
Abstract
There is substantial causal and consequential interaction between the ever-growing heart failure and renal failure patients. Half of the patients with heart failure (HF) have preserved left ventricular ejection fraction (HFpEF), which is difficult to diagnose and rising in prevalence relative to HF with reduced EF (HFpEF). To date, only weight reduction, exercise training, and diuretics have been shown to improve exercise tolerance and morbidity in HFpEF. This review aims to establish the baseline kidney-related concepts specific to the diagnosis and treatment of HFpEF patients and the different aspects of HFpEF and HFpEF in the clinical setting.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Many patients with heart failure (HF) have associated chronic kidney disease (CKD), in conjunction with a reduced glomerular filtration rate (GFR) or an increased protein/albumin excretion in urine. The risk of developing HF is substantially increased as the stage of CKD progresses [1]. Various mechanisms are responsible for this increased risk. First of all, certain common factors contribute to the development of both these entities including increased age, presence of diabetes mellitus, and hypertension [2]. Secondly, both CKD and HF can cause or worsen certain comorbidities such as anemia [3], coronary and peripheral atherosclerosis [4], and malnutrition [5]. Thirdly, kidney dysfunction contributes to HF by increased salt retention and volume expansion, by up-regulation of neurohormonal pathways, and by stimulating proinflammatory mechanisms [1]. HF, in turn, worsens CKD by decreasing renal perfusion and by activating catecholaminergic pathways as well as the renin–angiotensin–aldosterone system (RAS) [6,7,8].
All of the previous observations stem from studies pertaining to HF with reduced ejection fraction (HFrEF) or from studies where information regarding ejection fraction is lacking. However, recent evidence suggests that up to 30–50% of patients with HF have preserved left ventricular ejection fraction (HFpEF). Mortality in HFpEF is equally as high as in HFrEF [9, 10]. Impaired renal function is also a risk factor for developing HFpEF [11].
HFrEF and HFpEF exhibit numerous similarities as well as dissimilarities, with increased age, hypertension, and arterial stiffness being common factors underlying both conditions. In addition, both hospitalization and mortality rates of HFpEF are similar to those of HFrEF [12]. On the other hand, HFrEF is primarily associated with myocardial infarction [13], whereas combined ventricular–vascular stiffening (As-related abnormal left atrium–left ventricle coupling) is the main contributor of the increased prevalence of HFpEF. Additionally, despite an improvement in the prognosis of HFrEF in last decades, the effective treatment of HFpEF remains an unmet need [12].
Although the consequences of reduced ejection fraction are well known, there is a paucity of data regarding the effect of HFpEF on renal function. Treatment of HFrEF is relatively straightforward: loop diuretics in case of volume overload and mortality-reducing treatment with beta-blockers and angiotensin-converting enzyme inhibitors (ACE inhibitors) (or in instances of ACE inhibitor or angiotensin receptor blocker (ARB) intolerance), followed by mineralocorticoids). However, treatment options for HFpEF are less established given the lack of clear evidence-based data. Diuretics are useful for symptom relief in case of sodium and water retention in HFpEF patients, while blood pressure and comorbidities should be adequately managed [14]. In light of the previous data, the present review summarizes the relationship between renal function and HFpEF.
The epidemiology and magnitude of the problem
Impaired renal function may worsen prognosis in HFpEF [15, 16] due to mechanisms including increased inflammation and endothelial dysfunction, cardiomyocyte stiffening and growth, and interstitial fibrosis [9, 11, 17, 18].
HFpEF is common among patients with chronic kidney disease (CKD) [1, 19, 20] and is associated with increased mortality [19]. In HFpEF populations, CKD [defined as an estimated glomerular filtration rate (eGFR) <60 mL/min/1.73 m2] is observed in 26–49% of cases [9, 19]. Additionally, albuminuria is common in HFpEF [21]. When studying the impact of renal dysfunction on all-cause mortality in HFpEF, only lower eGFR at admission remained significantly predictive of all-cause mortality (hazard ratio (HR) 2.97 and 95% confidence interval (CI) 1.59–5.53) [22].
A recent meta-analysis showed that CKD was associated with an odds ratio (OR) of 3.22 (95% CI 2.66–3.90) for all-cause mortality in patients with an EF >40%, compared to ORs of 2.00 (95% CI 1.81–2.21) and 2.56 (95% CI 2.24–2.93) for an EF <30% and an EF between 30 and 40%, respectively. Thus, the presence of CKD results in a higher increase in mortality in patients with HFpEF compared to HFrEF [23]. In summary, a growing body of evidence suggests that CKD and HFpEF are closely interrelated when considering cardiovascular outcomes.
Pathogenesis of preserved ejection fraction
The pathophysiology of HFpEF is far from being completely understood. Increased age and age-related myocardial and vascular changes may play a major role, driving concentric remodeling and cardiovascular stiffness [24]. Importantly, comorbidities, such as hypertension, diabetes, and increased body mass index, are key components in the development of HFpEF.
Bhatia et al. studied 2802 patients in order to highlight the characteristics of patients with HF. Patients were categorized in three groups: those with an ejection fraction <40% (HFrEF), those with an ejection fraction of 40 to 50% (HF with borderline ejection fraction), and those with an ejection fraction >50% (HFpEF). Patients with HFpEF were more likely to be older and female and to have a history of hypertension and atrial fibrillation [25, 26], along with alterations in systemic vascular resistance (SVR). Anemia, a frequent comorbid condition of HFpEF, is also likely contributive to the observed lower SVRs in HFpEF patients [27]. However, in the setting of CKD, two of the most important pathophysiological features leading to HFpEF are thought to be endothelial dysfunction (ED) and chronic inflammation.
Under normal conditions, the endothelium displays anti-inflammatory properties which protect against thrombosis while regulating vascular tone. In CKD, persistent inflammation and ED are highly common [28]; thus, if the endothelium does not function properly (as in CKD), its anti-inflammatory and anti-thrombotic properties thereby decrease. This has both distant and paracrine effects, including ED-induced cardiac inflammation [29]. In addition, urinary sodium retention as well as altered levels of renal endocrine factors and serum calcium and phosphate has all been linked to ED [30, 31].
Imbalances in certain novel factors such as elevated fibroblast growth factor-23 (FGF-23) [32], decreased vitamin D [33], erythropoietin deficiency [34], and proteinuria [35] have been associated with ED. All of these mechanisms may explain the high incidence of ED in patients with CKD and HFpEF, ED being indeed common in patients with HFpEF [36, 37].
As suggested earlier, the second factor leading to HFpEF may be increased inflammation. Inflammation induces oxidative inactivation of nitric oxide (NO) since superoxide anions react with NO and form peroxynitrite; this is further supported by the recent finding of high nitrotyrosine expression in HFpEF myocardium [38]. Additionally, various inflammatory markers including interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α) are increased in HFpEF patients [39, 40]. Uremia associated with chronic inflammation also causes inhibition of endothelial proliferation through uremia-associated proinflammatory cytokines [41]. Furthermore, uremic toxins increase reactive oxygen species (ROS) in vascular endothelial cells, thereby causing oxidative stress [42].
In addition to ED and inflammation, hemodialysis (HD) treatment per se may also have an impact. In patients on HD, interaction with the dialysis membrane results in complement activation, leading to micro inflammation and may ultimately result in vascular stiffness and ED. It should be emphasized that uremia not only affects the endothelium but also causes vascular smooth muscle cell dysfunction [43].
Hypervolemia and sodium excess are commonly observed in CKD patients. Increased sodium can bind to the endothelial glycocalyx causing stiffened endothelial cell, decreased NO levels, and resulting in ED [44]. Since ED and HFpEF are interrelated [17], this mechanism may link sodium and hypervolemia in the development of HFpEF. Obviously, the role of sodium in the development of hypertension is mostly due to hypervolemia, a major common feature in CKD. A recent study demonstrated that HFpEF and HFrEF are both associated with hypervolemia [45].
In chronic renal failure, there is accumulation of advanced glycation end products (AGEs), due to decreased clearance and increased oxidative stress [46, 47]. AGEs may induce HFpEF by causing fibrosis due to cross-linking in the extracellular matrix, by activation of their receptor which has a proinflammatory effect, or by causing a delay in calcium uptake [48, 49]. AGEs also influence endothelial function by reducing NO availability [50].
In CKD patients, alterations in cardiovascular structure are commonly observed. In one study, renal dysfunction was identified as being associated with abnormal left ventricular (LV) geometry (defined as concentric hypertrophy, or eccentric hypertrophy, or concentric remodeling) leading to lower midwall fractional shortening (MWFS) [21]. Compared to patients without renal dysfunction, those with lower GFR and no albuminuria had a higher prevalence of abnormal LV geometry and lower MWFS as compared to those with only albuminuria. Conversely, albuminuria alone was associated with greater LV dimensions. Patients with combined renal impairment had mixed abnormalities (higher LV wall thicknesses, lower MWFS). The authors concluded that both eGFR and albuminuria are highly prevalent in HFpEF and are associated with cardiac remodeling, while adding the fact that the observed differences in cardiac structure/function between each type of renal damage suggest that both parameters of kidney function may play a distinct role in HFpEF [21].
Metabolic syndrome may also be associated with HFpEF. Zyatenkova et al. investigated epicardial fat thickness as a correlate of visceral fat thickness in patients with and without metabolic syndrome (MS). A total of 59 patients with HFpEF were included [29 without MS (group 1), 30 with MS (group 2)] in whom interventricular septum thickness, left ventricle wall thickness, and left ventricle myocardium mass, along with epicardial fat thickness and size of heart chambers, were assessed. Epicardial fat thickness was lower in non-MS patients compared to MS patients [51]. In another study, Karakurt et al. demonstrated that in patients with HFpEF, MS was associated with systolic and diastolic dysfunction [52,53,54].
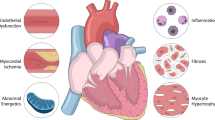
Thus, all of the aforementioned factors (ED, inflammation, hypervolemia, increased oxidative stress and AGE, dialysis-related factors, and MS) appear to activate a cascade of events resulting in cardiac hypertrophy and stiffness in HFpEF, different from HFrEF, and in which cardiomyocyte death and myocardial fibrosis are predominant features (Fig. 1). Such differences between HFpEF and HFrEF are addressed in greater detail in the following section.

Potential mechanisms for development of heart failure with preserved (HFpEF) in chronic kidney disease
Differences between preserved ejection fraction and reduced ejection fraction
Although it has been suggested that there is a true continuum between HFpEF and HFrEF, there are various differences between these two conditions (Table 1), suggesting that different phenotypes may be in play. These differences arise from pathogenetic, clinical, and therapeutic issues. In HFpEF, there is a systemic inflammatory state, causing increased coronary ROS formation and an increase in peroxynitrite. Consequently, there is a reduction in protein kinase G (PKG) activity [54], resulting in impaired relaxation, as well as myocardial stiffness leading to diastolic dysfunction [53]. Furthermore, autocrine and paracrine factors, such as apelin, transforming growth factor-β, and endothelin-1 from the endothelium, contribute to the development of cardiac hypertrophy [55]. As suggested earlier, cardiac hypertrophy is the main occurrence in HFpEF with little myocardial cell death, in contrast to HFrEF where cell death is more prominent and one of the most important mechanisms characterizing the latter [17, 53]. This distinction is one of the many differences between these two diseases, as outlined in the following.
As stated previously, renal dysfunction and HFpEF are closely related with regard to cardiovascular outcomes, and it is important to note that although conflicting data have been reported, not only baseline renal function but also changes in renal function over time should be considered. In acute decompensated HF patients, renal dysfunction at discharge was found an independent predictor of the primary outcome in patients with reduced EF, whereas it was not associated with the primary outcome in patients with preserved EF [56].
However, in another study, worsening renal function (WRF) (defined as an increase in creatinine of ≥0.3 mg/dl within 72 h after admission) was observed in 40% of HFpEF patients [20]. WRF in the setting of HFpEF was also found associated with poor prognosis [20, 57]. Higher blood pressure on admission and less fluid removal were significantly associated with higher risk of WRF in these patients [20], thus implying the importance of renal congestion in HF [58].
There are also contradictory data in this regard. Takei et al. investigated the effect of plasma volume reduction (PVR) in acute HF patients with both HFpEF and HFrEF. Estimated PVR (as a measure of greater volume reduction) was associated with WRF in HFpEF after adjusting for history of hypertension, diabetes mellitus, and estimated GFR (OD, 3.34; 95% CI, 1.52–7.33; P = 0.003). However, PVR had no effect on WRF in HFrEF. The authors concluded that the effect of estimated PVR differs according to HF type, and the estimated PVR during hospitalization is a predictor of WRF in patients with HFpEF but not in HF with reduced ejection fraction [59]. In contrast, in acutely decompensated HFrEF patients, hemoconcentration was strongly associated with WRF, but associated with lower mortality. Additionally; it is not clear whether less or more fluid removal is associated with WRF in HFpEF patients [60].
To explain the mechanisms regarding WRF in HFpEF patients, Abramov et al. reported that HFpEF patients had a lower total blood volume (i.e., intravascular volume] than those with HFrEF [61]. Similarly, Schwartzenberg et al. reported a greater stroke volume reduction in HFpEF than in HFrEF after vasodilatation treatment, indicating the presence of occult intravascular hypovolemia in HFpEF [62]. Another potential difference between HFpEF and HFrEF may be related to plasma volume. Volume distribution has been hypothesized to be the predominant cause of congestion in HFpEF, rather than volume overload which is typically the main cause of congestion in HFrEF. In HFpEF, congestion is mainly caused by the shift in fluid from the intravascular space to the extravascular space [63, 64]. For example, Takei et al. demonstrated that PVR was associated with WRF in HFpEF patients but not in HFrEF patients [59].
In addition to the development of WRF, the impact of WRF on prognosis may also differ between patients with HFpEF and HFrEF. At this point, it should, however, be acknowledged and emphasized that RAAS inhibition may similarly trigger WRF in both HFpEF and HFrEF [65]. However, in contrast to HFrEF patients, RAAS inhibitors mostly failed to demonstrate any clinical benefit in HFpEF patients [66,67,68], although it is hypothesized that the TOPCAT trial was a missed opportunity to demonstrate mineralocorticoid receptor antagonism (MRA) efficacy in HFpEF, owing to geographic differences (Eastern Europe vs. America), which may have led to a loss of (statistical) power [65, 69].
These assumptions become increasingly important given the fact that patients with HFrEF receiving an angiotensin-converting enzyme inhibitor, an ARB, or an MRA (spironolactone or eplerenone) display better clinical outcomes despite experiencing more frequent episodes of hyperkalemia and/or WRF shortly after RAAS inhibitor initiation or thereafter [70,71,72,73]. However, WRF after initiation of irbesartan treatment in HFpEF has been associated with excess risk. WRF is moreover associated with first occurrence of cardiovascular death or HF hospitalization [74].
In a study by McAlister et al., eGFR was found to be a stronger predictor of all-cause mortality in HFrEF than in HFpEF, and for any given eGFR category, mortality was higher in patients with HFrEF than in patients with HFpEF. The authors suggested that the reduction in eGFR is a marker of reduced cardiac output, which is a more important prognostic factor in patients with HFrEF than in those with HFpEF [75]. A dissociation between renal dysfunction and the risk of developing HFpEF has also been observed. In the PREVEND trial, poorer renal function, as assessed by cystatin C and albuminuria, was a strong risk factor for developing HFpEF, but not for HFrEF [11].
In addition to the previous concerns, there are also reports showing that WRF is a beneficial event in terms of prognosis in HF patients [58]. Thus, further studies are needed to determine whether WRF in itself is beneficial or harmful in HF patients stratified as HFpEF and HFrEF. In patients with HFpEF, end-systolic and arterial elastance measurements were found higher compared to patients with HFrEF, resulting in a greater drop in blood pressure with similar changes in central volume [24, 76, 77]. Therefore, the role of PVR should be re-examined based on these differences.
In one study, the prognostic role of renal resistive index (RRI) in HFpEF was investigated non-invasively through Doppler ultrasonic examination in 90 HFpEF patients and compared with 90 age- and sex-matched hypertensive patients without evidence of HF who served as controls. Mean RRI was substantially greater in HFpEF patients than in controls (p < 0.0001). On multivariable analysis, mean RRI was independently associated with HFpEF. In addition, increased mean RRI was an independent predictor of poor outcome (HR = 1.06, 95% CI 1.01–1.10, p = 0.007) while remaining significantly associated with outcome after adjustment for univariate predictors [78].
The role of ultrafiltration (UF) in patients with HFpEF for volume reduction also needs to be clarified. The Ultrafiltration Versus Intravenous Diuretics for Patients Hospitalized for Acute Decompensated Heart Failure (UNLOAD) trial showed that acute HF patients in general exhibited superior volume and weight reduction with UF compared with usual care [79]. However, there were no specific comments regarding HFpEF in this study, which included approximately 30% of patients with an ejection fraction higher than 40%. Jefferies et al. evaluated two patient cohorts admitted to a single institution for acute decompensated HF and treated with UF in HFrEF (left ventricular ejection fraction ≤40%; n = 87) and HFpEF (left ventricular ejection fraction >40%; n = 97) patients. The authors observed no significant differences in total weight loss (7.7% in HFrEF and 7.0% in HFpEF), electrolyte and renal disturbances, or in-hospital mortality (3.4% in HFrEF and 3.3% in HFpEF) between the two groups. Mortality at 90 days tended to be greater in HFrEF (24.1%) than in HFpEF (15.5%). However, as acknowledged by the authors, more studies are needed regarding this issue [80].
Novel biomarkers and therapeutic options
Before pursuing this section, we should acknowledge that no evidence-based therapy has been shown to improve outcomes for HFpEF patients. This may be related to the fact that underlying mechanisms are currently not known for HFpEF. However, there is growing evidence that classical neurohumoral activation occurring in HFrEF is not a main factor in HFpEF [81].
In patients with HF with preserved ejection fraction, biomarkers that reflect collagen homeostasis such as the soluble form of ST2 (an interleukin-1 receptor family member), matrix metalloproteinase-2, collagen III N-terminal propeptide, and galectin-3 have been correlated with the presence and severity of disease [82]. While baseline biomarkers failed to modify the response to LCZ696 in lowering N-terminal pro B-type natriuretic peptide, left atrial volume reduction, however, varied according to baseline levels of the soluble form of ST2 and galectin 3. Interestingly, galectin-3 has been shown to precede the development of both CKD and incident HFpEF [83]. Infusion of galectin-3 in a rat model of hypertensive HF was found to induce severe LV fibrosis and LV dysfunction [84]. Similarly, galectin-3 has been linked to the development of renal fibrosis, while inhibition of galectin-3 in rats was found to protect against hypertensive nephropathy and resulted in reduced proteinuria, improved renal function, and decreased renal damage [85, 86]. In patients with HFpEF, galectin-3 levels have been associated with severity of renal dysfunction, although not with cardiac structure, after correction for renal function [87]. The direct effect of galectin-3 on cardiac structure therefore remains unconfirmed; however, hypothetically, a profibrotic pathway, indicated by elevated levels of fibrosis markers such as galectin-3 activity, could also be involved in the relationship between renal dysfunction and HFpEF. Of note, galectin-3 was also found associated with prognosis as well as with eGFR in HFrEF patients [88].
ED may represent a potential target for therapy, by activating the cGMP pathway through compounds such as NO donors, guanylyl activators and stimulators, or phosphodiesterase 9A inhibitors. Another promising venue is sodium nitrite. In patients with HFpEF, a marked increase in the development of LV filling pressures has been reported, especially during exercise. Numerous lines of evidence indicate that abnormalities in nitric oxide (NO)–cyclic guanosine monophosphate (cGMP) signaling play a central role in causing these reserve depletions. Inorganic nitrite is now recognized as an alternative in vivo source of NO-cGMP that is independent of the traditional NO synthase pathway. Borlaug et al. tested the effect of infusion of sodium nitrite on HFpEF, in which the primary endpoint of exercise PCWP was substantially improved by nitrite compared with placebo (adjusted mean: 19 ± 5 mmHg vs. 28 ± 6 mmHg; p = 0.0003). Nitrite-enhanced cardiac output reserve also improved with exercise (+0.5 ± 0.7 vs. −0.4 ± 0.7 L/min; p = 0.002) and normalized the increase in cardiac output relative to oxygen consumption [89].
Another recent experimental study trial evaluated the effect of a dipeptidyl peptidase-4 (DPP-4) inhibitor class of incretin-based therapy in preventing HFpEF. As a result, DPP-4 inhibitors prevented the development of cardiac diastolic dysfunction, including a reduction in cardiac collagen I synthesis. These changes were independent of renal function [90]. LCZ696, a first-in-class angiotensin receptor neprilysin inhibitor (ARNI) composed of the ARB valsartan and the neprilysin inhibitor prodrugs acubitril, has also been tested in HFpEF [91]. In the PARAMOUNT trial, 301 patients with New York Heart Assocation (NYHA) class II–III, LVEF 45% or higher, and NT-proBNP >400 pg/mL were enrolled and randomly assigned (1:1) to receive either LCZ696 or valsartan alone for 36 weeks. The results showed that LCZ696 reduced the levels of NT-proBNP at 12 weeks (LCZ696: baseline, 783 pg/mL 12 weeks, 605 pg/ mL vs. valsartan: baseline, 862 pg/mL 12 weeks, 835 pg/mL) and reduced left atrial size and improved NYHA class at 36 weeks. LCZ696 was well tolerated with adverse effects similar to those of valsartan [92].
Lastly, the role of other novel risk factors such as FGF-23 in preserved EF needs to be more intensively studied. Only one study in the literature examined the relationship between FGF-23 and reduced and preserved HF. In this particular study, Koller et al. demonstrated that FGF-23 was independently associated with an increased risk of mortality in patients with HFrEF but not in those with HFpEF, suggesting a different pathophysiological role for both entities [93].
Conclusion
HF with preserved EF is as common as HF with reduced EF, although these two entities differ with respect to pathogenesis, cardiac mechanics, and treatment responses. Evidence-based treatment options for HFrEF such as beta-blockers and ACE inhibitors have not been tested in detail in patients with HFpEF. With regard to kidney function, HFpEF is also a risk factor for development of WRF. Given the absence of clear treatment strategies comparatively to patients with HFrEF, new research in HFpEF is undoubtedly necessary in order to more accurately define its pathogenesis and treatment options.
References
Smith DH, Thorp ML, Gurwitz JH et al (2013) Chronic kidney disease and outcomes in heart failure with preserved versus reduced ejection fraction: the Cardiovascular Research Network PRESERVE Study. Circ Cardiovasc Qual Outcomes 6:333–342
Lloyd-Jones DM, Larson MG, Leip EP et al (2002) Lifetime risk for developing congestive heart failure the Framingham Heart Study. Circulation 106:3068–3072
Colombo PC, Ganda A, Lin J et al (2012) Inflammatory activation: cardiac, renal, and cardio-renal interactions in patients with the cardiorenal syndrome. Heart Fail Rev 17:177–190
Go AS, Yang J, Ackerson LM et al (2006) Hemoglobin level, chronic kidney disease, and the risks of death and hospitalization in adults with chronic heart failure the anemia in chronic heart failure: outcomes and resource utilization (ANCHOR) study. Circulation 113:2713–2723
von Haehling S, Anker SD (2010) Cachexia as a major underestimated and unmet medical need: facts and numbers. J Cachex Sarcopenia Muscle 1:1–5
Curtis BM, Parfrey PS (2005) Congestive heart failure in chronic kidney disease: disease-specific mechanisms of systolic and diastolic heart failure and management. Cardiol Clin 23:275–284
Bruch C, Rothenburger M, Gotzmann M et al (2007) Chronic kidney disease in patients with chronic heart failure—impact on intracardiac conduction, diastolic function and prognosis. Int J Cardiol 118:375–380
Hillege HL, Girbes AR, de Kam PJ et al (2000) Renal function, neurohormonal activation, and survival in patients with chronic heart failure. Circulation 102:203–210
Ather S, Chan W, Bozkurt B et al (2012) Impact of noncardiac comorbidities on morbidity and mortality in a predominantly male population with heart failure and preserved versus reduced ejection fraction. J Am Coll Cardiol 59:998–1005
Lam CS, Donal E, Kraigher-Krainer E, Vasan RS (2011) Epidemiology and clinical course of heart failure with preserved ejection fraction. Eur J Heart Fail 13:18–28
Brouwers FP, de Boer RA, van der Harst P et al (2013) Incidence and epidemiology of new onset heart failure with preserved vs. reduced ejection fraction in a community-based cohort: 11-year follow-up of PREVEND. Eur Heart J 34:1424–1431
Athyros VG, Pagourelias ED, Gossios TD, Vasilikos VG (2015) Treating heart failure with preserved ejection fraction related to arterial stiffness. Can we kill two birds with one stone? Curr Vasc Pharmacol 13:368–380
Ferreira JP, Girerd N, Duarte K et al (2017) Serum chloride and sodium interplay in patients with acute myocardial infarction and heart failure with reduced ejection fraction: an analysis from the high-risk myocardial infarction database initiative. Circ Heart Fail 10. doi:10.1161/CIRCHEARTFAILURE.116.003500
van Riet EE, Hoes AW, Limburg A, Landman MA, Zuithoff PN, Rutten FH (2016) Effect of training general practitioners in drug treatment of newly detected heart failure patients with reduced or preserved ejection fraction: a cluster randomized trial. Int J Cardiol 217:174–182
Kao DP, Lewsey JD, Anand IS et al (2015) Characterization of subgroups of heart failure patients with preserved ejection fraction with possible implications for prognosis and treatment response. Eur J Heart Fail 17:925–935
Unger ED, Dubin RF, Deo R et al (2015) Association of chronic kidney disease with abnormal cardiac mechanics and adverse outcomes in patients with heart failure and preserved ejection fraction. Eur J Heart Fail 18:103–112
Maaten JM, Damman K, Verhaar MC et al (2016) Connecting heart failure with preserved ejection fraction and renal dysfunction: the role of endothelial dysfunction and inflammation. Eur J Heart Fail 18:588–98
Metra M, Cotter G, Gheorghiade M, Dei Cas L, Voors AA (2012) The role of the kidney in heart failure. Eur Heart J 33:2135–2142
Yancy CW, Lopatin M, Stevenson LW, De Marco T, Fonarow GC (2006) Clinical presentation, management, and in-hospital outcomes of patients admitted with acute decompensated heart failure with preserved systolic function: a report from the Acute Decompensated Heart Failure National Registry (ADHERE) Database. J Am Coll Cardiol 47:76–84
Sharma K, Hill T, Grams M et al (2015) Outcomes and worsening renal function in patients hospitalized with heart failure with preserved ejection fraction. Am J Cardiol 116:1534–1540
Gori M, Senni M, Gupta DK et al (2014) Association between renal function and cardiovascular structure and function in heart failure with preserved ejection fraction. Eur Heart J 35:3442–3451
Casado J, Montero M, Formiga F et al (2013) Clinical characteristics and prognostic influence of renal dysfunction in heart failure patients with preserved ejection fraction. Eur J Int Med 24:677–683
Damman K, Valente MA, Voors AA, O'Connor CM, Van Veldhuisen DJ, Hillege HL (2014) Renal impairment, worsening renal function, and outcome in patients with heart failure: an updated meta-analysis. Eur Heart J 35:455–469
Kawaguchi M, Hay I, Fetics B, Kass DA (2003) Combined ventricular systolic and arterial stiffening in patients with heart failure and preserved ejection fraction implications for systolic and diastolic reserve limitations. Circulation 107:714–720
Bhatia RS, Tu JV, Lee DS et al (2006) Outcome of heart failure with preserved ejection fraction in a population-based study. N Engl J Med 355:260–269
Owan TE, Hodge DO, Herges RM, Jacobsen SJ, Roger VL, Redfield MM (2006) Trends in prevalence and outcome of heart failure with preserved ejection fraction. N Engl J Med 355:251–259
Anand IS (2008) Heart failure and anemia: mechanisms and pathophysiology. Heart Fail Rev 13:379–386
Galle J, Quaschning T, Seibold S, Wanner C (2003) Endothelial dysfunction and inflammation: what is the link? Kidney Int 63:S45–S49
Murdoch CE, Chaubey S, Zeng L et al (2014) Endothelial NADPH oxidase-2 promotes interstitial cardiac fibrosis and diastolic dysfunction through proinflammatory effects and endothelial-mesenchymal transition. J Am Coll Cardiol 63:2734–2741
Di Marco GS, Hausberg M, Hillebrand U et al (2008) Increased inorganic phosphate induces human endothelial cell apoptosis in vitro. Am J Physiol-Renal Physiol 294:F1381–F1387
Stas S, Whaley-Connell A, Habibi J et al (2007) Mineralocorticoid receptor blockade attenuates chronic overexpression of the renin-angiotensin-aldosterone system stimulation of reduced nicotinamide adenine dinucleotide phosphate oxidase and cardiac remodeling. Endocrinology 148:3773–3780
Silswal N, Touchberry CD, Daniel DR et al (2014) FGF23 directly impairs endothelium-dependent vasorelaxation by increasing superoxide levels and reducing nitric oxide bioavailability. Am J Physiol-Endocrinol Metab 307:E426–E436
Chitalia N, Recio-Mayoral A, Kaski JC, Banerjee D (2012) Vitamin D deficiency and endothelial dysfunction in non-dialysis chronic kidney disease patients. Atherosclerosis 220:265–268
Jie KE, Zaikova MA, Bergevoet MW et al (2010) Progenitor cells and vascular function are impaired in patients with chronic kidney disease. Nephrol Dialysis Transplant 25:1875–1882
Yilmaz MI, Sonmez A, Saglam M et al (2008) ADMA levels correlate with proteinuria, secondary amyloidosis, and endothelial dysfunction. J Am Soc Nephrol 19:388–395
Lam CS, Brutsaert DL (2012) Endothelial dysfunction: a pathophysiologic factor in heart failure with preserved ejection fraction. J Am Coll Cardiol 60:1787–1789
Akiyama E, Sugiyama S, Matsuzawa Y et al (2012) Incremental prognostic significance of peripheral endothelial dysfunction in patients with heart failure with normal left ventricular ejection fraction. J Am Coll Cardiol 60:1778–1786
van Heerebeek L, Hamdani N, Falcão-Pires I et al (2012) Low myocardial protein kinase G activity in heart failure with preserved ejection fraction. Circulation:CIRCULATIONAHA. 111.076075.
Glezeva N, Baugh J (2014) Role of inflammation in the pathogenesis of heart failure with preserved ejection fraction and its potential as a therapeutic target. Heart Fail Rev 19:681–694
Kalogeropoulos A, Georgiopoulou V, Psaty BM et al (2010) Inflammatory markers and incident heart failure risk in older adults: the Health ABC (Health, Aging, and Body Composition) study. J Am Coll Cardiol 55:2129–2137
Moradi H, Sica DA, Kalantar-Zadeh K (2013) Cardiovascular burden associated with uremic toxins in patients with chronic kidney disease. Am J Nephrol 38:136–148
Tumur Z, Niwa T (2009) Indoxyl sulfate inhibits nitric oxide production and cell viability by inducing oxidative stress in vascular endothelial cells. Am J Nephrol 29:551–557
Yamamoto H, Tsuruoka S, Ioka T et al (2006) Indoxyl sulfate stimulates proliferation of rat vascular smooth muscle cells. Kidney Int 69:1780–1785
Oberleithner H, Riethmüller C, Schillers H, MacGregor GA, de Wardener HE, Hausberg M (2007) Plasma sodium stiffens vascular endothelium and reduces nitric oxide release. Proc Natl Acad Sci 104:16281–16286
Miller WL, Mullan BP (2016) Volume overload profiles in patients with preserved (HFpEF) and reduced (HFrEF) ejection fraction chronic heart failure: are there differences? A pilot study. JACC: Heart Failure.
Hartog JW, Smit AJ, van Son WJ et al (2004) Advanced glycation end products in kidney transplant patients: a putative role in the development of chronic renal transplant dysfunction. Am J Kidney Dis 43:966–975
Stenvinkel P, Alvestrand A (2002) Review articles: inflammation in end-stage renal disease: sources, consequences, and therapy. Semin Dial 15:329–337
Smit AJ, Lutgers H (2004) The clinical relevance of advanced glycation endproducts (AGE) and recent developments in pharmaceutics to reduce AGE accumulation. Curr Med Chem 11:2767–2784
Petrova R, Yamamoto Y, Muraki K et al (2002) Advanced glycation endproduct-induced calcium handling impairment in mouse cardiac myocytes. J Mol Cell Cardiol 34:1425–1431
Hartog JW, Voors AA, Bakker SJ, Smit AJ, Veldhuisen DJ (2007) Advanced glycation end-products (AGEs) and heart failure: pathophysiology and clinical implications. Eur J Heart Fail 9:1146–1155
Zyatenkova E, Drapkina O, Ivashkin V (2014) Characteristics of vessels wall, myocardium and epicardial fat in patients with heart failure with preserved ejection fraction with and without metabolic syndrome. Endosc Ultrasound 3:S1
Karakurt O, Oztekin S, Yazıhan N, Akdemir R (2011) Impaired right ventricular functions in metabolic syndrome patients with preserved left ventricular ejection fraction. Turk Kardiyol Dern Ars 39:549–556
Paulus WJ, Tschöpe C (2013) A novel paradigm for heart failure with preserved ejection fraction: comorbidities drive myocardial dysfunction and remodeling through coronary microvascular endothelial inflammation. J Am Coll Cardiol 62:263–271
Schulz E, Jansen T, Wenzel P, Daiber A, Münzel T (2008) Nitric oxide, tetrahydrobiopterin, oxidative stress, and endothelial dysfunction in hypertension. Antioxid Redox Signal 10:1115–1126
Kamo T, Akazawa H, Komuro I (2015) Cardiac nonmyocytes in the hub of cardiac hypertrophy. Circ Res 117:89–98
Kajimoto K, Sato N, Takano T, registry iotADHFS (2015) Association of anemia and renal dysfunction with in-hospital mortality among patients hospitalized for acute heart failure syndromes with preserved or reduced ejection fraction. Eur Heart J:2048872615593387.
Rusinaru D, Buiciuc O, Houpe D, Tribouilloy C (2011) Renal function and long-term survival after hospital discharge in heart failure with preserved ejection fraction. Int J Cardiol 147:278–282
Afsar B, Ortiz A, Covic A, Solak Y, Goldsmith D, Kanbay M (2016) Focus on renal congestion in heart failure. Clin Kidney J 9:39–47
Takei M, Kohsaka S, Shiraishi Y et al (2015) Effect of estimated plasma volume reduction on renal function for acute heart failure differs between patients with preserved and reduced ejection fraction. Circ Heart Fail 8:527–532
Testani JM, Chen J, McCauley BD, Kimmel SE, Shannon RP (2010) Potential effects of aggressive decongestion during the treatment of decompensated heart failure on renal function and survival. Circulation 122:265–272
Abramov D, Cohen RS, Katz SD, Mancini D, Maurer MS (2008) Comparison of blood volume characteristics in anemic patients with low versus preserved left ventricular ejection fractions. Am J Cardiol 102:1069–1072
Schwartzenberg S, Redfield MM, From AM, Sorajja P, Nishimura RA, Borlaug BA (2012) Effects of vasodilation in heart failure with preserved or reduced ejection fraction: implications of distinct pathophysiologies on response to therapy. J Am Coll Cardiol 59:442–451
Bhuiyan T, Maurer MS (2011) Heart failure with preserved ejection fraction: persistent diagnosis, therapeutic enigma. Curr Cardiovasc Risk Rep 5:440–449
Bench T, Burkhoff D, O’Connell JB et al (2009) Heart failure with normal ejection fraction: consideration of mechanisms other than diastolic dysfunction. Curr Heart Fail Rep 6:57–64
Rossignol P, Zannad F (2015) Regional differences in heart failure with preserved ejection fraction trials when nephrology meets cardiology but east does not meet west. Circulation 131:7–10
Yusuf S, Pfeffer MA, Swedberg K et al (2003) Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: the CHARM-Preserved Trial. Lancet 362:777–781
Massie BM, Carson PE, McMurray JJ et al (2008) Irbesartan in patients with heart failure and preserved ejection fraction. N Engl J Med 359:2456–2467
Pitt B, Pfeffer MA, Assmann SF et al (2014) Spironolactone for heart failure with preserved ejection fraction. N Engl J Med 370:1383–1392
Ferreira JP, Girerd N, Rossignol P, Zannad F (2015) Geographic differences in heart failure trials. Eur J Heart Fail 17:893–905
Rossignol P, Dobre D, Gregory D et al (2014) Incident hyperkalemia may be an independent therapeutic target in low ejection fraction heart failure patients: insights from the HEAAL study. Int J Cardiol 173:380–387
Lesogor A, Cohn JN, Latini R et al (2013) Interaction between baseline and early worsening of renal function and efficacy of renin–angiotensin–aldosterone system blockade in patients with heart failure: insights from the Val-HeFT study. Eur J Heart Fail 15:1236–1244
Rossignol P, Dobre D, McMurray JJ et al (2013) Incidence, determinants, and prognostic significance of hyperkalemia and worsening renal function in patients with heart failure receiving the mineralocorticoid receptor antagonist eplerenone or placebo additional to optimal medical therapy: results from the Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure (EMPHASIS-HF). Circ Heart Fail:CIRCHEARTFAILURE. 113.000792.
Rossignol P, Zannad F, Pitt B (2014) Time to retrieve the best benefits from renin angiotensin aldosterone system (RAAS) inhibition in heart failure patients with reduced ejection fraction: lessons from randomized controlled trials and registries. Int J Cardiol 177:731–733
Damman K, Perez AC, Anand IS et al (2014) Worsening renal function and outcome in heart failure patients with preserved ejection fraction and the impact of angiotensin receptor blocker treatment. J Am Coll Cardiol 64:1106–1113
McAlister FA, Ezekowitz J, Tarantini L et al (2012) Renal dysfunction in patients with heart failure with preserved versus reduced ejection fraction impact of the new chronic kidney disease-epidemiology collaboration group formula. Circ Heart Fail 5:309–314
Borlaug BA, Kass DA (2008) Ventricular-vascular interaction in heart failure. Heart Fail Clin 4:23–36
Lam CS, Roger VL, Rodeheffer RJ et al (2007) Cardiac structure and ventricular–vascular function in persons with heart failure and preserved ejection fraction from Olmsted County, Minnesota. Circulation 115:1982–1990
Ennezat PV, Maréchaux S, Six-Carpentier M et al (2011) Renal resistance index and its prognostic significance in patients with heart failure with preserved ejection fraction. Nephrol Dial Transplant 26:3908–3913
Costanzo MR, Guglin ME, Saltzberg MT et al (2007) Ultrafiltration versus intravenous diuretics for patients hospitalized for acute decompensated heart failure. J Am Coll Cardiol 49:675–683
Jefferies JL, Bartone C, Menon S, Egnaczyk GF, O’Brien TM, Chung ES (2013) Ultrafiltration in heart failure with preserved ejection fraction comparison with systolic heart failure patients. Circ Heart Fail 6:733–739
Butler J, Hamo CE, Udelson JE et al (2016) Exploring new endpoints for patients with heart failure with preserved ejection fraction. Circ Heart Fail 9. doi:10.1161/CIRCHEARTFAILURE.116.003358
Zile MR, Jhund PS, Baicu CF et al (2016) Plasma biomarkers reflecting profibrotic processes in heart failure with a preserved ejection fraction data from the prospective comparison of ARNI with ARB on Management of Heart Failure with Preserved Ejection Fraction Study. Circ Heart Fail 9:e002551
O’Seaghdha CM, Hwang S-J, Ho JE, Vasan RS, Levy D, Fox CS (2013) Elevated galectin-3 precedes the development of CKD. J Am Soc Nephrol 24:1470–1477
Sharma UC, Pokharel S, van Brakel TJ et al (2004) Galectin-3 marks activated macrophages in failure-prone hypertrophied hearts and contributes to cardiac dysfunction. Circulation 110:3121–3128
Frenay A-RS YL, van der Velde AR et al (2015) Pharmacological inhibition of galectin-3 protects against hypertensive nephropathy. Am J Physiol-Renal Physiol 308:F500–F509
Henderson NC, Mackinnon AC, Farnworth SL et al (2008) Galectin-3 expression and secretion links macrophages to the promotion of renal fibrosis. Am J Pathol 172:288–298
AbouEzzeddine OF, Haines P, Stevens S et al (2015) Galectin-3 in heart failure with preserved ejection fraction: a RELAX trial substudy (phosphodiesterase-5 inhibition to improve clinical status and exercise capacity in diastolic heart failure). JACC Heart Fail 3:245–252
Lopez-Andrès N, Rossignol P, Iraqi W et al (2012) Association of galectin-3 and fibrosis markers with long-term cardiovascular outcomes in patients with heart failure, left ventricular dysfunction, and dyssynchrony: insights from the CARE-HF (Cardiac Resynchronization in Heart Failure) trial. Eur J Heart Fail 14:74–81
Borlaug BA, Koepp KE, Melenovsky V (2015) Sodium nitrite improves exercise hemodynamics and ventricular performance in heart failure with preserved ejection fraction. J Am Coll Cardiol 66:1672–1682
Connelly KA, Bowskill BB, Advani SL et al (2014) Dipeptidyl peptidase-4 inhibition improves left ventricular function in chronic kidney disease. Clin Invest Med 37:E172
Lin LM, Wu Y, Wu MF, Lin JX (2016) Focus on the novel cardiovascular drug LZC696: from evidence to clinical consideration. Cardiovasc Drugs Ther 30:623–633
Solomon SD, Zile M, Pieske B et al (2012) The angiotensin receptor neprilysin inhibitor LCZ696 in heart failure with preserved ejection fraction: a phase 2 double-blind randomised controlled trial. Lancet 380:1387–1395
Koller L, Kleber ME, Brandenburg VM, Goliasch G, Richter B, Sulzgruber P, Scharnagl H, Silbernagel G, Grammer TB, Delgado G, Tomaschitz A, Pilz S, Berger R, Mörtl D, Hülsmann M, Pacher R, März W, Niessner A (2015) Fibroblast growth factor 23 is an independent and specific predictor of mortality in patients with heart failure and reduced ejection fraction. Circ Heart Fail 8(6):1059–1067
Acknowledgements
The authors thank Mr. Pierre Pothier for the editing of the manuscript.
Author information
Authors and Affiliations
Contributions
All authors approved the final version of manuscript.
Corresponding author
Ethics declarations
This article does not contain any studies with human participants or animals performed by any of the authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Funding
None declared.
Rights and permissions
About this article

Cite this article
Afsar, B., Rossignol, P., van Heerebeek, L. et al. Heart failure with preserved ejection fraction: a nephrologist-directed primer. Heart Fail Rev 22, 765–773 (2017). https://doi.org/10.1007/s10741-017-9619-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10741-017-9619-2