
Abstract
Three novel lanthanide-organic frameworks: [Ln2(pyba)3(μ3-OH)2(μ2-OH)(H2O)] n (Ln = Er (1), Y (2), Dy (3) Hpyba = 4-pyridin-4-yl-benzoic acid) have been hydrothermally synthesized and structurally characterized by single crystal X-ray diffraction. Structure analysis shows that each {Ln4(μ3-OH)4(μ2-OH)2} cluster units interconnect to form 1-D chains, which are further linked by π–π interactions to make a 3-D supramolecular network structure. Furthermore, the IR, PXRD and TGA of compounds 1–3 were also studied.
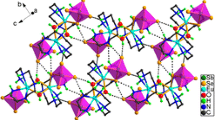
Graphical Abstract
Three novel lanthanide-organic frameworks: [Ln2(pyba)3(μ3-OH)2(μ2-OH)(H2O)] n (Ln = Er (1), Y (2), Dy (3), Hpyba = 4-pyridin-4-yl-benzoic acid) have been hydrothermally synthesized and structurally characterized. Single-crystal X-ray structure analysis shows that each {Ln4(μ3-OH)4(μ2-OH)2} cluster units interconnect to form 1-D chains, which are further linked by π–π interactions to make a 3-D supramolecular network.

Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In the last decade, the construction of metal–organic frameworks (MOFs) is of high interest in crystal engineering not only due to their intriguing variety of topologies and motifs but also because of their potential applications in ion-exchange, catalytic, adsorption, fluorescence, and magnetic properties [1–4]. However, most of the work has so far focused on the assembly of the transition metals (TMs), the analogous chemistry of lanthanides is still less developed [5–9]. It is may attribute to the tendency of the lanthanide (Ln) ions for high coordination, which favors the formation of condensed structures with or without ligands [10, 11]. Noticeably, selection of the multifunctional organic ligands is of vital importance in designing and assembling expected complexes. So far, various ligands are used to build these Ln-containing MOFs [12–20]. Our group has successfully prepared several Ln-containing MOFs by isonicotinic acid (Hin), a rigid linear ligand containing O and N donors on the opposite sides, which can construct the extended frameworks with high structural stability and special topologies [21–26]. Furthermore, we attempt to obtain more open frameworks under rationally hydrothermal conditions by replacing Hin with the lengthened ligand 4-pyridin-4-yl-benzoic acid (Hpyba). Till now, we have successfully obtained two novel 1-D stair-step chains built by the {Cu2X2 } cluster units, [Cu2X2(Hpyba)2] n (X = Br, I) [27] and a series of 1-D Ln-organic coordination polymers, [Ln(μ3-OH)(pybz)(pa)] n (Ln = Er, Tb, Gd; Hpa = 2-picolinic acid), based on [Ln4(μ3-OH)4] cubanes [28]. Here we report the first 1-D Ln-organic frameworks supported by the pyba ligands and based on {Ln4(μ3-OH)4(μ2-OH)2} tetrameric cluster units: [Ln2(pyba)3(μ3-OH)2(μ2-OH)(H2O)] n (Ln = Er (1), Y (2), Dy (3)).
Experimental
Materials and Methods
All chemicals were commercially purchased and used without further purification. IR spectra (KBr pellets) were recorded on an ABB Bomen MB102 spectrometer over a range 400–4000 cm−1. The thermogravimetric analysis was performed on a Mettler Toledo TGA/SDTA 851e analyzer in air atmosphere with a heating rate of 10 °C/min from 30 to 1000 °C. The elemental analysis was carried out using the combustion method on an Elemental Vario EL III CHNOS elemental analyzer.
Synthesis of [Er2(pyba)3(μ3-OH)2(μ2-OH)(H2O)] n (1)
A mixture of Er2O3 (1 mmol, 0.3824 g), Cu(Ac)2 · H2O (0.1 mmol, 0.0200 g), Hpyba (2 mmol, 0.3987 g), H2O (10 mL, 0.22 mmol) and three drops of HClO4 with the pH value of about 2.0 was sealed in a 30 mL Teflon-lined bomb at 190 °C for 7 days, and then cooled to room temperature, Yellow prismatic crystals of 1 were recovered by filtration, washed with distilled water and dried at ambient temperature (Yield, 32% based on Er2O3). Elemental Analysis: Calcd. for C36H29N3O10Er2: C 43.28, H 2.91, N 4.20. Found: C 43.21, H 2.87, N 4.15.
Synthesis of [Ln2(pyba)3(μ3-OH)2(μ2-OH)(H2O)] n (Ln = Y(2), Dy(3))
The crystals of 2 and 3 were prepared by a similar method as used in the synthesis of the crystals of 1 except that Er2O3 was replaced by Y2O3 and Dy2O3. The resulting colorless microcrystalline of 2 and 3 were obtained (yield: 28% based on Y2O3, 30% based on Dy2O3). X-ray diffraction data on single crystals show that the unit cell is very similar to 1. For 2: monoclinic, space group C2/c, a = 37.744(5) Å, b = 7.238(7) Å, c = 31.31(7) Å, β = 123.95(4)°, and V = 7096.7(3) Å3. For 3: monoclinic, space group C2/c, a = 37.561(4) Å, b = 7.205(6) Å, c = 31.787(4) Å, β = 123.65(4)°, and V = 7001.5(3) Å3. The experimental PXRD patterns of 2 and 3 correspond well with the simulated and experimental PXRD patterns of 1, further indicating that these compounds are isostructural (Fig. 1). Elemental Analysis: Calcd. for 2: C 51.37, H 3.45, N 4.82. Found: C 51.35, H 3.52, N 4.78; Calcd. for 3: C 43.40, H 2.93, N 4.21. Found: C 43.38, H 2.90, N 4.25.

The experimental and simulated PXRD patterns of compounds 1–3
X-ray Crystallography
The intensity data was collected on a Scxmini CCD diffractometer with a graphite-monochromatized Mo Kα (λ = 0.71073 Å) radiation at room temperature. All absorption corrections were performed using SADABS program [29]. The structure was solved by direct methods and refined by full-matrix least squares on F 2 with the SHELXL-97 program [30, 31]. Non-hydrogen atoms were refined anisotropically, while hydrogen atoms were introduced in calculated positions and refined isotropically. Details of crystal data, collection and refinement are listed in Table 1. Selected bond distance and angle data for 1 is listed in Table 2.
Result and Discussion
Synthesis Considerations
In order to make Ln-organic-TM frameworks, we add the Cu(CH3COO)2 · H2O into the reaction system. However, the Cu2+ cations have not been introduced into the reaction product. Only the Ln-organic frameworks 1–3 were obtained. But the starting materials, Cu(CH3COO)2 · H2O, is crucial for the formation of compounds 1–3, no single crystals were obtained in the absence of Cu(CH3COO)2 · H2O under the similar conditions.
Structure Description
X-ray crystal structure analyses and PXRD patterns reveal that compounds 1–3 are isomorphous and crystallize in monoclinic space group C2/c (Table 1 and Fig. 1). Therefore, only the structure of 1 is described in details. The asymmetric unit of 1 contains two unique Er3+ ions, one μ2-OH−, two μ3-OH−, one coordinated water molecule and three pybz− ligands with two different coordination modes in the ratio of 2:1 (Fig. 2). Er1 ion is eight-coordinated, and the coordination geometry is close to that of a bicapped trigonal prism: three OCOO (O4/O5 in mode I, O7 in mode II) from three pybz− ligands, a coordinated water (O1w), a μ2-hydroxyl (O2) and three μ3-bridging hydroxyl (O1/O9/O9a). Er2 ion surrounded by four OCOO (O3/O6 in mode I, O7/O8 in mode II) from three pybz− ligands, a μ2-bridging hydroxyl (O2) and three μ3-bridging hydroxyl (O1, O1a, O9). The coordination geometry for eight-coordinated Er2 ion is close to that of a bicapped trigonal prism as the same as Er1. The Er–O distances range from 2.229(3) to 2.538(3) Å.

a The asymmetric unit of 1 with the atom labeling (H atoms connected to carbon atoms are omitted for clarity). b Coordination modes of the pybz-ligands in 1
In the structure, one Er1 and two Er2 ions (Er1…Er2 3.7024 Å) are linked by three μ3-OH (O1/O1a/O9) groups and one μ2-OH (O2) to form a truncated cubane. Two such units connected each other to form a {Ln4(μ3-OH)4(μ2-OH)2} tetrameric cluster unit by sharing one four member ring (Er2–O1–Er2–O1). Then, these tetrameric cluster units are further linked by two μ3-OH (O9/O9a) groups, resulting in a 1-D chain (Fig. 3a), which differs from the reported 1-D chains built by [Er4(μ3-OH)4] cluster units [28]. The deprotonated pyba ligands are regularly appended up and down of the 1-D chain, linked via carboxylate oxygen atoms from ligands (Fig. 3b).

a View of the 1-D chain along the c-axis (H atoms connected to carbon atoms are omitted for clarity); b 1-D chain based on {Ln4(μ3-OH)4(μ2-OH)2} cluster units
The distance of every two chains is up to 19.1 Å parallel to the a-axis, which is much longer than the length of the pyba ligand; in addition, adjacent paralleled chains are linked together via weak π–π stacking, with the centeroid–centeroid distance between them is 3.643 Å (Fig. 4a), to form a 3-D open framework (Fig. 4b).

a The π–π stacking interactions between adjacent chains in 1; b the 3-D supramolecular network formed by the 1-D chains via π–π stacking interactions along the b-axis
IR Spectroscopy
As shown in Fig. 5, the characteristic features of pyba ligand dominate the IR spectrum. The strong and acuity absorption band around 3400 cm−1 were assigned as the characteristic peaks of OH vibration. The strong vibrations at 1556 and 1415 cm−1 are corresponding to the asymmetric and symmetric stretching vibrations of the carboxylate group, respectively. Bands in the 1000–1400 cm−1 range are attributed to v(C–N) and v(C–C) vibrations. The δ O–C–O vibration in plane occurs in middle intensity peaks around in 777 cm−1. The absence of strong bands ranging from 1690 to 1730 cm−1 indicates that the ligands are deproponated.

IR spectra of compounds 1–3
Thermal Properties
The thermogravimetric analysis was carried out in flowing dry air atmosphere with a heating rate of 10 °C min−1 in the temperature range of 30–1,000 °C. As shown in Fig. 6, the weight loss from 30 to 360 °C is attributed to the removal of the coordination water molecules (calcd. 1.8%, found 1.7%; 30–180 °C) and the dehydration of the hydroxyl groups (calcd. 2.7%, found 2.9%; 180–360 °C). Above 360 °C until 550 °C, one step of weight loss was observed, corresponding to the successive release of organic ligands. At 550 °C, the coordination networks of compound 1 decompose completely. Assuming that the residue corresponds to Er2O3, the observed weight (38.1%) is in good agreement with the calculated value (38.0%).

TG curve of compound 1
Conclusions
Three novel Ln-organic frameworks: [Ln2(pyba)3(μ3-OH)2(μ2-OH)(H2O)] n (Ln = Er (1), Y (2), Dy (3)) have been successfully made by hydrothermally method. Structure analysis shows that each {Ln4(μ3-OH)4(μ2-OH)2} units interconnect to form 1-D chains, which are further linked by π–π interactions to make a 3-D Ln-organic framework. This work provides a rational route for the construction of fascinating Ln-organic frameworks based on {Ln4(μ3-OH)4(μ2-OH)2} units. Further work will be focused on the construction of high-D Ln-organic-TM frameworks involving the combination of Ln clusters and TM clusters.
Supporting Information
CCDC-770644 contains the supplementary crystallographic data for 1. These data can be obtained free of charge via www.ccdc.cam.ac.uk/conts/retrieving.html (or from the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: (+44) 1223-336-033; or e-mail: deposit@ccdc.cam.ac.uk).
References
D. Gatteschi, L. Pardi, and A. Müller (1991). Nature 354, 463.
S. Kitagawa, R. Kitaura, and S. Noro (2004). Angew. Chem. Int. Ed. 43, 2334.
W.-H. Zhu, Z. M. Wang, and S. Gao (2007). Inorg. Chem. 46, 1337.
G. X. Liu, K. Zhu, and H. Chen (2008). CrystEngCommun. 10, 1527.
M. J. Zaworotko (1994). Chem. Soc. Rev. 23, 283.
M. Fujita, Y. J. Kwon, and S. Washizu (1994). J. Am. Chem. Soc. 116, 1151.
S. R. Batten and R. Robson (1998). Angew. Chem. Int. Ed. 37, 1460.
T. M. Reineke, M. O’Keeffe, and O. M. Yaghi (1999). Angew. Chem. Int. Ed. 38, 2590.
D. L. Long, A. J. Blake, and N. R. Champness (2001). Angew. Chem. Int. Ed. 40, 2443.
L. Pan, N. Zheng, and Y. J. Li (2001). Inorg. Chem. 40, 828.
L. Pan, E. B. Woodlock, and C. Zheng (2000). Inorg. Chem. 39, 4174.
T. M. Reineke, M. Eddaoudi, and O. M. Yaghi (1999). J. Am. Chem. Soc. 121, 1651.
T. Devic, C. Serre, and J. Marrot (2005). J. Am. Chem. Soc. 127, 12788.
L. Pan, K. M. Adams, and K. Kaneko (2003). J. Am. Chem. Soc. 125, 3062.
X. Zheng, C. Sun, and S. Gao (2004). Eur. J. Inorg. Chem. 3262.
Y. Q. Sun, J. Zhang, and G. Y. Yang (2005). Angew. Chem. Int. Ed. 44, 2.
N. Rosi, M. O’Keeffe, and O. M. Yaghi (2005). J. Am. Chem. Soc. 127, 1504.
G. Zhang, G. Yang, and J. S. Ma (2006). Cryst. Growth Des. 6, 933.
Y. G. Huang, F. L. Jiang, and M. C. Hong (2008). Cryst. Growth Des. 8, 166.
Z. Chen, B. Zhao, and P. Cheng (2008). Cryst. Growth Des. 8, 2291.
J. W. Cheng, J. Zhang, and G. Y. Yang (2006). Angew. Chem. Int. Ed. 45, 73.
J. W. Cheng, S. T. Zheng, and G. Y. Yang (2008). Chem. Eur. J. 14, 88.
M. B. Zhang, J. Zhang, and G. Y. Yang (2005). Angew. Chem. Int. Ed. 44, 1385.
J. W. Cheng, J. Zhang, and G. Y. Yang (2007). Inorg. Chem. 46, 10261.
J. W. Cheng, S. T. Zheng, and G. Y. Yang (2007). Inorg. Chem. 46, 10534.
J. W. Cheng, S. T. Zheng, and G. Y. Yang (2008). Inorg. Chem. 47, 4930.
X. L. Jia, J. Zhou, and G. Y. Yang (2009). J. Clust. Sci. 20, 555.
Z. L. Wang, W. H. Fang, and G. Y. Yang (2009). J. Clust. Sci. 20, 725.
G. M. Sheldrick, SHELXS97, Program for Siemens Area Detector Absorption Corrections (University of Göttingen. Germany, 1997).
G. M. Sheldrick, SHELXS 97, Program for Crystal Structure Solution (University of Göttingen. Germany, 1997).
G. M. Sheldrick, SHELXL97, Program for Crystal Structure Refinement (University of Göttingen, Germany, 1997).
Acknowledgements
The authors are thankful for the financial supports from the National Natural Science Fund for Distinguished Young Scholars of China (no. 20725101), the NNSF of China (no. 50872133), the 973 Program (no. 2006CB932904), the NSF of Fujian Province (nos. E0510030 and 2008F3120) and the Knowledge Innovation Program from CAS (no. KJCX2.YW.H01).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Fang, WH., Wang, ZL. & Yang, GY. Lanthanide-Organic Frameworks Based on {Ln4(μ3-OH)4(μ2-OH)2} Cluster Units. J Clust Sci 21, 187–195 (2010). https://doi.org/10.1007/s10876-010-0327-5
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10876-010-0327-5