
Abstract
The tumor microenvironment comprises a mass of heterogeneous cell types, including immune cells, endothelial cells, and fibroblasts, alongside cancer cells. It is increasingly becoming clear that the development of this support niche is critical to the continued uncontrolled growth of the cancer. The tumor microenvironment contributes to the maintenance of cancer stemness and also directly promotes angiogenesis, invasion, metastasis, and chronic inflammation. In this chapter, we describe on the role of fibroblasts, specifically termed cancer-associated fibroblasts (CAFs), in the promotion and maintenance of cancers. CAFs have a multitude of effects on the growth and maintenance of cancer, and here we focus on their roles in modulating immune cells and responses; CAFs both inhibit immune cell access to the tumor microenvironment and inhibit their functions within the tumor. Finally, we describe the potential modulation of CAF function as an adjunct to bolster the effectiveness of cancer immunotherapies.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


Keywords
6.1 Introduction
The critical role of the tumor microenvironment in carcinogenesis has long been recognized, with Virchow first noting that malignancy arose at sites of chronic inflammation in 1863 [1]. In 1889 a more holistic hypothesis, that of the “seed and soil,” was proposed by Paget suggesting that elements of the stroma were important for tumor development [2]. At a similar time, physicians noted that the status of the immune system may have important consequences for tumor development. Indeed sarcoma remissions had been observed following Streptococcus pyogenes infections , and in 1868 Busch induced a local infection and demonstrated a reduction in tumor burden, at least while the infection was ongoing [3]. Later several other physicians demonstrated remissions through localized applications of infectious agents [4,5,6]. However, these observations were soon overshadowed by other contemporary discoveries, namely, the identification of tumor suppressor genes and oncogenes as well as the advent of chemotherapy and radiotherapy. These led to increased focus on cell intrinsic mechanisms of carcinogenesis and more easily controlled therapeutic options, respectively, consigning the immune system and the stroma to the backburner.
6.2 Fibroblasts
There has, however, been a resurgence of interest in the tumor stroma in more recent times. In 1982 Bissell et al. outlined a modern formulation of Paget’s seed and soil hypothesis stating that the tumor microenvironment is as important for tumor development as the accumulation of enabling genetic mutations [7]. This was based on the several elegant experiments showing that normalization of the stromal microenvironment could suppress tumor development; Illmensee and Mintz showed that while teratocarcinoma cells could be repeatedly transplanted and grow as ascites tumors, they would contribute to normal tissues when injected into a blastocyst [8]. Bissell’s group also showed that normalization of integrin signaling in both 3D culture and in vivo could abrogate the malignant phenotype of genetically deranged breast cancer cell lines [9]. This focus on the extracellular matrix (ECM) and its role in modulating tumor cell behavior led to a general interest in cells responsible for generating and modulating the collagen matrix-fibroblasts. First described in 1858 based on their morphology and location [10], fibroblasts are non-epithelial, nonvascular, and non-hematopoietic cells in the connective tissue and are largely responsible for the synthesis of the ECM [11].
Fibroblasts are critical in both normal homeostasis and during wound healing. At steady state, fibroblasts are essential for epithelial homeostasis in many normal tissues having both direct interactions with the epithelial cells and secreting growth factors [12]. During wound healing macrophages produce transforming growth factor-β (TGF-β) and platelet-derived growth factor (PDGF) leading to activation of normal tissue fibroblasts [13, 14] to acquire a myofibroblast state defined by expression of α-smooth muscle actin (α-SMA) [15]. These myofibroblasts, first described in granulation tissue [16], play important roles in wound healing. Early histological studies showed that tumors appear similarly to healing wounds with Dvorak describing them as “wounds which do not heal” [17]; in the context of a healing wound angiogenesis , remodeling of the ECM and epithelial proliferation are all adaptive; however in the tumor microenvironment, these aid in tumor development. As may be expected in the tumor microenvironment, similar processes are ongoing as in healing wounds, and carcinoma-associated fibroblasts (CAFs) have also previously been defined by their expression of α-SMA [18]. Indeed the roles outlined above are all subverted during tumor progression to facilitate continued growth. In this context there has been great attention to the roles CAFs play compared to normal tissue fibroblasts; however, it has been challenging to study these cells in vivo due to the absence of ideal markers of these cells and the heterogeneity of the CAF population.
6.3 CAFs
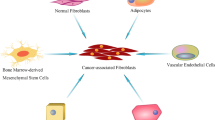
Markers used to identify CAFS are often controversial. Fibroblast-specific protein-1 (FSP-1) , one marker widely used to identify CAFs, has also been shown to be expressed by monocytes and invasive carcinoma cells [19, 20]; α-SMA on the other hand is expressed by pericytes and vascular smooth muscle cells [21, 22], while platelet-derived growth factor receptor-α (PDGFR-α) marks normal tissue fibroblasts [23] and some non-fibroblastic cells in the retinal pigment epithelium (unpublished observations and [24]). One promising marker for CAFs was fibroblast activation protein-α (FAP-α), which was identified by its reactivity with the F19 monoclonal antibody and reported to be selectively expressed on fibroblasts in healing wounds and in adenocarcinomas [25]. Further study appeared to support this with FAP-expressing cells being found in chronic inflammatory situations [26, 27]; however, when a reporter of FAP was generated, it was found to also be expressed on normal tissue fibroblasts, on fibroblastic reticular cells of the lymph node, and on some epithelial cells in the retinal pigment epithelium ([28, 29], unpublished observations) indicating that this too suffered from similar limitations regarding specificity. As such there has been a general lack of genetic systems by which to alter fibroblast function in vivo to dissect these roles more precisely. Furthermore, in many studies CAFs have been treated as a single entity, which is likely an oversimplification. Even normal tissue fibroblasts display remarkable heterogeneity with fibroblasts isolated from skin at different sites being as different transcriptomically as different leukocyte subsets [30]. Kidd et al. demonstrated in 2012 that fibroblast subsets defined by different markers originated from different sites with FSP-1+ fibroblasts deriving from the bone marrow, while other stromal cells may originate from adjacent tissues [31]. Indeed CAFs have many potential origins but most are thought to arise from local progenitors. CAFs are most commonly derived from tissue-resident fibroblasts [18, 32, 33], which are induced to undergo activation in response to the tumor microenvironment produced by neoplastic cells [34,35,36,37,38]. CAFs can also be derived from stellate cells [39, 40], migration of adipose or bone marrow-derived stromal cells [31, 41, 42], and endothelial and epithelial cells, through endothelial- or epithelial-to-mesenchymal transition [43, 44]. Despite these caveats there have been many successful ingenious studies elucidating the critical roles CAFs play in tumor development.
6.4 Driving Cancer Cell Proliferation
As stated earlier fibroblasts support epithelial homeostasis through secretion of growth factors, and CAFs too have been shown to have a direct effect on cancer cell growth in some systems. Orimo et al. showed that CXCL12 produced by CAFs drove cancer cell growth through CXCR4 expressed on the cancer cells [45]. CAFs have also been shown to produce numerous growth factors in different systems including insulin-like growth factors [46], connective tissue growth factor (CTGF) [47], platelet-derived growth factor (PDGF) [48], and hepatocyte growth factor [49], which have been shown to stimulate tumor cell growth in vitro. There is also evidence that CAFs can stimulate cell growth by releasing growth factors from the ECM through expression of matrix metalloproteases (MMPs). This indirect effect of MMP expression is a character shared with tumor-associated macrophages (TAMs) and other stromal cells [50]. CAFs do not simply affect the growth of cancer cells; however, they also modulate their phenotype making them more carcinogenic. Wang et al. showed that injection of an SV40-immortalized but not transformed prostate cancer cell line with CAFs led to poorly differentiated carcinomas developing, while there was minimal growth and no tumor development when this was carried out with normal prostate fibroblasts . When these cells were injected along with urogenital mesenchyme, epithelial cell growth was observed although there was no tumor development. As a result this series of experiments suggested that while the CAFs did stimulate growth, they had other pro-tumorigenic effects distinct from stimulating cell division [51].
6.5 Maintaining Cancer Stemness
Recent work has suggested that CAFs may also have a role in maintaining the “stemness” of cancer stem cells. Work in the intestinal crypt has shown that Wnt signaling is important in the maintenance of stem cells and crypt homeostasis [52]. Vermeulen et al. used a reporter of β-catenin-driven transcription to show that there was heterogenous Wnt signaling within colon cancer spheroidal cultures despite all cancer cells having mutations in the APC gene. The cells with higher levels of Wnt signaling were shown to be “cancer stem cells,” that is, they had enhanced clonogenicity and were able to recreate a tumor similar to the initial malignancy if injected into an immune-deficient mouse . As there was heterogeneity in the spheroids, it was apparent that there was some cell autonomous regulation of Wnt signaling, and this has been shown previously in stem cells in the crypts [53]. However Wnt signaling in both the normal and the cancer stem cells is also modulated by surrounding cells [54]. Subsequently it was shown that HGF produced by CAFs induced high levels of Wnt signaling and increased “stemness” [55]. Thus in at least one system, CAFs have been reported to be important for the maintenance of cancer “stemness.”
6.6 Driving Angiogenesis
In their study mentioned earlier, Orimo et al. isolated fibroblasts from human breast carcinomas and normal fibroblasts from the same patients. These fibroblasts were co-injected with breast carcinoma cells into nude mice, and it was shown that CAFs promoted tumor growth significantly more than did the normal fibroblasts. This was shown to be due, at least partially, to the high levels of CXCL12 secreted by CAFs which recruited endothelial progenitors, increasing vascularization of the tumors [45]. Furthermore Yang et al. subsequently showed that CAFs isolated from human prostate cancer similarly promoted xenograft growth due to the action of CTGF. It was found that CTGF expression was induced by TGFβ and that overexpression of CTGF in 3T3 fibroblasts also led to these cells increasing tumor size and microvessel density in a xenograft model [56]. CAFs can also promote angiogenesis in a less direct method by releasing active growth factors from the ECM due to their expression of MMPs. CAFs are a source of MMP9 [57] and MMP13 [58], which have both been shown to be involved in angiogenesis. MMP9 and MMP13 have both been shown to release vascular endothelial growth factors (VEGF) from the ECM increasing angiogenesis in the tumor [59, 60]. This work is complicated by the fact that in integrin α1 knock out mice which lack integrin α1β1, an inhibitor of MMP synthesis, there is decreased tumor vascularization due to the increased production of angiostatin [61]. Angiostatin is produced by MMP9 and MMP7 acting on circulating plasminogen. As such MMPs have been shown to have conflicting roles in angiogenesis. As stated previously MMPs are not restricted to CAFs as TAMs, and other stromal cells are also important sources of these molecules [62].
6.7 Promoting Invasion and Metastasis
CAFs may also exert their effects through modulation of ECM composition; Levental et al. showed that CAFs express lysyl oxidase, which leads to collagen cross-linking and increased tissue stiffness. This increased stiffness was associated with changes in integrin signaling and progression to invasive disease, and treatment of the mammary fat pad with lysyl oxidase could promote growth and invasion of premalignant cells highlighting the critical role for these stromal cells [63]. CAFs have also been shown to express multiple MMPs that, by altering interactions between tumor cells and the extracellular matrix, alter tumor cell phenotype [57, 64, 65]. MMP activity has been implicated in all of the functions of CAFs so far, and it has been demonstrated that activation of MMPs is sufficient to produce a CAF-like phenotype in fibroblasts [66]. In this study, by deleting all four tissue inhibitors of metalloproteinases (TIMPs) , the authors demonstrated that exosomes from the CAFs induced cancer cell motility and upregulated stem cell markers. These effects were dependent on the metalloproteinase ADAM10 [66]. Fibroblast exosomes have been shown to drive Wnt-planar cell polarity signaling in a CD81-dependent manner. This promoted breast cancer cell invasive behavior [67]. Interestingly communication between cancer cells and stromal components using exosomes appears to occur in both directions to promote metastasis with transfer of miR-105 from cancer cells to endothelial cells leading to increased vascular permeability and metastasis [68]. Furthermore cancer-derived exosomes appear to have roles in transmitting invasive behaviors between different cancer cell clones [69]. Intriguingly it appears that stromal cells may even play a more direct role in metastasis, with tumor cells which were part of heterotypic cell clusters demonstrating increased robustness and increased potential to develop metastases. Furthermore using a parabiosis model CAFs from the original tumor could transiently be found in metastases indicating these cells could be important in establishing metastases [70].
6.8 Promoting Resistance to Therapy
CAFs have also been implicated in driving resistance to chemotherapy . Previous work using the KPC mouse has shown that the chemotherapeutic drug gemcitabine is excluded due to dense ECM and high intratumoral tissue pressure. Enzymatically disrupting the ECM led to increased infiltration of the tumor and increased response [71, 72]. While this is a presumed effect of CAFs due to their role in producing the dense desmoplastic stroma in these tumors, fibroblast-derived exosomes have been shown to directly promote resistance to chemotherapy. One study demonstrated that exosomes carried numerous RNAs which stimulated RIG-I in breast cancer cells and along with NOTCH3 signaling driven by the CAFs themselves converged on STAT1 signaling which led to expansion of tumor cells resistant to both chemotherapy and radiotherapy [73].
6.9 CAFs and Inflammation
There have been also been unbiased approaches to understand how CAFs differ from normal fibroblasts in an attempt to define their roles in cancer. FAP+ cells sorted from normal tissues were found by RNA-seq to be similar to one another, while different CAF populations characterized by co-expression or lack of CD34 were found to be more similar to one another validating this as an approach [74]. Hanahan conducted a more complete analysis sorting PDGFR-α+ cells from a range of normal tissues and from tumors and demonstrated a CAF-specific gene signature [23]. This gene signature was characterized by CAFs producing greater amounts of CXCL1, CXCL2, IL-1β, and IL-6 (among others) than fibroblasts in normal tissues. This inflammatory gene signature implies that CAFs could be important players within the tumor inflammatory environment. Previous work has shown that inflammatory mediators play important roles in carcinogenesis: IL-6 may also protect tumor cells from apoptosis in a STAT3-dependent mechanism [75] and can drive angiogenesis [76], and IL-1β has also been shown to drive IL-23 expression and thus to promote skin carcinogenesis [77]. Despite these more direct effects, the inflammatory signature also suggests that CAFs may be modulating the immune system in the tumor microenvironment.
6.10 Tumor Immunology
While the role for the tumor stroma in tumor development was becoming more widely accepted, the role for the immune system in tumor development was controversial until more recently. Indeed in 2000 Hanahan and Weinberg wrote a highly influential review about the hallmarks of cancer noting that there was an overly narrow focus on the genetically deranged cancer cells and that heterotypic signaling with normal stromal cells explained at least some of the aforementioned hallmarks of cancer [78]; however, it wasn’t until 2011 when they penned an update that evading the immune system appeared as a critical hallmark of tumor development [79]. Interest in the immune response to cancer reemerged as more focused approaches to immune modulation began to demonstrate results. After the initial demonstration that blockade of CTLA-4 could induce rejection of a transplanted primary tumor and lead to protection from a rechallenge [80], immune therapies gradually made their way to the clinic. Recently numerous immunotherapeutic approaches to tumor therapy have shown dramatic responses in patients resulting in numerous approvals of checkpoint blockade targeting CTLA-4 and PD-1 [81,82,83,84,85,86,87], as well as the use of chimeric antigen receptor (CAR) T cells to target tumor stroma [88]. It is now clear that vast complement of immune cells populate tumors, including dendritic cells, macrophages, neutrophils, natural killer cells, mast cells, B cells, CD8+ T cells, CD4+ T cells (and the many subsets thereof), and regulatory T cells (Treg cells). These cells have multiple roles in tumor and can have both pro- and antitumoral effects. These effects are, at least in part, directed by the microenvironment and are a product of CAFs and the inflammatory milieu they contribute to.
6.11 Stromal and Tumor Immune Responses
While these two areas have experienced renewed interest, interactions between these fields have only begun to emerge more recently. One of the most well-defined roles CAFs play in suppressing the antitumoral immune response regards entry of T cells. T cell infiltration into cancer nests within the tumor has long been recognized as an important predictor of patient survival, with increased infiltration of CD8+ T cells into cancer-dense regions strongly associated with improved outcomes for the patient [89,90,91,92]. Indeed, recent clinical trials using checkpoint blockade inhibitors and/or adoptive T cell therapy do not show strong results in tumors typically associated with high stromal burden, such as pancreatic ductal adenocarcinoma, prostate cancer, ovarian cancer, and colorectal cancer [82, 83, 93], in part due to a failure of CD8+ T cell to infiltrate cancer nests [94]. Normally, once activated, T cells leave the lymph node and migrate toward the inflammatory site, where they exit the blood stream and enter the tumor. The tumor vasculature inhibits extravasation of T cells from the blood stream into the tumor mass, resulting in accumulation of T cells within the stroma while still allowing movement of monocytes and neutrophils into the tumor. Indeed, overexpression of endothelin B receptor on tumor vasculature decreases lymphocyte adherence to endothelium [95], while the disorganization of tumor vasculature that is typically associated with tumor progression [96] limits T cell extravasation and entry into the tumor parenchyma [97].
6.12 CAFs and T Cell Infiltration
CAFs themselves may also directly limit T cell infiltration of cancer nests within the tumor. CAFs both deposit and degrade extracellular matrix (ECM) components and thus remodel the ECM during cancer progression; a severe desmoplastic reaction correlates with poor prognosis in many cancers [98,99,100,101]. The remodeling of the ECM allows CAFs to determine the migration and localization of cells within the tumor. Live cell imaging of human lung cancer [102] demonstrated poor T cell infiltration and motility in the dense collagen surrounding cancer nests, while more active T cell behavior was observed in regions with looser matrix deposition. Treatment with collagenase degraded the dense matrix surrounding cancer nests, and increased T cell infiltration and contact with cancer cells, suggesting that the nature of matrix deposition can have profound effects on the efficacy of antitumor T cell immunity. Recently, Fearon and colleagues demonstrated that, through production of CXCL12, pancreatic cancer-associated CAFs inhibit T cell infiltration of pancreatic carcinoma [74]. In this study, administration of checkpoint blockade inhibitors alone did not alter the course of tumor progression, mirroring that observed for human pancreatic cancer [93]. However, administration of the CXCR4 antagonist AMD3100 alongside checkpoint inhibitors significantly diminished tumor growth and allowed T cell infiltration and killing of cancer cells. Overexpression of CXCL12 by CAFs [103] has been shown to promote cancer growth through direct effects on the cancer cells, promoting cancer cell proliferation, migration, and invasion [104,105,106,107,108,109]. However, since no change in tumor growth was observed in the absence of a T cell response, CXCL12 blockade must be affecting the immune regulation rather than the tumor-promoting aspect of CXCL12-CXCR4 signaling. Importantly, these studies have shown that the pre-existing T cell response is capable of inducing tumor regression when suppression is alleviated, suggesting that vaccination again tumor antigens will not be necessary to harness the antitumor immune response for immunotherapy.
6.13 Suppressing Intratumoral T Cells
Once T cells enter a tumor, they must migrate toward the cancer cells, engage the T cell receptor, and then deliver cytotoxic- and/or cytokine-mediated kill signals. There are several obstacles that T cells must overcome in the tumor microenvironment in order to achieve their aim. The tumor microenvironment is full of suppressive signals for T cells, including secreted factors, suppressive immune cells, and the immunosuppressive actions of CAFs. CAFs have been shown to exert a directly immunosuppressive mechanism of action. Depletion of FAP-expressing stromal cells using a diphtheria toxin-mediated model results in failure of tumor growth that is entirely dependent on the presence of an intact immune response, demonstrating that CAFs are a critical regulator of tumoral immunosuppression of the T cell response [110]. Tumor killing in this model was dependent on interferon-gamma and TNF-alpha, and was induced by hypoxic necrosis, all hallmarks of T cell-mediated immunity. Some factors produced by CAFs have also been identified; CAFs, along with myeloid cells and cancer cells, are important sources of tumoral indoleamine 2,3-dioxygenase (IDO) , an enzyme that depletes local tryptophan by catabolizing tryptophan through the kynurenine pathway. Overexpression of IDO in the tumor microenvironment promotes tumor growth through immune resistance, as both T cell- and NK cell-mediated antitumoral responses are severely dysfunctional in the presence of IDO [111, 112]. CAFs also secrete TGFβ, and tumoral TGFβ expression is associated with significant pro-tumoral effect. TGFβ reduces CD8+ T cell and NK cell function [113,114,115], promotes the polarization of macrophages and neutrophils to a pro-tumoral type 2 phenotype [116,117,118], and enhances Treg cell and Th17 cell differentiation [119,120,121]. It is important to note that CAFs are not the sole source of TGFβ in the tumor microenvironment, with both neoplastic cells and myeloid-derived cells expressing high amounts of this cytokine. Accordingly, TGFβ blockade significantly inhibits tumor progression and enhances tumor immunotherapy [122, 123]. In addition to these effects, CAFs have been shown to promote the accumulation of immature myeloid cells, or MDSCs [124], which produce large amounts of suppressive cytokines, such as TGFβ, into the tumor microenvironment.
6.14 Skewing T Cell Recruitment
Alteration of CAFs by cancer cells has also been shown to alter the immune cell components within the tumor. For example, expression of the lymph node chemoattractant chemokine CCL21 in cancer cells, which is induced in invasive cancer cells [125], resulted stromal organization reminiscent of fibroblastic reticular cells (FRCs) of the lymph node. This FRC-like stromal organization recruited CCR7-expressing DCs, naïve T cells, and Treg cells to the tumor, leading to enhanced tumor growth through suppressive mechanisms including Treg cells and secreted molecules such as IDO [126]. Indeed, suppression was so profound it was able to prevent rejection of non-syngeneic allografts, demonstrating the immense capability of the tumor microenvironment to prevent T cell-mediated eradication of tumors. While this has not been shown to occur in human tumors, it points to the intriguing possibility that fibroblast populations as well as being heterogeneous are incredibly plastic, and our understanding of CAFs may be increased by investigating their roles in normal tissues like the lymph node.
6.15 Conclusions
There has been great interest in the potential of targeting CAFs in order to improve the response to immunotherapy in the clinic. While the responses to checkpoint blockade have been impressive with a proportion of patients showing complete, durable remissions [87], there are still many patients who do not respond. While direct ablation of CAFs led to tumor regression with immunogenic tumors [74, 110], the lack of specificity for the tumor led to significant side effects including cachexia and anemia when these cells were eliminated through administration of diphtheria toxin or by targeting with a FAP-specific chimeric antigen receptor T cell [29, 127]. As stated earlier there is a lack of markers that are specific for CAFs, and as such this type of approach is currently not feasible, and so greater understanding of the mechanisms by which CAFs exert their effects is needed in order to develop more targeted therapeutics. A promising example is the previously mentioned study by Feig et al. demonstrating that AMD3100 could potentiate the activity of checkpoint blockade [74]. As previously stated the heterogeneity of normal tissue fibroblasts [30] would suggest that while Hanahan found a CAF signature across different tumor types [23], there is still likely to be great heterogeneity in the mechanisms of immune suppression that CAFs are employing in different contexts. As such it is unlikely that there is any single CAF mechanism of action that can be targeted across all tumor types. It is critical that future work continues to elucidate the essential roles CAFs are playing in immune modulation in different tumor types, and it is also crucial that findings in mouse models are extended into patient samples to examine whether these potential therapeutic directions are viable.
References
Balkwill F, Mantovani A. Inflammation and cancer: back to Virchow? Lancet. 2001;357(9255):539–45.
Paget S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1989;8(2):98–101.
Busch W. Aus der sitzung der medicinischen section vom 13. Berliner Klin Wochenschrift. 1868;5:137.
Fehleisen F. Ueber die Zuchtung der Erysipelkokken auf kunstlichem Nahrboden und die Ubertragbarkeit auf den Menschen. Dtsch Med Wochenschr. 1882;8:553–4.
Bruns P. Die Heilwirkung des Erysipelas auf Geschwuelste. Beitr Klin Chir. 1888;3:443
Coley WB. A preliminary note on the treatment of inoperable sarcoma by the toxic product of erysipelas. Postgrad Dent. 1893;8:278.
Bissell MJ, Hall HG, Parry G. How does the extracellular matrix direct gene expression? J Theor Biol. 1982;99(1):31–68.
Illmensee K, Mintz B. Totipotency and normal differentiation of single teratocarcinoma cells cloned by injection into blastocysts. Proc Natl Acad Sci U S A. 1976;73(2):549–53.
Weaver VM, Petersen OW, Wang F, Larabell CA, Briand P, Damsky C, et al. Reversion of the malignant phenotype of human breast cells in three-dimensional culture and in vivo by integrin blocking antibodies. J Cell Biol. 1997;137(1):231–45.
Virchow R. Die Cellularpathologie in Ihrer Begruendung auf Physiologische und Pathologische Gewebelehre. Hirschwald, A, Berlin. 1858.
Tarin D, Croft CB. Ultrastructural features of wound healing in mouse skin. J Anat. 1969;105(Pt 1):189–90.
Bellusci S, Grindley J, Emoto H, Itoh N, Hogan BL. Fibroblast growth factor 10 (FGF10) and branching morphogenesis in the embryonic mouse lung. Development. 1997;124(23):4867–78.
Wahl SM, McCartney-Francis N, Allen JB, Dougherty EB, Dougherty SF. Macrophage production of TGF-beta and regulation by TGF-beta. Ann N Y Acad Sci. 1990;593:188–96.
Bonner JC, Osornio-Vargas AR, Badgett A, Brody AR. Differential proliferation of rat lung fibroblasts induced by the platelet-derived growth factor-AA, -AB, and -BB isoforms secreted by rat alveolar macrophages. Am J Respir Cell Mol Biol. 1991;5(6):539–47.
Skalli O, Ropraz P, Trzeciak A, Benzonana G, Gillessen D, Gabbiani G. A monoclonal antibody against alpha-smooth muscle actin: a new probe for smooth muscle differentiation. J Cell Biol. 1986;103(6 Pt 2):2787–96.
Ryan GB, Cliff WJ, Gabbiani G, Irle C, Montandon D, Statkov PR, et al. Myofibroblasts in human granulation tissue. Hum Pathol. 1974;5(1):55–67.
Dvorak HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315(26):1650–9.
Ronnov-Jessen L, Petersen OW. Induction of alpha-smooth muscle actin by transforming growth factor-beta 1 in quiescent human breast gland fibroblasts. Implications for myofibroblast generation in breast neoplasia. Lab Investig. 1993;68(6):696–707.
Le Hir M, Hegyi I, Cueni-Loffing D, Loffing J, Kaissling B. Characterization of renal interstitial fibroblast-specific protein 1/S100A4-positive cells in healthy and inflamed rodent kidneys. Histochem Cell Biol. 2005;123(4–5):335–46.
Strutz F, Okada H, Lo CW, Danoff T, Carone RL, Tomaszewski JE, et al. Identification and characterization of a fibroblast marker: FSP1. J Cell Biol. 1995;130(2):393–405.
Tomasek JJ, Gabbiani G, Hinz B, Chaponnier C, Brown RA. Myofibroblasts and mechano-regulation of connective tissue remodelling. Nat Rev Mol Cell Biol. 2002;3(5):349–63.
Ronnov-Jessen L, Petersen OW, Bissell MJ. Cellular changes involved in conversion of normal to malignant breast: importance of the stromal reaction. Physiol Rev. 1996;76(1):69–125.
Erez N, Truitt M, Olson P, Arron ST, Hanahan D. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-kappaB-dependent manner. Cancer Cell. 2010;17(2):135–47.
Vinores SA, Henderer JD, Mahlow J, Chiu C, Derevjanik NL, Larochelle W, et al. Isoforms of platelet-derived growth factor and its receptors in epiretinal membranes: immunolocalization to retinal pigmented epithelial cells. Exp Eye Res. 1995;60(6):607–19.
Garin-Chesa P, Old LJ, Rettig WJ. Cell surface glycoprotein of reactive stromal fibroblasts as a potential antibody target in human epithelial cancers. Proc Natl Acad Sci U S A. 1990;87(18):7235–9.
Bauer S, Jendro MC, Wadle A, Kleber S, Stenner F, Dinser R, et al. Fibroblast activation protein is expressed by rheumatoid myofibroblast-like synoviocytes. Arthritis Res Ther. 2006;8(6):R171.
Levy MT, McCaughan GW, Abbott CA, Park JE, Cunningham AM, Muller E, et al. Fibroblast activation protein: a cell surface dipeptidyl peptidase and gelatinase expressed by stellate cells at the tissue remodelling interface in human cirrhosis. Hepatology. 1999;29(6):1768–78.
Denton AE, Roberts EW, Linterman MA, Fearon DT. Fibroblastic reticular cells of the lymph node are required for retention of resting but not activated CD8+ T cells. Proc Natl Acad Sci U S A. 2014;111(33):12139–44.
Roberts EW, Deonarine A, Jones JO, Denton AE, Feig C, Lyons SK, et al. Depletion of stromal cells expressing fibroblast activation protein-alpha from skeletal muscle and bone marrow results in cachexia and anemia. J Exp Med. 2013;210(6):1137–51.
Chang HY, Chi JT, Dudoit S, Bondre C, van de Rijn M, Botstein D, et al. Diversity, topographic differentiation, and positional memory in human fibroblasts. Proc Natl Acad Sci U S A. 2002;99(20):12877–82.
Kidd S, Spaeth E, Watson K, Burks J, Lu H, Klopp A, et al. Origins of the tumor microenvironment: quantitative assessment of adipose-derived and bone marrow-derived stroma. PLoS One. 2012;7(2):e30563.
Kojima Y, Acar A, Eaton EN, Mellody KT, Scheel C, Ben-Porath I, et al. Autocrine TGF-beta and stromal cell-derived factor-1 (SDF-1) signaling drives the evolution of tumor-promoting mammary stromal myofibroblasts. Proc Natl Acad Sci U S A. 2010;107(46):20009–14.
Quante M, Tu SP, Tomita H, Gonda T, Wang SS, Takashi S, et al. Bone marrow-derived myofibroblasts contribute to the mesenchymal stem cell niche and promote tumor growth. Cancer Cell. 2011;19(2):257–72.
Giannoni E, Bianchini F, Masieri L, Serni S, Torre E, Calorini L, et al. Reciprocal activation of prostate cancer cells and cancer-associated fibroblasts stimulates epithelial-mesenchymal transition and cancer stemness. Cancer Res. 2010;70(17):6945–56.
Lohr M, Schmidt C, Ringel J, Kluth M, Muller P, Nizze H, et al. Transforming growth factor-beta1 induces desmoplasia in an experimental model of human pancreatic carcinoma. Cancer Res. 2001;61(2):550–5.
Shao ZM, Nguyen M, Barsky SH. Human breast carcinoma desmoplasia is PDGF initiated. Oncogene. 2000;19(38):4337–45.
Yang G, Rosen DG, Zhang Z, Bast RC Jr, Mills GB, Colacino JA, et al. The chemokine growth-regulated oncogene 1 (Gro-1) links RAS signaling to the senescence of stromal fibroblasts and ovarian tumorigenesis. Proc Natl Acad Sci U S A. 2006;103(44):16472–7.
Lieubeau B, Garrigue L, Barbieux I, Meflah K, Gregoire M. The role of transforming growth factor beta 1 in the fibroblastic reaction associated with rat colorectal tumor development. Cancer Res. 1994;54(24):6526–32.
Bachem MG, Schneider E, Gross H, Weidenbach H, Schmid RM, Menke A, et al. Identification, culture, and characterization of pancreatic stellate cells in rats and humans. Gastroenterology. 1998;115(2):421–32.
Yin C, Evason KJ, Asahina K, Stainier DY. Hepatic stellate cells in liver development, regeneration, and cancer. J Clin Invest. 2013;123(5):1902–10.
Ishii G, Sangai T, Oda T, Aoyagi Y, Hasebe T, Kanomata N, et al. Bone-marrow-derived myofibroblasts contribute to the cancer-induced stromal reaction. Biochem Biophys Res Commun. 2003;309(1):232–40.
Direkze NC, Hodivala-Dilke K, Jeffery R, Hunt T, Poulsom R, Oukrif D, et al. Bone marrow contribution to tumor-associated myofibroblasts and fibroblasts. Cancer Res. 2004;64(23):8492–5.
Iwano M, Plieth D, Danoff TM, Xue C, Okada H, Neilson EG. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest. 2002;110(3):341–50.
Zeisberg EM, Potenta S, Xie L, Zeisberg M, Kalluri R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer Res. 2007;67(21):10123–8.
Orimo A, Gupta PB, Sgroi DC, Arenzana-Seisdedos F, Delaunay T, Naeem R, et al. Stromal fibroblasts present in invasive human breast carcinomas promote tumor growth and angiogenesis through elevated SDF-1/CXCL12 secretion. Cell. 2005;121(3):335–48.
Yee D, Rosen N, Favoni RE, Cullen KJ. The insulin-like growth factors, their receptors, and their binding proteins in human breast cancer. Cancer Treat Res. 1991;53:93–106.
Frazier KS, Grotendorst GR. Expression of connective tissue growth factor mRNA in the fibrous stroma of mammary tumors. Int J Biochem Cell Biol. 1997;29(1):153–61.
Ponten F, Ren Z, Nister M, Westermark B, Ponten J. Epithelial-stromal interactions in basal cell cancer: the PDGF system. J Invest Dermatol. 1994;102(3):304–9.
Nakamura T, Matsumoto K, Kiritoshi A, Tano Y, Nakamura T. Induction of hepatocyte growth factor in fibroblasts by tumor-derived factors affects invasive growth of tumor cells: in vitro analysis of tumor-stromal interactions. Cancer Res. 1997;57(15):3305–13.
Kessenbrock K, Plaks V, Werb Z. Matrix metalloproteinases: regulators of the tumor microenvironment. Cell. 2010;141(1):52–67.
Wang Y, Sudilovsky D, Zhang B, Haughney PC, Rosen MA, Wu DS, et al. A human prostatic epithelial model of hormonal carcinogenesis. Cancer Res. 2001;61(16):6064–72.
Korinek V, Barker N, Moerer P, van Donselaar E, Huls G, Peters PJ, et al. Depletion of epithelial stem-cell compartments in the small intestine of mice lacking Tcf-4. Nat Genet. 1998;19(4):379–83.
Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459(7244):262–5.
Yen TH, Wright NA. The gastrointestinal tract stem cell niche. Stem Cell Rev. 2006;2(3):203–12.
Vermeulen L, De Sousa EMF, van der Heijden M, Cameron K, de Jong JH, Borovski T, et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol. 2010;12(5):468–76.
Yang F, Tuxhorn JA, Ressler SJ, McAlhany SJ, Dang TD, Rowley DR. Stromal expression of connective tissue growth factor promotes angiogenesis and prostate cancer tumorigenesis. Cancer Res. 2005;65(19):8887–95.
Boire A, Covic L, Agarwal A, Jacques S, Sherifi S, Kuliopulos A. PAR1 is a matrix metalloprotease-1 receptor that promotes invasion and tumorigenesis of breast cancer cells. Cell. 2005;120(3):303–13.
Vosseler S, Lederle W, Airola K, Obermueller E, Fusenig NE, Mueller MM. Distinct progression-associated expression of tumor and stromal MMPs in HaCaT skin SCCs correlates with onset of invasion. Int J Cancer. 2009;125(10):2296–306.
Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, et al. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol. 2000;2(10):737–44.
Lederle W, Hartenstein B, Meides A, Kunzelmann H, Werb Z, Angel P, et al. MMP13 as a stromal mediator in controlling persistent angiogenesis in skin carcinoma. Carcinogenesis. 2010;31(7):1175–84.
Pozzi A, Moberg PE, Miles LA, Wagner S, Soloway P, Gardner HA. Elevated matrix metalloprotease and angiostatin levels in integrin alpha 1 knockout mice cause reduced tumor vascularization. Proc Natl Acad Sci U S A. 2000;97(5):2202–7.
Mantovani A, Schioppa T, Porta C, Allavena P, Sica A. Role of tumor-associated macrophages in tumor progression and invasion. Cancer Metastasis Rev. 2006;25(3):315–22.
Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139(5):891–906.
Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer. 2006;6(5):392–401.
Lochter A, Galosy S, Muschler J, Freedman N, Werb Z, Bissell MJ. Matrix metalloproteinase stromelysin-1 triggers a cascade of molecular alterations that leads to stable epithelial-to-mesenchymal conversion and a premalignant phenotype in mammary epithelial cells. J Cell Biol. 1997;139(7):1861–72.
Shimoda M, Principe S, Jackson HW, Luga V, Fang H, Molyneux SD, et al. Loss of the Timp gene family is sufficient for the acquisition of the CAF-like cell state. Nat Cell Biol. 2014;16
Luga V, Zhang L, Viloria-Petit AM, Ogunjimi AA., Inanlou MR, Chiu E, et al. Exosomes mediate stromal mobilization of autocrine Wnt-PCP signaling in breast cancer cell migration. Cell. Elsevier Inc. 2012;151(7):1542–56.
Zhou W, Fong MY, Min Y, Somlo G, Liu L, Palomares MR, et al. Cancer-secreted miR-105 destroys vascular endothelial barriers to promote metastasis. Cancer Cell. Elsevier Inc. 2014;25(4):501–15.
Zomer A, Maynard C, Verweij FJ, Kamermans A, Schäfer R, Beerling E, et al. In vivo imaging reveals extracellular vesicle-mediated phenocopying of metastatic behavior. Cell. 2015;161(5):1046–57.
Duda DG, Duyverman AM, Kohno M, Snuderl M, Steller EJ, Fukumura D, et al. Malignant cells facilitate lung metastasis by bringing their own soil. Proc Natl Acad Sci U S A. 2010;107(50):21677–82.
Jacobetz MA, Chan DS, Neesse A, Bapiro TE, Cook N, Frese KK, et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. Gut. 2013;62(1):112–20.
Provenzano PP, Cuevas C, Chang AE, Goel VK, Von Hoff DD, Hingorani SR. Enzymatic targeting of the stroma ablates physical barriers to treatment of pancreatic ductal adenocarcinoma. Cancer Cell. Elsevier Inc. 2012;21(3):418–29.
Boelens MC, Wu TJ, Nabet BY, Xu B, Qiu Y, Yoon T, et al. Exosome transfer from stromal to breast cancer cells regulates therapy resistance pathways. Cell. Elsevier Inc. 2014;159(3):499–513.
Feig C, Jones JO, Kraman M, Wells RJ, Deonarine A, Chan DS, et al. Targeting CXCL12 from FAP-expressing carcinoma-associated fibroblasts synergizes with anti-PD-L1 immunotherapy in pancreatic cancer. Proc Natl Acad Sci U S A. 2013;110(50):20212–7.
Naugler WE, Karin M. The wolf in sheep’s clothing: the role of interleukin-6 in immunity, inflammation and cancer. Trends Mol Med. 2008;14(3):109–19.
Nilsson MB, Langley RR, Fidler IJ. Interleukin-6, secreted by human ovarian carcinoma cells, is a potent proangiogenic cytokine. Cancer Res. 2005;65(23):10794–800.
Langowski JL, Zhang X, Wu L, Mattson JD, Chen T, Smith K, et al. IL-23 promotes tumour incidence and growth. Nature. 2006;442(7101):461–5.
Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70.
Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–74.
Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science (80- ). 1996;271(5256):1734–6.
Hodi FS, O’Day SJ, McDermott DF, Weber RW, Sosman JA, Haanen JB, et al. Improved survival with ipilimumab in patients with metastatic melanoma. N Engl J Med. 2010;363(8):711–23.
Brahmer JR, Tykodi SS, Chow LQ, Hwu WJ, Topalian SL, Hwu P, et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N Engl J Med. 2012;366(26):2455–65.
Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med. 2012;366(26):2443–54.
Powles T, Eder JP, Fine GD, Braiteh FS, Loriot Y, Cruz C, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature. 2014;515(7528):558–62.
Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti-PD-L1 antibody MPDL3280A in cancer patients. Nature. 2014;515(7528):563–7.
Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature. 2014;515(7528):568–71.
Sharma P, Allison JP. The future of immune checkpoint therapy. Science (80- ). 2015;348(6230):56–61.
Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science (80- ). 2015;348(6230):62–8.
Naito Y, Saito K, Shiiba K, Ohuchi A, Saigenji K, Nagura H, et al. CD8+ T cells infiltrated within cancer cell nests as a prognostic factor in human colorectal cancer. Cancer Res. 1998;58(16):3491–4.
Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G, et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med. 2003;348(3):203–13.
Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F, et al. Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci U S A. 2005;102(51):18538–43.
Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science (80- ). 2006;313(5795):1960–4.
Royal RE, Levy C, Turner K, Mathur A, Hughes M, Kammula US, et al. Phase 2 trial of single agent Ipilimumab (anti-CTLA-4) for locally advanced or metastatic pancreatic adenocarcinoma. J Immunother. 2010;33(8):828–33.
Breart B, Lemaitre F, Celli S, Bousso P. Two-photon imaging of intratumoral CD8+ T cell cytotoxic activity during adoptive T cell therapy in mice. J Clin Invest. 2008;118(4):1390–7.
Buckanovich RJ, Facciabene A, Kim S, Benencia F, Sasaroli D, Balint K, et al. Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nat Med. 2008;14(1):28–36.
Ryschich E, Schmidt J, Hammerling GJ, Klar E, Ganss R. Transformation of the microvascular system during multistage tumorigenesis. Int J Cancer. 2002;97(6):719–25.
Hamzah J, Jugold M, Kiessling F, Rigby P, Manzur M, Marti HH, et al. Vascular normalization in Rgs5-deficient tumours promotes immune destruction. Nature. 2008;453(7193):410–4.
Cardone A, Tolino A, Zarcone R, Borruto Caracciolo G, Tartaglia E. Prognostic value of desmoplastic reaction and lymphocytic infiltration in the management of breast cancer. Panminerva Med. 1997;39(3):174–7.
Maeshima AM, Niki T, Maeshima A, Yamada T, Kondo H, Matsuno Y. Modified scar grade: a prognostic indicator in small peripheral lung adenocarcinoma. Cancer. 2002;95(12):2546–54.
Tsujino T, Seshimo I, Yamamoto H, Ngan CY, Ezumi K, Takemasa I, et al. Stromal myofibroblasts predict disease recurrence for colorectal cancer. Clin Cancer Res. 2007;13(7):2082–90.
Kawase A, Ishii G, Nagai K, Ito T, Nagano T, Murata Y, et al. Podoplanin expression by cancer associated fibroblasts predicts poor prognosis of lung adenocarcinoma. Int J Cancer. 2008;123(5):1053–9.
Salmon H, Franciszkiewicz K, Damotte D, Dieu-Nosjean MC, Validire P, Trautmann A, et al. Matrix architecture defines the preferential localization and migration of T cells into the stroma of human lung tumors. J Clin Invest. 2012;122(3):899–910.
Allinen M, Beroukhim R, Cai L, Brennan C, Lahti-Domenici J, Huang H, et al. Molecular characterization of the tumor microenvironment in breast cancer. Cancer Cell. 2004;6(1):17–32.
Domanska UM, Timmer-Bosscha H, Nagengast WB, Oude Munnink TH, Kruizinga RC, Ananias HJ, et al. CXCR4 inhibition with AMD3100 sensitizes prostate cancer to docetaxel chemotherapy. Neoplasia. 2012;14(8):709–18.
Li X, Ma Q, Xu Q, Liu H, Lei J, Duan W, et al. SDF-1/CXCR4 signaling induces pancreatic cancer cell invasion and epithelial-mesenchymal transition in vitro through non-canonical activation of Hedgehog pathway. Cancer Lett. 2012;322(2):169–76.
Wang Z, Ma Q, Liu Q, Yu H, Zhao L, Shen S, et al. Blockade of SDF-1/CXCR4 signalling inhibits pancreatic cancer progression in vitro via inactivation of canonical Wnt pathway. Br J Cancer. 2008;99(10):1695–703.
Hall JM, Korach KS. Stromal cell-derived factor 1, a novel target of estrogen receptor action, mediates the mitogenic effects of estradiol in ovarian and breast cancer cells. Mol Endocrinol. 2003;17(5):792–803.
Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME, et al. Involvement of chemokine receptors in breast cancer metastasis. Nature. 2001;410(6824):50–6.
Scotton CJ, Wilson JL, Scott K, Stamp G, Wilbanks GD, Fricker S, et al. Multiple actions of the chemokine CXCL12 on epithelial tumor cells in human ovarian cancer. Cancer Res. 2002;62(20):5930–8.
Kraman M, Bambrough PJ, Arnold JN, Roberts EW, Magiera L, Jones JO, et al. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-alpha. Science (80- ). 2010;330(6005):827–30.
Li T, Yang Y, Hua X, Wang G, Liu W, Jia C, et al. Hepatocellular carcinoma-associated fibroblasts trigger NK cell dysfunction via PGE2 and IDO. Cancer Lett. 2012;318(2):154–61.
Uyttenhove C, Pilotte L, Theate I, Stroobant V, Colau D, Parmentier N, et al. Evidence for a tumoral immune resistance mechanism based on tryptophan degradation by indoleamine 2,3-dioxygenase. Nat Med. 2003;9(10):1269–74.
Thomas DA, Massague J. TGF-beta directly targets cytotoxic T cell functions during tumor evasion of immune surveillance. Cancer Cell. 2005;8(5):369–80.
Gorelik L, Flavell RA. Immune-mediated eradication of tumors through the blockade of transforming growth factor-beta signaling in T cells. Nat Med. 2001;7(10):1118–22.
Lee JC, Lee KM, Kim DW, Heo DS. Elevated TGF-beta1 secretion and down-modulation of NKG2D underlies impaired NK cytotoxicity in cancer patients. J Immunol. 2004;172(12):7335–40.
Gidekel Friedlander SY, Chu GC, Snyder EL, Girnius N, Dibelius G, Crowley D, et al. Context-dependent transformation of adult pancreatic cells by oncogenic K-Ras. Cancer Cell. 2009;16(5):379–89.
Tsunawaki S, Sporn M, Ding A, Nathan C. Deactivation of macrophages by transforming growth factor-beta. Nature. 1988;334(6179):260–2.
Allavena P, Sica A, Garlanda C, Mantovani A. The Yin-Yang of tumor-associated macrophages in neoplastic progression and immune surveillance. Immunol Rev. 2008;222:155–61.
Trapani JA. The dual adverse effects of TGF-beta secretion on tumor progression. Cancer Cell. 2005;8(5):349–50.
Ghiringhelli F, Puig PE, Roux S, Parcellier A, Schmitt E, Solary E, et al. Tumor cells convert immature myeloid dendritic cells into TGF-beta-secreting cells inducing CD4+CD25+ regulatory T cell proliferation. J Exp Med. 2005;202(7):919–29.
Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, et al. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature. 2006;441(7090):231–4.
Kim S, Buchlis G, Fridlender ZG, Sun J, Kapoor V, Cheng G, et al. Systemic blockade of transforming growth factor-beta signaling augments the efficacy of immunogene therapy. Cancer Res. 2008;68(24):10247–56.
Terabe M, Ambrosino E, Takaku S, O’Konek JJ, Venzon D, Lonning S, et al. Synergistic enhancement of CD8+ T cell-mediated tumor vaccine efficacy by an anti-transforming growth factor-beta monoclonal antibody. Clin Cancer Res. 2009;15(21):6560–9.
Mace TA, Ameen Z, Collins A, Wojcik S, Mair M, Young GS, et al. Pancreatic cancer-associated stellate cells promote differentiation of myeloid-derived suppressor cells in a STAT3-dependent manner. Cancer Res. 2013;73(10):3007–18.
Shields JD, Fleury ME, Yong C, Tomei AA, Randolph GJ, Swartz MA. Autologous chemotaxis as a mechanism of tumor cell homing to lymphatics via interstitial flow and autocrine CCR7 signaling. Cancer Cell. 2007;11(6):526–38.
Shields JD, Kourtis IC, Tomei AA, Roberts JM, Swartz MA. Induction of lymphoidlike stroma and immune escape by tumors that express the chemokine CCL21. Science (80- ). 2010;328(5979):749–52.
Tran E, Chinnasamy D, Yu Z, Morgan RA, Lee CC, Restifo NP, et al. Immune targeting of fibroblast activation protein triggers recognition of multipotent bone marrow stromal cells and cachexia. J Exp Med. 2013;210(6):1125–35.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Denton, A.E., Roberts, E.W., Fearon, D.T. (2018). Stromal Cells in the Tumor Microenvironment. In: Owens, B., Lakins, M. (eds) Stromal Immunology. Advances in Experimental Medicine and Biology, vol 1060. Springer, Cham. https://doi.org/10.1007/978-3-319-78127-3_6
Download citation
DOI: https://doi.org/10.1007/978-3-319-78127-3_6
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-78125-9
Online ISBN: 978-3-319-78127-3
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)