less well explored. In the non-pregnant state, the gland is thought to contain only low lymphoid numbers, however during pregnancy and lactation the mammary gland is colonized by T-cells and B cells, respectively [33]. A more recent histological study has shown that in both normal human breast sections and in those with lobulitis/mastitis, there is a consistent presence of CD8+ lymphocytes and CD11c+ dendritic cells (DCs) [34]. The exact role of these immune cells in the steady state is unclear.
Considerably more is known about the immune cell infiltrate during involution, compared to other developmental or reproductive states, largely due to detailed gene expression profiling of the mouse mammary gland during development, with a specific focus on involution. By utilization of Affymetrix RNA microarrays on mammary glands from Balb/c mice, Stein [35] identified two neutrophil associated genes (Cxcl1 and Lrg that were highly up-regulated on the first day of involution, indicating that neutrophils are infiltrating the mammary gland immediately upon initiation of involution. In addition, macrophage and lymphocyte recruitment and differentiation-related genes (Cxcl14, Cd68, Ctss, Mps1 and Lgals3) began to increase on the third and fourth days of involution, associated with infiltration of macrophages and lymphocytes [35]. These immune cells are thought to play a pivotal role in mammary gland involution by assisting in the removal of milk proteins and the large numbers of apoptotic cells to allow the mammary gland to return to its pre-pregnant state. Pathway analysis showed that the immune profile was akin to an acute inflammatory response [36]. In addition, the levels of plasma cells and eosinophils increase during mammary gland involution, however their specific role in this process is not yet known [37,38].
Direct interaction between fibroblasts and immune cells is predicted, but little has been reported. One of the main functions of fibroblasts is to secrete ECM that acts to support normal development. Immune cells such as macrophages, mast cells and eosinophils are in high numbers in the developing pubertal mammary gland where matrix deposition and remodelling occurs to facilitate ductal growth. The macrophages present during involution have been well studied and are polarized to a tissue remodeling state, and their presence is associated with collagen deposition and matrix remodeling [39,40]. Further studies that characterise the immune cells present at each developmental stage and also the specific collagens and MMPs present at this time, will allow us to determine the depth of the stromal influence over immune cell activity during normal development. As discussed later, more is known about the stromal influence within cancer and other cases of chronic inflammation.
Contribution of Fibroblasts and Immune Cells to Chronic Inflammation and Cancer
The Role of Fibroblasts in Tumor Development
Activated fibroblasts, characterized by their high expression of alpha smooth muscle actin, are present in large numbers in malignant breast cancers and their presence is correlated with poor clinical outcome [41]. A recent study by Lisanti and colleagues indicated that paracrine oxidative stress caused by oncogenes are most likely responsible for both the formation and accumulation of these activated cancer-associated fibroblasts (CAFs) [42]. CAFs contribute to cancer cell survival and progression by secreting high levels of nutrient rich ECM, promoting persistent chronic inflammation within the tumor microenvironment and inducing epithelial-mesenchymal transition of tumor cells. One of the critical challenges a tumor faces as it expands is to generate sufficient energy to meet the ever-increasing demands of continued tumor growth. To overcome this, tumor cells induce the functional activation of CAFs by promoting oxidative stress through their upregulation of transforming growth factor beta (TGF-β) signaling [43,44]. As a result, these catabolic CAFs provide tumor cells with excess nutrients, secreting essential molecules such as L-lactate, ketones, glutamine, fatty acids and amino acids. These fuels are taken up by the nearby anabolic tumor cells and converted to energy through the tricarboxylic acid cycle and oxidative phosphorylation, ultimately sustaining tumor cell survival and promoting tumor cell proliferation [45]. This metabolic coupling is an important component of tumor development, since tumor cells would struggle to survive in their hypoxic environment without this support.
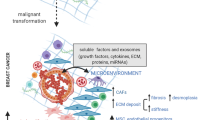
As well as supplying metabolic support, the tumor microenvironment also has an indispensable role in promoting chronic inflammation at the tumor site. Such chronic inflammation triggers the release of pro-inflammatory cytokines that disrupt the normal cytokine balance and promote tumor cell growth through stimulation of angiogenesis and lymphangiogenesis, while also inhibiting activation of cytotoxic immune cells. A recent article proposed that constitutively active CAFs are responsible for this switch from acute resolving inflammation, which occurs during a natural wound healing response, to persistent chronic inflammation in the presence of a tumor [46]. CAFs continually secrete high levels of pro-inflammatory cytokines, such as IL-1β, IL-8, IL-10, tumor necrosis factor-alpha (TNFα), monocyte chemoattractant protein-1 (CCL2), stromal derived factor-1 (CXCL12) and interferon-beta (IFNβ) that have a range of effects on the immune system [46,47] (Fig. 1). For example, secretion of CCL2 promotes the chemotactic recruitment of monocytes and T lymphocytes to the tumor site, ultimately modulating the release of anti-inflammatory cytokines as well as inhibiting the formation of tumor-reactive T cells [48]. Together, the release of these cytokines promotes the generation of a Th2 pro-inflammatory immune response [49,50].

Cancer Associated Fibroblasts (CAFs) and the chronic persistence of inflammation. CAFs mediate the persistent chronic inflammation in the tumour microenvironment via their production of growth factors, cytokines and chemokines. The CAFs stimulate the recruitment and activation of monocytes, macrophages and lymphocytes. Specifically T cells become Th2 rather than Th1 polarized and the macrophages become M2 rather than M1 polarized, both of which are pro-tumorigenic. The CAFs also express Vegf that leads to the accumulation of MDSC and Treg cells. Fibroblast activated protein blocks the T cell effector functions, including CD4+ and CD8+ T cells. Another growth factor released by CAFs, TGFB, is able to inhibit the cytotoxic effects of CD8+ T cells and NK cells, and also control the ECM production and degradation through production of collagenases, proteases, collagens, fibronectin, chondroitin dermatan sulfate and proteoglycans. Below each of the immune cells we have depicted with a positive or negative symbol whether they stimulate or inhibit tumorigenesis.
The Contribution of Fibroblasts to Chronic Inflammation
In addition to their role in stimulating tumor development, fibroblasts have been implicated in the pathogenesis of chronic inflammatory diseases, such as rheumatoid arthritis and chronic obstructive pulmonary disease (COPD) [51–53]. It has been suggested that fibroblasts mediate the transition from acute to chronic inflammation by inappropriately providing recruitment, survival and retention signals to infiltrating leukocytes, thus inhibiting the normal resolution of inflammation [46,54].
COPD is characterized by a chronic obstruction of expiratory flow affecting peripheral airways, associated with chronic bronchitis and emphysema, which includes mucus hypersecretion and goblet cell and submucosal gland hyperplasia as well as the destruction of airway parenchyma. Both occur along with fibrosis, tissue damage and inflammation of the small airways. Fibroblasts isolated from lungs of patients with COPD secrete increased levels of pro-inflammatory factors compared with normal fibroblasts [55].
Rheumatoid arthritis (RA) is a chronic autoimmune disease of unknown origin that primarily affects the joints and ultimately leads to their destruction. Activated fibroblasts in RA produce a variety of cytokines, chemokines and matrix-degrading enzymes that regulate the inflammatory and endothelial cells known to be responsible for the progressive destruction of articular cartilage and bone. The cytokines and chemokines produced within the rheumatoid synovium recruit T cells, macrophages and neutrophils that, in turn, attract more inflammatory cells and ultimately enhance the activated state of the rheumatoid arthritic synovial fibroblasts and osteoclasts [56].
Periodontal diseases range from simple gum inflammation to serious diseases that result in major damage to the soft tissue and bone supporting the teeth. Bacteria induce tissue destruction indirectly by activating host defense cells that produce and release mediators that stimulate the effectors of connective tissue breakdown [57]. T cells activated by the bacteria produce pro-inflammatory cytokines that activate fibroblasts and macrophages to produce enzymes that degrade the ECM, leading to tissue destruction. The activation of T cells also facilitates bone resorption [58].
Benign prostatic hyperplasia (BPH), or non-cancerous enlargement of the prostate, is a common condition associated with aging. It is characterized by the proliferation of fibroblasts/myofibroblasts and epithelial cells within the peri-urethral or transitional zone of the prostate gland. The role of fibroblasts in BPH has been highlighted by studies showing an age-related loss of the anti-proliferative effect of prostate fibroblasts on prostate epithelial cells [59]. Prostate fibroblasts in BPH secrete cytokines and chemokines that foster an inflammatory proliferative microenvironment [60]. The transcriptome of the aging prostate stroma is characterized by the up-regulation of several genes that encode secreted inflammatory mediators, including CXC-type chemokines (CXCL1, CXCL2, CXCL5, CXCL6, CXCL12), interleukins (IL-11, IL-33), and transcripts with cytokine homology (CYTL1). The majority of these cytokines and chemokines are pro-angiogenic, and chemotactic factors for neutrophils. Activated neutrophils and macrophages secrete cytokines, such as IL-1, IL-6, and IL-8 that are chemotactic for various types of lymphocytes involved in the chronic inflammatory response [61,62].
Immune Cells and Cancer
The interplay between the tumor and the immune system during tumor progression is called immunoediting, involving immune-mediated tumor cell cytotoxicity, immune cell and tumor cell equilibrium, and the development of immunologically undetectable tumour cell variants. Immunoediting comprises three phases: elimination, equilibrium and escape [63,64], but here we will also discuss the initial phase of transformation. The earliest stage of tumor development is the transformation of normal somatic cells. Tumor cells arise due to mutations in either proto-oncogenes or tumor suppressor genes, ultimately leading to the disruption of intrinsic or extrinsic mechanisms regulating cell growth and proliferation [65]. A small proportion of tumor cells may survive the initial phase of immunoediting, the elimination phase, allowing them to enter a state of dynamic equilibrium where they are continually subjected to potent and selective pressure from lymphocytes and IFNγ [66]. This selective pressure is predicted to maintain but not eliminate all tumor cells [67]. In the escape phase of immunoediting, new genetic variants of tumor cells are able to proliferate without recognition by the immune system, resulting in expansion of the tumor to a clinically detectable size. The role of immune cells in cancer has been subjected to many studies. Innate immune cells such as macrophages are enriched in the premalignant and malignant setting and contribute to tumor formation by regulating the ECM, angiogenesis and proliferation. Immune cells supply both direct and indirect mitogenic growth mediators that stimulate proliferation of neoplastic cells and other stromal cells [68]. They also express diverse classes of proteolytic enzymes that cleave and modify the structure of the ECM [69].
Historically breast cancer was not considered to be immunogenic, as the incidence of breast cancer is not increased in transplant patients who are immune suppressed. In contrast, non-melanoma skin cancers, lip cancer, Kaposi sarcoma, non-Hodgkin’s lymphoma and cancers of the anogenital tract are increased [70]. Similarly, patients with HIV have an increased risk of the same cancers [71]. Together these data indicated that breast cancer incidence was not altered in the absence of a functional immune system. However there are now irrefutable data demonstrating that the immune cell infiltrate of a breast tumor affects both its growth and metastasis. The data for a role of the immune system in breast cancer initiation are sparse, however limited studies indicate that even at these early stages of transformation, immune cells may be involved. Emerging research indicates that some breast cancer subtypes may be more immunogenic than others. Lymphocytic infiltration is associated with good prognosis in rapidly dividing tumors and triple negative tumors [72,73], but not in other subtypes. This may be due to the high genomic instability and poor differentiation of these tumors.
Immune Cell Involvement in the Early Stages of Breast Cancer Development
As mentioned above, limited data on the role of immune cells in the very early stages of breast cancer are available. A large global analysis of immune cells in 53 mastectomy samples ranging from normal to invasive ductal carcinoma has been reported [74]. The number of CD20+ B cells, CD68+ monocytes and macrophages, CD3+ T cells and granzyme B+ cytotoxic T cells was assessed. The most significant finding was a 30-fold increase in CD3+ T cells during the transition from normal to benign stage of disease. This increase was sustained during cancer development and progression. There were also smaller increases in CD20+ B cells and CD68+ macrophages [74] Together these data indicate that both the innate and adaptive immune systems are important in the initiation of breast cancer. This research is supported by more recent data showing a significant increase in leukocyte infiltration with tumor progression. Normal breast had minimal leukocyte infiltration, whilst ductal carcinoma in-situ (DCIS) and invasive ductal carcinoma (IDC) showed a higher percentage (28 % and 44 % respectively) of strong staining [75].
Immune Cell Involvement in Established Cancers
Many studies have correlated prognosis with the influx of immune cells into primary breast cancers. The levels of tumor associated macrophages (TAMs), CD4+ T helper cells and CD8+ cytotoxic T cells have been shown to predict breast cancer outcome. Patients whose tumors had a CD68hi, CD4hi and CD8low immune profile (rather than CD68low CD4low and CD8high) had decreased overall survival and relapse-free survival [76]. This immune profile also held true when assessed in two independent tissue cohorts [77,78] and agrees with earlier work in predicting disease-free survival by measuring the ratio of CD4+ T cells to CD8+ T cells [79].
The orientation of immune cell subsets dictates their pro-tumor versus anti-tumor activity. TAM infiltration is associated with worse prognosis in many cancers [80,55]. For example, IL-10, TGFβ and other cytokines from the tumor microenvironment switch macrophages from an M1-like (pro-inflammatory or classically activated) macrophages to M2-like (anti-inflammatory or alternatively activated) TAMs. M2-like TAMs support tumor growth through promotion of angiogenesis, matrix remodeling and metastasis, and by suppression of adaptive immunity [81,82]. There are also a number of subtypes within the T helper cell population, including Th1, Th2, Th9, Th17, Th22 and ThFH cells, that differ in their functions, signature cytokine profile and cell targets [83]. T helper cells present during acute inflammation are primarily Th1 polarized, enhancing anti-tumor responses by secreting cytokines such as IFNγ, TNFα and IL-2 [79]. These cytokines promote antigen processing in the proteasome, induce major histocompatibility complex (MHC) expression, stimulate activation of macrophages and increase antigen display by tumor cells. In contrast, T helper cells present during chronic inflammation and cancer are predominantly Th2 polarized, assisting in tumor growth and progression by expressing IL-4, IL-5, IL-6, IL-10 and IL-13 that can inhibit T cell mediated cytotoxicity and enhance chronic B cell immunity [79].
Generally the presence of increased numbers of cytotoxic T cells in a tumor is associated with better prognosis [79,84]. Under the influence of immunosuppressive cytokines however, the levels of cytotoxic T cell numbers are reduced and anti-tumor Th1 T helper cells are converted to tumor-tolerant Th2 T helper cells [85].
DCs express MHC Class II and can present their antigenic peptides to CD4+ T cells. They are the most professional of the antigen presenting cells (APCs), since they can quickly internalize tumor antigens, degrade them into peptides and present them on both MHC class I and II molecules to CD4+ and CD8+ T cells, respectively. They essentially prime tumor specific effector T cells to attack the tumor. DCs are found in most tumors and are thought to play an important role in shaping the host response to the tumor. Interactions between DCs and dying cells are determined by a balance of several (often opposing) molecular interactions that regulate recognition, uptake, processing and ultimately, the presentation of cellular antigens to the immune system. DC maturation and survival are impaired in the immunosuppressive tumor microenvironment. While the percentage of myeloid-derived DCs does not relate to clinical outcome in breast cancer patients, the infiltration of plasmacytoid DCs (pDCs) into primary breast tumors is correlated with poor clinical outcome, thereby indicating that pDCs contribute to breast cancer progression [86].
Turning to consider metastatic breast cancer, the role of the immune system has recently been reviewed extensively [87]. Suffice to say that suppression of metastasis in mice has been shown by two groups to be dependent, at least in part, on CD8+ T cells [88,89]. In humans, breast tumor infiltrating CD8+ T cells predict favorable prognosis [90,91]. The innate immune system also contributes to metastasis, with natural killer (NK) cell deficient SCID mice developing higher levels of breast cancer metastasis [92] [93], as do NOD/SCID/IL2R-/- (NSG) mice [88]. NK cells isolated from patients with advanced breast cancer have impaired function [94].
Macrophages and neutrophils are also major mediators of metastasis. While it was generally predicted that TAMs would promote metastatic disease, inhibition of macrophages by targeting CSF-1/CSF-1R signaling did not impact on either primary tumor growth or metastasis in the MMTV-PyMT transgenic mouse [76], and in our study, blockade of CSF-1R signaling in two mammary transplant models actually increased metastasis without altering primary tumor growth [95]. However, metastasis was reduced in the Csf-1 op/op mouse [96], demonstrating that genetic loss of Csf-1 results in a different response to that of pharmacological inhibition of CSF-1 signaling. In the mammary transplant models, the pharmacological blockade of Csf-1R signaling led to an increase in metastasis-promoting neutrophils that could be overcome by treatment of the mice with a neutralizing antibody targeting the granulocyte-CSF receptor [95].
Are the Fibroblasts Orchestrating the Immune Cell Response During Tumorigenisis?
There are many differences between normal and chronically inflamed or activated fibroblasts, including their ability to influence leukocyte survival, differentiation and accumulation through their expression of pro-inflammatory mediators [97,46,98,99]. The interrelationship between stromal cells and immune cells in cancer is supported by gene expression studies showing that a stromal microenvironment with expression of genes proposed to lead to an anti-tumor immune cell environment can accurately predict tumor prognosis [100]. CAFs manipulate the inflammatory microenvironment through two distinct mechanisms. They have a pro-inflammatory expression profile that recruits macrophages, neutrophils and other stimulatory immune cells (Fig. 1) [101,48,102] and they can suppress tumor detection and rejection by the host immune system by immunoediting [103]. For example, melanoma derived CAFs, but not normal skin stromal cells, can impede NK cell cytotoxicity via cell to cell contact and release of PGE2 [104].
Through IL-4 secretion, CAFs can help to polarize macrophages toward an M2 phenotype [105]. CAF mediated recruitment of T cells is facilitated by secretion of chemokines and cytokines, including CXCL9, CXCL10 and CXCL12/SDF-1α [106]. In addition to promoting the recruitment of T cells into tumors, CAFs sway the balance of tumor promoting lymphocytes such as regulatory T cells (Tregs) and T helper subtypes Th2 and Th17, versus cytotoxic T cells and tumor suppressing Th1 cells (Fig. 1) [107]. CAFs can also recruit and expand Th17 cells in tumors. Th17 is a newly defined T helper cell population that expresses IL-17 [108]. These T helper cells regulate leukocyte recruitment and activation and contribute to the pathogenesis of autoimmune diseases and inflammation. Fibroblasts found in the chronic inflammatory milieu of rheumatoid arthritis promote the stimulation of Treg cells via IL-15 secretion (Fig. 1) [109].
CAFs also produce vascular endothelial growth factor (VEGF) that is known to impact on immune cells. VEGF causes immunosuppression by affecting T cell progenitors and by altering the maturation and function of DCs and APCs. High expression of VEGF in tumors can lead to increased infiltration of Tregs and myeloid derived suppressor cells (MDSCs) (Fig. 1) [110,111]. T cell function can also be inhibited through the expression of the immunosuppressive cytokine TGFβ1 by CAFs. TGFβ1 suppresses the acquisition and function of effector T cells [112], leading to inhibition of the anti-tumor activity of NK cells and CD8+ cytotoxic T cells (Fig. 1).
The role of CAFs was analysed in the K14-HPV16 mouse model of multistep squamous skin carcinogenesis [113,114] where the pre-neoplastic stage is well characterised. During this stage, there is extensive remodelling of the underlying dermal stroma, facilitating both angiogenesis and eventual tumor cell invasion. This extensive stromal remodelling begins at the dysplastic stage and is characterized by a chronic inflammatory response. When stromal cells from normal and dysplastic skin of K14-HPV16 transgenic mice were assessed using genMAPP pathway profiling, inflammatory response and immune cell chemotaxis pathways were implicated [97]. This predominantly pro-inflammatory gene signature consists of two chemokines that are chemoattractants for neutrophils and macrophages (CXCL1 and CXCL2) as well as the pro-inflammatory cytokines IL-1β and IL-6, the latter being implicated in the link between inflammation and cancer [115,116]. This pro-inflammatory gene signature was also present in breast and pancreatic cancers [97], which are characterised by extensive desmoplastic stroma [117–119] and was identified subsequently in inflammation-induced gastric cancer [120]. It was shown that the CAF derived IL-6 caused differentiation of CD14+CD1A− monocytes into macrophages rather than into antigen presenting DC cells that would aid in tumor destruction [121].
A recent study of breast cancer has indicated that CAFs regulate tumor prognosis largely through their ability to modulate the immune microenvironment [100]. Finak assessed 53 primary breast tumors, taking care to measure stromal and cancer cell expression signatures separately. Importantly, it was the differential gene expression from the stroma that was linked to clinical outcome [100]. The 200 most variable genes organised the 53 stromal samples into three clusters. Cluster 1 had a reduced rate of recurrence and longer relapse-free survival compared to Cluster 2 that had an increased rate of recurrence and a shorter relapse free survival, whilst Cluster 3 was mixed [100]. The good outcome cluster was independent of ER, HER2 and lymph node status as well as age, grade and tumor size and the type of therapy. The gene set predominantly expressed in the good outcome cluster was enriched for elements of Th1 immune response including T cell receptor complex (CD8a, CD247, CD3D), MHC class I protein binding and granzyme A and B activity. They also observed elevated levels of CD8a and CD247 positive cells in tumor stroma from individuals from good outcome cluster compared to those in the poor outcome cluster, indicating an increased recruitment of activated T cells and NK cells to tumors in the good outcome group. Instead of the immune cell signature in tumor stroma being associated with good outcome, the poor outcome stroma showed markers of increased hypoxia and angiogenesis and a decrease in chemokines that stimulate NK migration and mediate pro-survival signals in T lymphocytes [100].
Another study by Peng and colleagues took CAFs and NFs from early stage breast cancers and assessed their gene expression profiles. They found that the ATM pathway, a set of cell cycle genes and immune associated signaling were altered. The immune genes that were abnormally expressed in CAFs included those associated with NK cell mediated cytotoxicity, Fc gamma R mediated phagocytosis, antigen processing and presentation, immune network for IgA production, TCR activation (Lck and Fyn tyrosine kinases) and B lymphocyte cell surface molecules [122]. Neither study attempted to characterize the relationship between the CAF and immune profiles any further. They did not use flow cytometry or immunohistochemistry to show the immune cell infiltrate, not did they show in vitro that NFs and/or CAFs could physically attract certain immune cell subsets.
Immunotherapies and Stromal Specific Therapies in Clinical Trials
Immune Based Therapies
As described by the immunoediting hypothesis, tumor cells eventually evade immune recognition and grow unrestricted. This phenomenon has led to the idea of targeting tumor-immune interactions to enhance the anti-tumor immune response. Such immunotherapies can be direct, with a number of monoclonal antibodies specifically targeting tumor interactions with T cells recently being developed, as well as indirect, with recent evidence indicating that some conventional chemotherapeutics have off-target immune effects, both of which lead to enhanced anti-tumor immunity. Some recent examples of both direct and indirect immunotherapies and their possible role in breast cancer treatment are discussed below.
Cytotoxic T lymphocyte antigen 4 (CTLA-4) is a surface receptor present on activated T cells, acting as a negative regulator in the expansion and activity of effector T cells [123]. CTLA-4 expression is up-regulated on a number of cancer cells including melanomas [124], lymphomas [125], breast cancers [126] and prostate cancers [127], resulting in enhanced immune escape of tumor cells. Consequently, anti-CTLA-4 monoclonal antibodies (mAb) have been developed to reverse this T cell inhibition. Two anti-CTLA-4 mAb (ipilimumab and tremelimumab) were originally developed, passing both Phase I/II clinical trials. Phase III clinical trials revealed treatment with 3 mg/kg every 3 weeks for 4 treatments improved the median overall survival of melanoma patients by 3.7 months [128], while treatment of 15 mg/kg tremelimumab every 90 days improved the median overall survival of patients by only 1.9 months [129]. As a result, tremelimumab was dropped from further trials, while ipilimumab was FDA approved for the treatment of advanced melanoma in March 2011. Although originally tested in melanoma and prostate cancer patients, ipilimumab recently began Phase I/II clinical trials for the treatment of breast cancer.
The most recent immunotherapeutic inhibitors to be developed are the anti-PD-1 and anti-PD-L1 mAbs. Tumor cells express PD-L1 and PD-L2 that interact with the PD-1 receptor on the surface of T cells, preventing their activation and cytotoxic activity and resulting in immune evasion. Both PD-1 receptor and PD-L1 ligand mAbs have been developed to block this interaction between tumor cells and T cells, thereby promoting T cell activation and anti-tumor cytotoxicity [130]. These mAbs are currently in Phase II clinical trials for the treatment of over 30 types of cancers, with Phase III trials being planned for non-small cell lung carcinoma, melanoma and renal cell cancer. Of these, lambrolizumab (MK-3475) looks to be the most promising, with a 52 % response rate observed in advanced stage 4 melanoma patients treated with 10 mg/kg every 2 weeks. Of these patients, reduced tumor burden was observed in 77 %, while 10 % of patients had a complete response [131]. Interestingly, cytotoxic chemotherapies such as paclitaxel, etoposide and 5-fluorouracil have been shown to induce PD-L1 expression on breast cancer cells in vitro [132]. Furthermore, Balb/c mice with subcutaneously established 4 T1 tumors treated with anti-PD-1 mAb, and anti–GITR mAb (which targets glucocorticoid-induced tumor necrosis factor receptor family–related protein on Treg cells), and with either cisplatin or paclitaxel, showed significantly reduced tumor growth and 80 % tumor free-survival compared to chemotherapy alone [133]. These results validate the PD-1 pathway as an effective target to prevent tumor immune resistance, providing a new opportunity for anti-cancer immunotherapy in a wide range of malignancies, including breast cancer.
Trastuzumab is an FDA approved mAb targeting the HER2 receptor present on a subpopulation of breast cancers. Trastuzumab is efficacious both as a single agent [134] and in combination with chemotherapy (Marty et al, 2005). Its mechanisms of action include degradation of the HER2 receptor on cancer cells [135] and inhibition of the MAPK and PI3K/Akt pathways to promote cell cycle arrest [136,137]. Interestingly, trastuzumab efficiency has also been correlated with lymphocyte infiltration within the tumor, with an increase in CD3+ T cells, CD68+ macrophages and NK1.1+ NK cells in patients treated with trastuzumab [138,139]. This result indicates that trastuzumab may also act by attracting immune cells to the tumor site to induce antibody-dependent cellular cytotoxicity [138].
Fibroblast Based Therapies
There have been fewer developments in fibroblast directed therapies. Fibroblast activated protein (FAP) is a serine protease involved in extracellular matrix remodeling [140] and has high expression in cancer associated stroma, in wound healing and in fibrotic conditions such as liver fibrosis. It shows little if any expression in normal fibroblasts or other normal tissues [141,142,140]. In several preclinical studies, targeting FAP genetically or with vaccines or pharmacological agents, resulted in impaired tumor progression in mice [143–145,107,146–148]. However the clinical studies targeting FAP with monoclonal antibodies F19 and its humanized version Sibrotuzumab [149–151], or the FAP enzyme inhibitor Talabostat did not show clinical efficacy. The lack of other fibroblast-based therapies is possibly due to the paucity of specific CAF markers. There are two Phase 1 clinical trials in progress that are using fibroblasts to attempt to stimulate the immune system to fight cancer. The first, NCT00793208, is using lethally irradiated, semi allogenic human fibroblasts transfected with DNA from the patients own tumor to treat non small cell lung cancer. The trial will evaluate the safety, immunogenicity and feasibility of this new vaccine. The second trial, NCT00058799, is assessing whether autologous fibroblasts engineered in culture to express genes known to prime the immune system towards killing cancer cells (CD40L and IL-2) can do so when given to the patient along with small amounts of their own acute leukemic cancer cells.
Concluding Remarks
Both CAFs and the immune cell microenvironment within a tumor affect the ability of breast cancer to grow and metastasize. Emerging data indicate that even the early stages of breast cancer initiation may be shaped by the immediate microenvironment. We have discussed the evidence that indicates that stromal fibroblasts are directly mediating the immune cell microenvironment in cancer and we discuss the possible connections that may exist in normal mammary gland development. Future research assessing how fibroblasts directly mediate immune cell activation and orientation will surely aid in the development of more targeted therapies using both immune targets and activation markers within fibroblasts.
Abbreviations
- APC:
-
Antigen presenting cell
- BPH:
-
Benign prostatic hyperplasia
- CAF:
-
Cancer associated fibroblast
- CCL2:
-
Monocyte chemoattractant protein 1
- COPD:
-
Chronic obstructive pulmonary disease
- CSF-1:
-
Colony stimulating factor 1
- CTLA-4:
-
Cytotoxic T lymphocyte antigen 4
- CXCL12:
-
Stromal derived factor 1
- DC:
-
Dendritic cell
- DCIS:
-
Ductal carcinoma in-situ
- DTR:
-
Diphtheria toxin receptor
- ECM:
-
Extracellular matrix
- EFG:
-
Epidermal growth factor
- FAP:
-
Fibroblast activated protein
- FGF:
-
Fibroblast growth factor
- FGFR:
-
Fibroblast growth factor receptor
- GH:
-
Growth hormone
- IDC:
-
Invasive ductal carcinoma
- IFN-γ:
-
Interferon gamma
- IFN- β:
-
Interferon beta
- mAb:
-
Monoclonal antibody
- MDSC:
-
Myeloid Derived suppressor cell
- MHC:
-
Major histocompatibility complex
- MMP:
-
Matrix metalloproteinase
- NF:
-
Normal fibroblast
- NK:
-
Natural killer cell
- pDC:
-
Plasmacytoid dendritic cell
- RA:
-
Rheumatoid arthritis
- TAM:
-
Tumor associated macrophage
- TEB:
-
Terminal end bud
- TGF-β:
-
Transforming growth factor beta
- TIMP:
-
Tissue inhibitor of matrix metalloproteinase
- TNF-α:
-
Tumor necrosis factor alpha
- Treg:
-
Regulatory T cell
- VEGF:
-
Vascular endothelial growth factor
References
Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74. doi:10.1016/j.cell.2011.02.013.
Watson CJ, Khaled WT. Mammary development in the embryo and adult: a journey of morphogenesis and commitment. Development. 2008;135(6):995–1003. doi:10.1242/dev.005439.
Hinck L, Silberstein GB. Key stages in mammary gland development: the mammary end bud as a motile organ. Breast Cancer Res. 2005;7(6):245–51. doi:10.1186/bcr1331.
Richert MM, Schwertfeger KL, Ryder JW, Anderson SM. An atlas of mouse mammary gland development. J Mammary Gland Biol Neoplasia. 2000;5(2):227–41.
Balogh GA, Heulings R, Mailo DA, Russo PA, Sheriff F, Russo IH, et al. Genomic signature induced by pregnancy in the human breast. Int J Oncol. 2006;28(2):399–410.
Li M, Liu X, Robinson G, Bar-Peled U, Wagner KU, Young WS, et al. Mammary-derived signals activate programmed cell death during the first stage of mammary gland involution. Proc Natl Acad Sci U S A. 1997;94(7):3425–30.
De Boeck A, Hendrix A, Maynard D, Van Bockstal M, Daniels A, Pauwels P, et al. Differential secretome analysis of cancer-associated fibroblasts and bone marrow-derived precursors to identify microenvironmental regulators of colon cancer progression. Proteomics. 2013;13(2):379–88. doi:10.1002/pmic.201200179.
Postlethwaite AE, Shigemitsu H, Kanangat S. Cellular origins of fibroblasts: possible implications for organ fibrosis in systemic sclerosis. Curr Opin Rheumatol. 2004;16(6):733–8.
Fries KM, Blieden T, Looney RJ, Sempowski GD, Silvera MR, Willis RA, et al. Evidence of fibroblast heterogeneity and the role of fibroblast subpopulations in fibrosis. Clin Immunol Immunopathol. 1994;72(3):283–92.
Berry DP, Harding KG, Stanton MR, Jasani B, Ehrlich HP. Human wound contraction: collagen organization, fibroblasts, and myofibroblasts. Plast Reconstr Surg. 1998;102(1):124–31. discussion 32-4.
Lu P, Ewald AJ, Martin GR, Werb Z. Genetic mosaic analysis reveals FGF receptor 2 function in terminal end buds during mammary gland branching morphogenesis. Dev Biol. 2008;321(1):77–87. doi:10.1016/j.ydbio.2008.06.005.
Wiesen JF, Young P, Werb Z, Cunha GR. Signaling through the stromal epidermal growth factor receptor is necessary for mammary ductal development. Development. 1999;126(2):335–44.
Gallego MI, Binart N, Robinson GW, Okagaki R, Coschigano KT, Perry J, et al. Prolactin, growth hormone, and epidermal growth factor activate Stat5 in different compartments of mammary tissue and exert different and overlapping developmental effects. Dev Biol. 2001;229(1):163–75. doi:10.1006/dbio.2000.9961.
Jackson D, Bresnick J, Rosewell I, Crafton T, Poulsom R, Stamp G, et al. Fibroblast growth factor receptor signalling has a role in lobuloalveolar development of the mammary gland. J Cell Sci. 1997;110(Pt 11):1261–8.
Schedin P, Mitrenga T, McDaniel S, Kaeck M. Mammary ECM composition and function are altered by reproductive state. Mol Carcinog. 2004;41(4):207–20. doi:10.1002/mc.20058.
Talhouk RS, Bissell MJ, Werb Z. Coordinated expression of extracellular matrix-degrading proteinases and their inhibitors regulates mammary epithelial function during involution. J Cell Biol. 1992;118(5):1271–82.
De Marzo AM, Marchi VL, Epstein JI, Nelson WG. Proliferative inflammatory atrophy of the prostate: implications for prostatic carcinogenesis. Am J Pathol. 1999;155(6):1985–92. doi:10.1016/S0002-9440(10)65517-4.
Kuper CF, Schuurman H, Bos-Kuijpers M, Bloksma N. Predictive testing for pathogenic autoimmunity: the morphological approach. Toxicol Lett. 2000;112–113:433–42.
van Kempen LC, de Visser KE, Coussens LM. Inflammation, proteases and cancer. Eur J Cancer. 2006;42(6):728–34. doi:10.1016/j.ejca.2006.01.004.
Sapi E. The role of CSF-1 in normal physiology of mammary gland and breast cancer: an update. Exp Biol Med. 2004;229(1):1–11.
Stanley ER, Berg KL, Einstein DB, Lee PS, Pixley FJ, Wang Y, et al. Biology and action of colony–stimulating factor-1. Mol Reprod Dev. 1997;46(1):4–10. doi:10.1002/(SICI)1098-2795(199701)46:1<4::AID-MRD2>3.0.CO;2-V.
Gouon-Evans V, Lin EY, Pollard JW. Requirement of macrophages and eosinophils and their cytokines/chemokines for mammary gland development. Breast Cancer Res. 2002;4(4):155–64.
Ingman WV, Wyckoff J, Gouon-Evans V, Condeelis J, Pollard JW. Macrophages promote collagen fibrillogenesis around terminal end buds of the developing mammary gland. Dev Dyn. 2006;235(12):3222–9. doi:10.1002/dvdy.20972.
Gyorki DE, Asselin-Labat ML, van Rooijen N, Lindeman GJ, Visvader JE. Resident macrophages influence stem cell activity in the mammary gland. Breast Cancer Res. 2009;11(4):R62. doi:10.1186/bcr2353.
Chua AC, Hodson LJ, Moldenhauer LM, Robertson SA, Ingman WV. Dual roles for macrophages in ovarian cycle-associated development and remodelling of the mammary gland epithelium. Development. 2010;137(24):4229–38. doi:10.1242/dev.059261.
O'Brien J, Martinson H, Durand-Rougely C, Schedin P. Macrophages are crucial for epithelial cell death and adipocyte repopulation during mammary gland involution. Development. 2012;139(2):269–75. doi:10.1242/dev.071696.
Lilla JN, Werb Z. Mast cells contribute to the stromal microenvironment in mammary gland branching morphogenesis. Dev Biol. 2010;337(1):124–33. doi:10.1016/j.ydbio.2009.10.021.
Wolters PJ, Pham CT, Muilenburg DJ, Ley TJ, Caughey GH. Dipeptidyl peptidase I is essential for activation of mast cell chymases, but not tryptases, in mice. J Biol Chem. 2001;276(21):18551–6. doi:10.1074/jbc.M100223200.
Lilla JN, Joshi RV, Craik CS, Werb Z. Active plasma kallikrein localizes to mast cells and regulates epithelial cell apoptosis, adipocyte differentiation, and stromal remodeling during mammary gland involution. J Biol Chem. 2009;284(20):13792–803. doi:10.1074/jbc.M900508200.
Rothenberg ME, MacLean JA, Pearlman E, Luster AD, Leder P. Targeted disruption of the chemokine eotaxin partially reduces antigen-induced tissue eosinophilia. J Exp Med. 1997;185(4):785–90.
Colbert DC, McGarry MP, O'Neill K, Lee NA, Lee JJ. Decreased size and survival of weanling mice in litters of IL-5-/ -mice are a consequence of the IL-5 deficiency in nursing dams. Contemp Top Lab Anim Sci/J Am Assoc Lab AnimSci. 2005;44(3):53–5.
Weisz-Carrington P, Roux ME, Lamm ME. Plasma cells and epithelial immunoglobulins in the mouse mammary gland during pregnancy and lactation. J Immunol. 1977;119(4):1306–7.
Degnim AC, Brahmbhatt RD, Radisky DC, Hoskin TL, Stallings-Mann M, Laudenschlager M, et al. Immune cell quantitation in normal breast tissue lobules with and without lobulitis. Breast Cancer Res Treat. 2014;144(3):539–49. doi:10.1007/s10549-014-2896-8.
Stein T, Morris JS, Davies CR, Weber-Hall SJ, Duffy MA, Heath VJ, et al. Involution of the mouse mammary gland is associated with an immune cascade and an acute-phase response, involving LBP, CD14 and STAT3. Breast Cancer Res. 2004;6(2):R75–91. doi:10.1186/bcr753.
Stein T, Salomonis N, Gusterson BA. Mammary gland involution as a multi-step process. J Mammary Gland Biol Neoplasia. 2007;12(1):25–35. doi:10.1007/s10911-007-9035-7.
Clarkson RW, Wayland MT, Lee J, Freeman T, Watson CJ. Gene expression profiling of mammary gland development reveals putative roles for death receptors and immune mediators in post-lactational regression. Breast Cancer Res. 2004;6(2):R92–109. doi:10.1186/bcr754.
Monks J, Rosner D, Geske FJ, Lehman L, Hanson L, Neville MC, et al. Epithelial cells as phagocytes: apoptotic epithelial cells are engulfed by mammary alveolar epithelial cells and repress inflammatory mediator release. Cell Death Differ. 2005;12(2):107–14. doi:10.1038/sj.cdd.4401517.
Coussens LM, Pollard JW. Leukocytes in mammary development and cancer. Cold Spring Harbor perspectives in biology. 2011;3(3). doi:10.1101/cshperspect.a003285.
O'Brien J, Schedin P. Macrophages in breast cancer: do involution macrophages account for the poor prognosis of pregnancy-associated breast cancer? J Mammary Gland Biol Neoplasia. 2009;14(2):145–57. doi:10.1007/s10911-009-9118-8.
Surowiak P, Suchocki S, Gyorffy B, Gansukh T, Wojnar A, Maciejczyk A, et al. Stromal myofibroblasts in breast cancer: relations between their occurrence, tumor grade and expression of some tumour markers. Folia Histochem Cytobiol/ Pol Acad Sci, Pol Histochem Cytochem Soc. 2006;44(2):111–6.
Lisanti MP, Martinez Outschoorn UE, Sotgia F. Oncogenes induce the cancer-associated fibroblast phenotype: Metabolic symbiosis and "fibroblast addiction" are new therapeutic targets for drug discovery. Cell Cycle. 2013;12(17):2723–32. doi:10.4161/cc.25695.
Taddei ML, Giannoni E, Raugei G, Scacco S, Sardanelli AM, Papa S, et al. Mitochondrial Oxidative Stress due to Complex I Dysfunction Promotes Fibroblast Activation and Melanoma Cell Invasiveness. J Signal Transduct. 2012;2012:684592. doi:10.1155/2012/684592.
Ronnov-Jessen L, Petersen OW. Induction of alpha-smooth muscle actin by transforming growth factor-beta 1 in quiescent human breast gland fibroblasts. Implications for myofibroblast generation in breast neoplasia. Lab Investig: J Tech Methods and Pathol. 1993;68(6):696–707.
Whitaker-Menezes D, Martinez-Outschoorn UE, Lin Z, Ertel A, Flomenberg N, Witkiewicz AK, et al. Evidence for a stromal-epithelial "lactate shuttle" in human tumors: MCT4 is a marker of oxidative stress in cancer-associated fibroblasts. Cell Cycle. 2011;10(11):1772–83.
Buckley CD, Pilling D, Lord JM, Akbar AN, Scheel-Toellner D, Salmon M. Fibroblasts regulate the switch from acute resolving to chronic persistent inflammation. Trends Immunol. 2001;22(4):199–204.
Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6(5):392–401. doi:10.1038/nrc1877.
Silzle T, Kreutz M, Dobler MA, Brockhoff G, Knuechel R, Kunz-Schughart LA. Tumor-associated fibroblasts recruit blood monocytes into tumor tissue. Eur J Immunol. 2003;33(5):1311–20. doi:10.1002/eji.200323057.
Cambien B, Pomeranz M, Millet MA, Rossi B, Schmid-Alliana A. Signal transduction involved in MCP-1-mediated monocytic transendothelial migration. Blood. 2001;97(2):359–66.
Gu L, Tseng S, Horner RM, Tam C, Loda M, Rollins BJ. Control of TH2 polarization by the chemokine monocyte chemoattractant protein-1. Nature. 2000;404(6776):407–11. doi:10.1038/35006097.
Baglole CJ, Maggirwar SB, Gasiewicz TA, Thatcher TH, Phipps RP, Sime PJ. The aryl hydrocarbon receptor attenuates tobacco smoke-induced cyclooxygenase-2 and prostaglandin production in lung fibroblasts through regulation of the NF-kappaB family member RelB. J Biol Chem. 2008;283(43):28944–57. doi:10.1074/jbc.M800685200.
Flavell SJ, Hou TZ, Lax S, Filer AD, Salmon M, Buckley CD. Fibroblasts as novel therapeutic targets in chronic inflammation. Br J Pharmacol. 2008;153 Suppl 1:S241–6. doi:10.1038/sj.bjp.0707487.
Martey CA, Pollock SJ, Turner CK, O'Reilly KM, Baglole CJ, Phipps RP, et al. Cigarette smoke induces cyclooxygenase-2 and microsomal prostaglandin E2 synthase in human lung fibroblasts: implications for lung inflammation and cancer. Am J Physiol Lung Cell Mol Physiol. 2004;287(5):L981–91. doi:10.1152/ajplung.00239.2003.
Smith GM, Biggs J, Norris B, Anderson-Stewart P, Ward R. Detection of a soluble form of the leukocyte surface antigen CD48 in plasma and its elevation in patients with lymphoid leukemias and arthritis. J Clin Immunol. 1997;17(6):502–9.
Zhang J, Wu L, Qu JM, Bai CX, Merrilees MJ, Black PN. Pro-inflammatory phenotype of COPD fibroblasts not compatible with repair in COPD lung. J Cell Mol Med. 2012;16(7):1522–32. doi:10.1111/j.1582-4934.2011.01492.x.
Huber LC, Distler O, Tarner I, Gay RE, Gay S, Pap T. Synovial fibroblasts: key players in rheumatoid arthritis. Rheumatology. 2006;45(6):669–75. doi:10.1093/rheumatology/kel065.
Page RC. The role of inflammatory mediators in the pathogenesis of periodontal disease. J Periodontal Res. 1991;26(3 Pt 2):230–42.
Kook SH, Jang YS, Lee JC. Human periodontal ligament fibroblasts stimulate osteoclastogenesis in response to compression force through TNF-alpha-mediated activation of CD4+ T cells. J Cell Biochem. 2011;112(10):2891–901. doi:10.1002/jcb.23205.
Begley L, Monteleon C, Shah RB, Macdonald JW, Macoska JA. CXCL12 overexpression and secretion by aging fibroblasts enhance human prostate epithelial proliferation in vitro. Aging Cell. 2005;4(6):291–8. doi:10.1111/j.1474-9726.2005.00173.x.
Begley LA, Kasina S, MacDonald J, Macoska JA. The inflammatory microenvironment of the aging prostate facilitates cellular proliferation and hypertrophy. Cytokine. 2008;43(2):194–9. doi:10.1016/j.cyto.2008.05.012.
Feghali CA, Wright TM. Cytokines in acute and chronic inflammation. Front Biosci: J Virtual Libr. 1997;2:d12–26.
Takata H, Tomiyama H, Fujiwara M, Kobayashi N, Takiguchi M. Cutting edge: expression of chemokine receptor CXCR1 on human effector CD8+ T cells. J Immunol. 2004;173(4):2231–5.
Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity's roles in cancer suppression and promotion. Science. 2011;331(6024):1565–70. doi:10.1126/science.1203486.
Vesely MD, Kershaw MH, Schreiber RD, Smyth MJ. Natural innate and adaptive immunity to cancer. Annu Rev Immunol. 2011;29:235–71. doi:10.1146/annurev-immunol-031210-101324.
Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10(8):789–99. doi:10.1038/nm1087.
Dunn GP, Old LJ, Schreiber RD. The three Es of cancer immunoediting. Annu Rev Immunol. 2004;22:329–60. doi:10.1146/annurev.immunol.22.012703.104803.
Smyth MJ, Dunn GP, Schreiber RD. Cancer immunosurveillance and immunoediting: the roles of immunity in suppressing tumor development and shaping tumor immunogenicity. Adv Immunol. 2006;90:1–50. doi:10.1016/S0065-2776(06)90001-7.
Balkwill F, Charles KA, Mantovani A. Smoldering and polarized inflammation in the initiation and promotion of malignant disease. Cancer Cell. 2005;7(3):211–7. doi:10.1016/j.ccr.2005.02.013.
Lu H, Hoshiba T, Kawazoe N, Koda I, Song M, Chen G. Cultured cell-derived extracellular matrix scaffolds for tissue engineering. Biomaterials. 2011;32(36):9658–66. doi:10.1016/j.biomaterials.2011.08.091.
Chapman JR, Webster AC, Wong G. Cancer in the transplant recipient. Cold Spring Harbor perspectives in medicine. 2013;3(7). doi:10.1101/cshperspect.a015677.
Grulich AE, van Leeuwen MT, Falster MO, Vajdic CM. Incidence of cancers in people with HIV/AIDS compared with immunosuppressed transplant recipients: a meta-analysis. Lancet. 2007;370(9581):59–67. doi:10.1016/S0140-6736(07)61050-2.
Aaltomaa S, Lipponen P, Eskelinen M, Kosma VM, Marin S, Alhava E, et al. Lymphocyte infiltrates as a prognostic variable in female breast cancer. Eur J Cancer. 1992;28A(4–5):859–64.
Loi S, Sirtaine N, Piette F, Salgado R, Viale G, Van Eenoo F, et al. Prognostic and predictive value of tumor-infiltrating lymphocytes in a phase III randomized adjuvant breast cancer trial in node-positive breast cancer comparing the addition of docetaxel to doxorubicin with doxorubicin-based chemotherapy: BIG 02-98. J Clin Oncol: Off J Am Soc Clin Oncol. 2013;31(7):860–7. doi:10.1200/JCO.2011.41.0902.
Hussein MR, Hassan HI. Analysis of the mononuclear inflammatory cell infiltrate in the normal breast, benign proliferative breast disease, in situ and infiltrating ductal breast carcinomas: preliminary observations. J Clin Pathol. 2006;59(9):972–7. doi:10.1136/jcp.2005.031252.
Erez N, Glanz S, Raz Y, Avivi C, Barshack I. Cancer associated fibroblasts express pro-inflammatory factors in human breast and ovarian tumors. Biochem Biophys Res Commun. 2013;437(3):397–402. doi:10.1016/j.bbrc.2013.06.089.
DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, et al. Leukocyte complexity predicts breast cancer survival and functionally regulates response to chemotherapy. Cancer Discov. 2011;1(1):54–67. doi:10.1158/2159-8274.CD-10-0028.
Brennan DJ, Gallagher WM. Prognostic ability of a panel of immunohistochemistry markers - retailoring of an 'old solution'. Breast Cancer Res. 2008;10(1):102. doi:10.1186/bcr1854.
Paulsson J, Sjoblom T, Micke P, Ponten F, Landberg G, Heldin CH, et al. Prognostic significance of stromal platelet-derived growth factor beta-receptor expression in human breast cancer. Am J Pathol. 2009;175(1):334–41. doi:10.2353/ajpath.2009.081030.
Kohrt HE, Nouri N, Nowels K, Johnson D, Holmes S, Lee PP. Profile of immune cells in axillary lymph nodes predicts disease-free survival in breast cancer. PLoS Med. 2005;2(9):e284. doi:10.1371/journal.pmed.0020284.
Mukhtar RA, Nseyo O, Campbell MJ, Esserman LJ. Tumor-associated macrophages in breast cancer as potential biomarkers for new treatments and diagnostics. Expert Rev Mol Diagn. 2011;11(1):91–100. doi:10.1586/erm.10.97.
Schmieder A, Michel J, Schonhaar K, Goerdt S, Schledzewski K. Differentiation and gene expression profile of tumor-associated macrophages. Semin Cancer Biol. 2012;22(4):289–97. doi:10.1016/j.semcancer.2012.02.002.
Siveen KS, Kuttan G. Role of macrophages in tumour progression. Immunol Lett. 2009;123(2):97–102. doi:10.1016/j.imlet.2009.02.011.
Dong C, Martinez GJ. T cell Subsets. In: Centre UoTHS, editor. Texas: Nature Publishing Group; 2010.
Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10(9):942–9. doi:10.1038/nm1093.
Knutson KL, Disis ML. Tumor antigen-specific T helper cells in cancer immunity and immunotherapy. Cancer Immunol Immunother. 2005;54(8):721–8. doi:10.1007/s00262-004-0653-2.
Treilleux I, Blay JY, Bendriss-Vermare N, Ray-Coquard I, Bachelot T, Guastalla JP, et al. Dendritic cell infiltration and prognosis of early stage breast cancer. Clin Cancer Res: Off J Am Assoc Cancer Res. 2004;10(22):7466–74. doi:10.1158/1078-0432.CCR-04-0684.
Slaney CY, Rautela J, Parker BS. The emerging role of immunosurveillance in dictating metastatic spread in breast cancer. Cancer Res. 2013;73(19):5852–7.
Bidwell BN, Slaney CY, Withana NP, Forster S, Cao Y, Loi S, et al. Silencing of Irf7 pathways in breast cancer cells promotes bone metastasis through immune escape. Nat Med. 2012;18(8):1224–31. doi:10.1038/nm.2830.
Faraji F, Pang Y, Walker RC, Nieves Borges R, Yang L, Hunter KW. Cadm1 is a metastasis susceptibility gene that suppresses metastasis by modifying tumor interaction with the cell-mediated immunity. PLoS Genet. 2012;8(9):e1002926. doi:10.1371/journal.pgen.1002926.
Mahmoud SM, Paish EC, Powe DG, Macmillan RD, Grainge MJ, Lee AH, et al. Tumor-infiltrating CD8+ lymphocytes predict clinical outcome in breast cancer. J Clin Oncol: Off J Am Soc Clin Oncol. 2011;29(15):1949–55. doi:10.1200/JCO.2010.30.5037.
Kim ST, Jeong H, Woo OH, Seo JH, Kim A, Lee ES, et al. Tumor-infiltrating lymphocytes, tumor characteristics, and recurrence in patients with early breast cancer. Am J Clin Oncol. 2013;36(3):224–31. doi:10.1097/COC.0b013e3182467d90.
Olkhanud PB, Baatar D, Bodogai M, Hakim F, Gress R, Anderson RL, et al. Breast cancer lung metastasis requires expression of chemokine receptor CCR4 and regulatory T cells. Cancer Res. 2009;69(14):5996–6004. doi:10.1158/0008-5472.CAN-08-4619.
Stagg J, Divisekera U, McLaughlin N, Sharkey J, Pommey S, Denoyer D, et al. Anti-CD73 antibody therapy inhibits breast tumor growth and metastasis. Proc Natl Acad Sci U S A. 2010;107(4):1547–52. doi:10.1073/pnas.0908801107.
Mamessier E, Sylvain A, Thibult ML, Houvenaeghel G, Jacquemier J, Castellano R, et al. Human breast cancer cells enhance self tolerance by promoting evasion from NK cell antitumor immunity. J Clin Invest. 2011;121(9):3609–22. doi:10.1172/JCI45816.
Swierczak A, Cook A, Lenzo J, Restall C, Doherty J, Anderson R et al. The promotion of breast cancer metastasis caused by inhibition of CSF-1R/CSF-1 signaling is blocked by targeting the G-CSF receptor. Cancer Immunol Res. e-print online April 29 2014.
Lin EY, Nguyen AV, Russell RG, Pollard JW. Colony-stimulating factor 1 promotes progression of mammary tumors to malignancy. J Exp Med. 2001;193(6):727–40.
Erez N, Truitt M, Olson P, Arron ST, Hanahan D. Cancer-Associated Fibroblasts Are Activated in Incipient Neoplasia to Orchestrate Tumor-Promoting Inflammation in an NF-kappaB-Dependent Manner. Cancer Cell. 2010;17(2):135–47. doi:10.1016/j.ccr.2009.12.041.
Celis JE, Moreira JM, Cabezon T, Gromov P, Friis E, Rank F, et al. Identification of extracellular and intracellular signaling components of the mammary adipose tissue and its interstitial fluid in high risk breast cancer patients: toward dissecting the molecular circuitry of epithelial-adipocyte stromal cell interactions. Mol Cell Proteomics. 2005;4(4):492–522. doi:10.1074/mcp.M500030-MCP200.
Dirat B, Bochet L, Dabek M, Daviaud D, Dauvillier S, Majed B, et al. Cancer-associated adipocytes exhibit an activated phenotype and contribute to breast cancer invasion. Cancer Res. 2011;71(7):2455–65. doi:10.1158/0008-5472.CAN-10-3323.
Finak G, Bertos N, Pepin F, Sadekova S, Souleimanova M, Zhao H, et al. Stromal gene expression predicts clinical outcome in breast cancer. Nat Med. 2008;14(5):518–27. doi:10.1038/nm1764.
Elkabets M, Gifford AM, Scheel C, Nilsson B, Reinhardt F, Bray MA, et al. Human tumors instigate granulin-expressing hematopoietic cells that promote malignancy by activating stromal fibroblasts in mice. J Clin Invest. 2011;121(2):784–99. doi:10.1172/JCI43757.
Boersma BJ, Reimers M, Yi M, Ludwig JA, Luke BT, Stephens RM, et al. A stromal gene signature associated with inflammatory breast cancer. Int J Cancer J Int Du Cancer. 2008;122(6):1324–32. doi:10.1002/ijc.23237.
Stover DG, Bierie B, Moses HL. A delicate balance: TGF-beta and the tumor microenvironment. J Cell Biochem. 2007;101(4):851–61. doi:10.1002/jcb.21149.
Balsamo M, Scordamaglia F, Pietra G, Manzini C, Cantoni C, Boitano M, et al. Melanoma-associated fibroblasts modulate NK cell phenotype and antitumor cytotoxicity. Proc Natl Acad Sci U S A. 2009;106(49):20847–52. doi:10.1073/pnas.0906481106.
Comito G, Giannoni E, Segura CP, Barcellos-de-Souza P, Raspollini MR, Baroni G et al. Cancer-associated fibroblasts and M2-polarized macrophages synergize during prostate carcinoma progression. Oncogene. 2013. doi:10.1038/onc.2013.191.
Barnas JL, Simpson-Abelson MR, Yokota SJ, Kelleher RJ, Bankert RB. T cells and stromal fibroblasts in human tumor microenvironments represent potential therapeutic targets. Cancer Microenviron: Off J Int Cancer Microenviron Soc. 2010;3(1):29–47. doi:10.1007/s12307-010-0044-5.
Liao D, Luo Y, Markowitz D, Xiang R, Reisfeld RA. Cancer associated fibroblasts promote tumor growth and metastasis by modulating the tumor immune microenvironment in a 4T1 murine breast cancer model. PLoS One. 2009;4(11):e7965. doi:10.1371/journal.pone.0007965.
Korn T, Bettelli E, Oukka M, Kuchroo VK. IL-17 and Th17 Cells. Annu Rev Immunol. 2009;27:485–517. doi:10.1146/annurev.immunol.021908.132710.
Benito-Miguel M, Garcia-Carmona Y, Balsa A. Perez de Ayala C, Cobo-Ibanez T, Martin-Mola E et al. A dual action of rheumatoid arthritis synovial fibroblast IL-15 expression on the equilibrium between CD4 + CD25+ regulatory T cells and CD4 + CD25- responder T cells. J Immunol. 2009;183(12):8268–79.
Ohm JE, Gabrilovich DI, Sempowski GD, Kisseleva E, Parman KS, Nadaf S, et al. VEGF inhibits T-cell development and may contribute to tumor-induced immune suppression. Blood. 2003;101(12):4878–86. doi:10.1182/blood-2002-07-1956.
Wada J, Suzuki H, Fuchino R, Yamasaki A, Nagai S, Yanai K, et al. The contribution of vascular endothelial growth factor to the induction of regulatory T-cells in malignant effusions. Anticancer Res. 2009;29(3):881–8.
Ahmadzadeh M, Rosenberg SA. TGF-beta 1 attenuates the acquisition and expression of effector function by tumor antigen-specific human memory CD8 T cells. J Immunol. 2005;174(9):5215–23.
Arbeit JM, Munger K, Howley PM, Hanahan D. Progressive squamous epithelial neoplasia in K14-human papillomavirus type 16 transgenic mice. J Virol. 1994;68(7):4358–68.
Coussens LM, Hanahan D, Arbeit JM. Genetic predisposition and parameters of malignant progression in K14-HPV16 transgenic mice. Am J Pathol. 1996;149(6):1899–917.
Bollrath J, Phesse TJ, von Burstin VA, Putoczki T, Bennecke M, Bateman T, et al. gp130-mediated Stat3 activation in enterocytes regulates cell survival and cell-cycle progression during colitis-associated tumorigenesis. Cancer Cell. 2009;15(2):91–102. doi:10.1016/j.ccr.2009.01.002.
Grivennikov S, Karin E, Terzic J, Mucida D, Yu GY, Vallabhapurapu S, et al. IL-6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis-associated cancer. Cancer Cell. 2009;15(2):103–13. doi:10.1016/j.ccr.2009.01.001.
Korc M. Pancreatic cancer-associated stroma production. Am J Surg. 2007;194(4 Suppl):S84–6. doi:10.1016/j.amjsurg.2007.05.004.
Mahadevan D, Von Hoff DD. Tumor-stroma interactions in pancreatic ductal adenocarcinoma. Mol Cancer Ther. 2007;6(4):1186–97. doi:10.1158/1535-7163.MCT-06-0686.
Walker RA. The complexities of breast cancer desmoplasia. Breast Cancer Res. 2001;3(3):143–5.
Quante M, Tu SP, Tomita H, Gonda T, Wang SS, Takashi S, et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell. 2011;19(2):257–72. doi:10.1016/j.ccr.2011.01.020.
Chomarat P, Banchereau J, Davoust J, Palucka AK. IL-6 switches the differentiation of monocytes from dendritic cells to macrophages. Nat Immunol. 2000;1(6):510–4. doi:10.1038/82763.
Peng Q, Zhao L, Hou Y, Sun Y, Wang L, Luo H, et al. Biological characteristics and genetic heterogeneity between carcinoma-associated fibroblasts and their paired normal fibroblasts in human breast cancer. PLoS One. 2013;8(4):e60321. doi:10.1371/journal.pone.0060321.
Walunas TL, Lenschow DJ, Bakker CY, Linsley PS, Freeman GJ, Green JM, et al. CTLA-4 can function as a negative regulator of T cell activation. Immunity. 1994;1(5):405–13.
van Elsas A, Hurwitz AA, Allison JP. Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J Exp Med. 1999;190(3):355–66.
Van Ginderachter JA, Liu Y, Geldhof AB, Brijs L, Thielemans K, De Baetselier P, et al. B7-1, IFN gamma and anti-CTLA-4 co-operate to prevent T-cell tolerization during immunotherapy against a murine T-lymphoma. Int J Cancer J Int Du Cancer. 2000;87(4):539–47.
Hurwitz AA, Yu TF, Leach DR, Allison JP. CTLA-4 blockade synergizes with tumor-derived granulocyte-macrophage colony-stimulating factor for treatment of an experimental mammary carcinoma. Proc Natl Acad Sci U S A. 1998;95(17):10067–71.
Kwon ED, Hurwitz AA, Foster BA, Madias C, Feldhaus AL, Greenberg NM, et al. Manipulation of T cell costimulatory and inhibitory signals for immunotherapy of prostate cancer. Proc Natl Acad Sci U S A. 1997;94(15):8099–103.
Hodi FS, O'Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–23. doi:10.1056/NEJMoa1003466.
Ribas A, Hodi FS, Callahan M, Konto C, Wolchok J. Hepatotoxicity with combination of vemurafenib and ipilimumab. N Engl J Med. 2013;368(14):1365–6. doi:10.1056/NEJMc1302338.
Topalian SL, Drake CG, Pardoll DM. Targeting the PD-1/B7-H1(PD-L1) pathway to activate anti-tumor immunity. Curr Opin Immunol. 2012;24(2):207–12. doi:10.1016/j.coi.2011.12.009.
Hamid O, Robert C, Daud A, Hodi FS, Hwu WJ, Kefford R, et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. N Engl J Med. 2013;369(2):134–44. doi:10.1056/NEJMoa1305133.
Zhang P, Su DM, Liang M, Fu J. Chemopreventive agents induce programmed death-1-ligand 1 (PD-L1) surface expression in breast cancer cells and promote PD-L1-mediated T cell apoptosis. Mol Immunol. 2008;45(5):1470–6. doi:10.1016/j.molimm.2007.08.013.
Lu L, Xu X, Zhang B, Zhang R, Ji H, Wang X. Combined PD-1 blockade and GITR triggering induce a potent antitumor immunity in murine cancer models and synergizes with chemotherapeutic drugs. J Transl Med. 2014;12:36. doi:10.1186/1479-5876-12-36.
Vogel CL, Cobleigh MA, Tripathy D, Gutheil JC, Harris LN, Fehrenbacher L, et al. Efficacy and safety of trastuzumab as a single agent in first-line treatment of HER2-overexpressing metastatic breast cancer. J Clin Oncol: Off J Am Soc Clin Oncol. 2002;20(3):719–26.
Klapper LN, Waterman H, Sela M, Yarden Y. Tumor-inhibitory antibodies to HER-2/ErbB-2 may act by recruiting c-Cbl and enhancing ubiquitination of HER-2. Cancer Res. 2000;60(13):3384–8.
Junttila TT, Akita RW, Parsons K, Fields C, Lewis Phillips GD, Friedman LS, et al. Ligand-independent HER2/HER3/PI3K complex is disrupted by trastuzumab and is effectively inhibited by the PI3K inhibitor GDC-0941. Cancer Cell. 2009;15(5):429–40. doi:10.1016/j.ccr.2009.03.020.
Nagata Y, Lan KH, Zhou X, Tan M, Esteva FJ, Sahin AA, et al. PTEN activation contributes to tumor inhibition by trastuzumab, and loss of PTEN predicts trastuzumab resistance in patients. Cancer Cell. 2004;6(2):117–27. doi:10.1016/j.ccr.2004.06.022.
Arnould L, Gelly M, Penault-Llorca F, Benoit L, Bonnetain F, Migeon C, et al. Trastuzumab-based treatment of HER2-positive breast cancer: an antibody-dependent cellular cytotoxicity mechanism? Br J Cancer. 2006;94(2):259–67. doi:10.1038/sj.bjc.6602930.
Denkert C, Loibl S, Noske A, Roller M, Muller BM, Komor M, et al. Tumor-associated lymphocytes as an independent predictor of response to neoadjuvant chemotherapy in breast cancer. J Clin Oncol: Off J Am Soc Clin Oncol. 2010;28(1):105–13. doi:10.1200/JCO.2009.23.7370.
Kelly T, Huang Y, Simms AE, Mazur A. Fibroblast activation protein-alpha: a key modulator of the microenvironment in multiple pathologies. Int Rev Cell Mol Biol. 2012;297:83–116. doi:10.1016/B978-0-12-394308-8.00003-0.
Rettig WJ, Garin-Chesa P, Beresford HR, Oettgen HF, Melamed MR, Old LJ. Cell-surface glycoproteins of human sarcomas: differential expression in normal and malignant tissues and cultured cells. Proc Natl Acad Sci U S A. 1988;85(9):3110–4.
Garin-Chesa P, Old LJ, Rettig WJ. Cell surface glycoprotein of reactive stromal fibroblasts as a potential antibody target in human epithelial cancers. Proc Natl Acad Sci U S A. 1990;87(18):7235–9.
Lee KN, Jackson KW, Christiansen VJ, Lee CS, Chun JG, McKee PA. Antiplasmin-cleaving enzyme is a soluble form of fibroblast activation protein. Blood. 2006;107(4):1397–404. doi:10.1182/blood-2005-08-3452.
Loeffler M, Kruger JA, Niethammer AG, Reisfeld RA. Targeting tumor-associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. J Clin Invest. 2006;116(7):1955–62. doi:10.1172/JCI26532.
Ostermann E, Garin-Chesa P, Heider KH, Kalat M, Lamche H, Puri C, et al. Effective immunoconjugate therapy in cancer models targeting a serine protease of tumor fibroblasts. Clin Cancer Res: Off J Am Assoc Cancer Res. 2008;14(14):4584–92. doi:10.1158/1078-0432.CCR-07-5211.
Santos AM, Jung J, Aziz N, Kissil JL, Pure E. Targeting fibroblast activation protein inhibits tumor stromagenesis and growth in mice. J Clin Invest. 2009;119(12):3613–25. doi:10.1172/JCI38988.
Kraman M, Bambrough PJ, Arnold JN, Roberts EW, Magiera L, Jones JO, et al. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science. 2010;330(6005):827–30. doi:10.1126/science.1195300.
Wen Y, Wang CT, Ma TT, Li ZY, Zhou LN, Mu B, et al. Immunotherapy targeting fibroblast activation protein inhibits tumor growth and increases survival in a murine colon cancer model. Cancer Sci. 2010;101(11):2325–32. doi:10.1111/j.1349-7006.2010.01695.x.
Welt S, Divgi CR, Scott AM, Garin-Chesa P, Finn RD, Graham M, et al. Antibody targeting in metastatic colon cancer: a phase I study of monoclonal antibody F19 against a cell-surface protein of reactive tumor stromal fibroblasts. J Clin Oncol: Off J Am Soc Clin Oncol. 1994;12(6):1193–203.
Hofheinz RD. al-Batran SE, Hartmann F, Hartung G, Jager D, Renner C et al. Stromal antigen targeting by a humanised monoclonal antibody: an early phase II trial of sibrotuzumab in patients with metastatic colorectal cancer. Onkologie. 2003;26(1):44–8.
Scott AM, Wiseman G, Welt S, Adjei A, Lee FT, Hopkins W, et al. A Phase I dose-escalation study of sibrotuzumab in patients with advanced or metastatic fibroblast activation protein-positive cancer. Clin Cancer Res: Off J Am Assoc Cancer Res. 2003;9(5):1639–47.
Acknowledgments
We would like to thank Dr Nicole Haynes for critical discussions and assistance with this review. We would also like to thank the National Breast Cancer Foundation (NBCF) of Australia for supporting Robin Anderson and Kara Britt with NBCF fellowships. Ashleigh Unsworth is supported by an Australian Postgraduate Award.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Unsworth, A., Anderson, R. & Britt, K. Stromal Fibroblasts and the Immune Microenvironment: Partners in Mammary Gland Biology and Pathology?. J Mammary Gland Biol Neoplasia 19, 169–182 (2014). https://doi.org/10.1007/s10911-014-9326-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10911-014-9326-8