
Abstract
Plant miRNAs are found to be present throughout the genome and they regulate gene expression either by cleaving mRNA or inhibiting the translational process at the post transcriptional level. Drought is one of the major limiting factors that negatively affect productivity of plants. Cardamom cultivation is having good production potential but the plants are vulnerable to biotic and abiotic stress factors. To date, nothing is known about the regulatory roles of miRNAs in response to drought stress in cardamom. Ion Torrent sequencing of two small RNA libraries prepared from control (C) and treated (T) plants raised under well irrigated and drought stressed treatments respectively created 3,938,342 and 4,083,181 primary reads. A total of 150 conserved and 20 novel microRNAs were identified from both the control and treated libraries. Discovery of 17 differentially expressed miRNAs under drought stress suggests that these miRNAs might have involved in various biological processes to improve plant tolerance to water stress. Several target genes for drought stress regulating miRNAs were identified including miR156l and miR169c which cleave the target mRNA involved in response to water deprivation. miR530b and miR156a target mRNAs which respond to water deprivation and inhibit the translational process. The expression patterns of some of the miRNAs and their targets were validated by qRT-PCR. This study is the first report of drought responsive miRNAs and their targets in cardamom. The outcome of this research could provide insights into the miRNA based regulatory mechanisms in response to drought stress in monocot plants.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
MicroRNAs (miRNAs) which are functionally significant and approximately 19–24 nucleotide (nt) long non coding RNAs, act as post-transcriptional gene expression regulators by involving in different processes like apoptosis, stress response, differentiation and different disease conditions (Shweta et al. 2015). This novel method of tuning gene expression in eukaryotes was noticed in the 1990s and got fascinated by the scientific community (Lin et al. 2014). Plant miRNAs are found to be present throughout the genome and are produced from exons, introns, intergenic regions and repetitive transposable elements (Agharbaoui et al. 2015). In plants, miRNA gene got transcribed by RNA polymerase II enzyme to form Primary miRNA. Dicer-like 1 (DCL 1), hyponastic leaves 1 (HYL 1) and serrate proteins act on it and generate precursor miRNAs which got cleaved to form miRNA::miRNA* duplex and transported from nucleus to cytoplasm through HASTY (HST1) (Li et al. 2015). The duplex is loaded on to RNA induced silencing complex (RISC). Either 5p or 3p single stranded miRNA binds with Argonaute (AGO) in the RISC while the other strand gets degraded. The miRNA-RISC targets complementary messenger RNA (mRNA) (Zhang et al. 2014; Tang et al. 2015). They regulate gene expression either by cleaving mRNA or inhibiting the translational process at the post transcriptional level (Yang et al. 2015b). Sometimes, they can also involve in methylation at the transcriptional level (Brodersen et al. 2008). A single miRNA can target many mRNAs and an mRNA can be targeted by multiple miRNAs (Zandkarimi et al. 2015; Kelly et al. 2015).
Both conserved and species specific miRNAs are reported to be present in many animal and plant species. Studies have shown that conserved miRNAs control common characters and species specific miRNAs regulate distinctive features (Zhang 2015). Arabidopsis is the first plant in which the miRNAs were reported in 2002 (Lima et al. 2012). Later it was found to be present in many plants including monocots, dicots, algae, ferns and moss (Song et al. 2015). Plant miRNAs exert function mainly by degrading mRNA, but recently, studies have reported that translational attenuation is also very frequent. Plant miRNAs are involved in different developmental process by binding with their own targets and also interacting with each other in a complex system of network (Wang et al. 2015). The present version of miRBase (a biological database that acts as an archive of microRNA sequences and annotations; Release 21: June 2014, accessible at http://www.mirbase.org) consists of 7057 plant miRNA entries from 73 species (Boke et al. 2015).
Sequencing is the base for awareness about the number, diversity, expression and probable roles of small non coding RNAs in plants (Sun et al. 2014). Sanger sequencing or chain termination method has been the dominant method of DNA sequencing which was developed by Frederick Sanger in 1977. The human genome project was also accomplished by utilising this method by the team work of 20 groups from United States, Britain, France, China, Japan and Germany (Lander 2001). This method needs a DNA template, primer, DNA polymerase, 2′-deoxynucleotides (dNTPs) and 2′, 3′-dideoxynucleotides (ddNTPs). Oligonucleotide chain elongation gets terminated by the incorporation of ddNTPs and polyacrylamide gel electrophoresis (PAGE) is used to separate the products generated with different lengths. The sequence of DNA strand is obtained by analysing the ddNTPs at 3′ terminal (Morozova and Marra 2008). Genomic research has become revolutionized with the launch of next-generation sequencing (NGS) technologies since 2005. NGS is more economical than Sanger sequencing and the researchers are able to carry out many experiments which were formerly problematic. Human genome project took around 13 years to get completed by Sanger sequencing with an estimated cost of $2.7 billion whereas NGS revealed the sequence of human genome within 5 months for around $1.5 million (Voelkerding et al. 2009). Roche 454, Illumina Inc, Life technologies, Ion torrent and Pacific Biosciences are some of the companies offering next generation sequencing technologies which utilises different platforms (Yang et al. 2015a).
Conventional methods like Sanger sequencing are not much reliable in detecting species specific miRNAs which are expressed in very small amounts (Song et al. 2010). NGS provides enormous amount of data to address such needs. However, fast processing power and efficient tools are needed to manage this big data (Patel and Jain 2012). With the advancement in computer capacity and algorithm, data interpretation becomes easier. Sometimes errors might occur during library construction and sequencing procedures which can affect the quality of the raw reads. Quality control check has to be done for the raw reads obtained from NGS platforms. It gives an idea of whether the data has any issues and can be conscious of before starting analysis (Trivedi et al. 2014). The blend of next generation sequencing and bioinformatics analysis has paved the way for discovery of many species specific miRNAs of plants from several species (Tang et al. 2015). NGS data are now being produced in non-model organisms at a greater speed as this high throughput studies can be performed at a reasonable budget.
Elettaria cardamomum Maton is a perennial, herbaceous rhizomatous plant which belongs to family Zingiberaceae. It is one of the valuable spice crops and is mentioned as ‘Queen of spices’. After vanilla and saffron, it is the costly spice in world market. The dried ripe fruit or capsule is the commercially important part of cardamom plant. It is used as flavouring agent in culinary purposes and is also valued for its medicinal properties (Kader et al. 2015). There is a possibility of decreasing the risks of cancer, dyslipidemia, hepatic steatosis and hyperglycemia by including cardamom in our regular diet (Nitasha et al. 2015; Qiblawi et al. 2015). Its origin is believed to be in the moist evergreen forests of Western Ghats of South India (Ravindran and Madhusoodanan 2002). Besides India, cardamom is cultivated in Sri Lanka, Guatemala, Papua New Guinea and Tanzania.
Cardamom cultivation is having good production potential but the plants are vulnerable to many pests, diseases and abiotic stress factors like droughts, floods, extreme temperatures, salinity, nutrition starvation, oxidative and heavy metal stress. Drought is one of the major limiting factors that negatively affect the crop productivity of the plant. Cardamom planters have significant worry about shortage of water that occurs as a consequence of climate change (Murugan et al. 2011). Increased transpiration rate and limited absorption of water from cold soil also result in water stress. To resist this drought stress, plants execute several mechanisms at the molecular and physiological levels (Bej and Basak 2014; Akdogan et al. 2015). Knowledge on the mechanism behind plant response to drought will be useful for better productivity (Ferdous et al. 2015). Studies have shown that post transcription regulation by miRNAs exerts a role in response of plants to drought stress (Li et al. 2013). Drought regulated miRNAs have been reported in many plant species like switch grass (Hivrale et al. 2016), Tobacco (Yin et al. 2015), rice (Zhao et al. 2007), wheat (Akdogan et al. 2015), sugarcane (Lin et al. 2014), Arabidopsis (Song et al. 2013), foxtail millet (Yi et al. 2015), chickpea (Hajyzadeh et al. 2015), banana (Muthusamy et al. 2014), Medicago truncatula (Wang et al. 2011), Vigna unguiculata (Barrera-Figueroa et al. 2011), Glycine max (Li et al. 2011b), Solanum tuberosum (Hwang et al. 2011), etc. In rice, which is a model plant, miR393 was found to be drought responsive and targets OsTIR1 and OsAFB2 (Xia et al. 2012) which are auxin receptor gene homologs. Another study also reported that miR169 shows upregulation in rice under water stress condition (Jeong and Green 2013). Because of drought stress, a rapid increase in expression on miR169g was found in roots of rice than in shoots (Zhao et al. 2007). miR169 targets NFYA5 mRNA which encodes a subunit of the transcription factor named as nuclear factor Y (Li et al. 2008). miR156, miR159, miR168, miR170, miR171, miR172, miR319, miR396, miR397, miR408, miR529, miR896, miR1030, miR1035, miR1050, miR1088 and miR1126 showed downregulation and miR159, miR319, miR395, miR474, miR845, miR851, miR854, miR896, miR901, miR903, miR1026 and miR1125 showed upregulation under drought stress in rice (Zhou et al. 2010).
Methods
Plant materials and drought stress treatment
Wild cardamom (accession no. TBG-C75) collected from the natural forest area in Therakkudi in the Edamalayar forest range (N10°13′13.20″ & E76°47′03.7″) in Kerala state of India was selected for the study. This population was recorded previously as remaining of the wild cardamom in Western Ghats (Kuriakose et al. 2009). The collected plants are maintained in a greenhouse with daily watering (voucher specimen deposited in JNTBGRI Herbarium as TBGT86201). Out of those plants, one group was labelled as ‘control’ and another as ‘treated’. Water was withheld to the plants labelled as ‘treated’ in order to begin the experiment. Stress indications like leaf rolling appeared 3 weeks after water withholding and at this drought condition the absolute moisture content of the soil was calculated to be < 4.5%. Normal watering was given only to the control plant. Completely opened uppermost leaves and stems from each control and treated plant were collected, frozen in liquid nitrogen and used for RNA isolation immediately.
Small RNA library preparation and sequencing
Total RNA was isolated from leaves and stems of control and drought treated cardamom using the combined miRNeasy Mini Kit and CTAB method (Nadiya et al. 2015). 100 mg of leaf and stem tissues were first subjected to CTAB method until the RNA got precipitated. RNA pellet was recovered and the isolation was further proceeded with miRNeasy Mini Kit (Qiagen, Germany) according to manufacturer’s protocol. Quantification of the isolated RNA was done using Biophotometer (Eppendorf, Germany). RNA quality was analysed through 1.2% agarose gel and Agilent 2100 BioAnalyzer. Ion total RNA seq Kit V2 was used for small RNA sequence library construction following manufacturer’s instructions after pooling the total RNA from leaf and stem tissues in both control and treated samples. The purified libraries were used for sequencing analysis with the Ion Torrent sequencer by Centre for Cellular and Molecular Platforms (C-CAMP, Bangalore). Standard procedure was followed for small RNA library construction and sequencing. Total RNA was run on polyacrylamide gel electrophoresis (PAGE) and the band corresponding to the size of 17–27 nt was cut out, so that only the miRNA fragments got extracted. Then 5p and 3p sequencing adaptors were ligated to the size selected RNA fragments. RT-PCR amplification was carried out to produce cDNA library, purified by PAGE and was used for subsequent sequencing (Ding et al. 2016). The sequencing data were deposited in the NCBI Sequence Read Archive (SRA, http://www.ncbi.nlm.nih.gov/sra/) as accession numbers SRX2273832 and SRX2273833.
Data pre-processing and miRNA identification
Adaptors were removed from the raw sequencing reads using Cutadapt with error rate (-e) set to 0.1 (Martin 2011). The remaining reads were checked against snRNAs, snoRNAs, rRNAs and tRNAs from NCBI database and the perfect matches were eliminated using Bowtie alignment tool (Langmead et al. 2009). Reads with 17–27 nt length were kept for further analysis. The identical sequences were collapsed into a single sequence by FASTQ/A Collapser tool available in the FASTX-Toolkit. Nucleotide distribution chart was created using FASTQ/A Statistics and FASTQ/A nucleotide distribution tools. Pipeline used for the analysis of miRNA sequencing data is depicted in Fig. 1.

Pipeline used for the analysis of miRNA sequencing data
A Blastn search was performed against plant mature miRNAs from miRBase database to identify conserved miRNAs in cardamom small RNA libraries. A maximum of three mismatches were allowed. The remaining reads were mapped onto the transcriptome sequence of Curcuma longa, the closest relative of cardamom, for the identification of novel miRNAs, as the whole genome or transcriptome sequence of cardamom is not available. The aligned reads were used as input to predict novel miRNAs with the software miRDeep-P (Yang and Li 2011). miRDeep-P is a miRNA finding software package equipped with plant specific parameters and has been reported in many studies as a novel miRNA prediction tool (Jain et al. 2014).
miRNA target prediction and functional annotation
Targets of all the cardamom miRNAs determined in this study were predicted using the psRNATarget (http://plantgrn.noble.org/psRNATarget/) software with default parameters (Dai and Zhao 2011). ‘User-submitted small RNAs/preloaded transcripts’ option was fixed and selected Arabidopsis thaliana as the reference genome for this analysis. Singular Enrichment Analysis (SEA) tool from AgriGO toolkit (http://bioinfo.cau.edu.cn/agriGO/) was used for gene ontology enrichment. Supported species was selected as A. thaliana (Du et al. 2010).
Differential expression analysis of miRNAs under drought stress
The frequency of miRNAs in control and treatment libraries was normalized to transcripts per million (TPM) by the formula, normalized expression = (actual miRNA count/total count of clean reads) × 1,000,000. If the normalized expression of a miRNA in both the control and treatment libraries showed a value less than one, it is removed due to the very low level of expression (Wang et al. 2011). Fold change of miRNA expression between treatment and control library was determined by using the formula, Fold change = log2 (normalized expression of miRNA in the treatment library/normalized expression of miRNA in the control library). miRNAs with difference in expression levels higher than 1.5-fold were considered to be differentially expressed under drought stress. Fold change with positive values indicate upregulation and negative values represent downregulation of miRNAs in the drought treated library.
Validation of cardamom miRNAs by quantitative real time PCR (qRT PCR) method
Results obtained by small RNA sequencing were experimentally proved using qRT PCR method. Total RNA was isolated from leaves and stems of control and drought treated cardamom using the combined miRNeasy Mini Kit and CTAB method as described above and complementary DNA (cDNA) was prepared using miScript II RT Kit (Qiagen, Germany). Gene specific real time PCR primers for randomly selected eight drought responsive miRNAs were designed using miRprimer2 software (Supplementary File 1). 5.8S rRNA was selected as the internal control. Each reaction was performed on a StepOnePlus real time PCR system (Applied Biosystems, USA). The relative expression level of miRNAs were calculated using CT and 2−ΔΔCT method (Schmittgen et al. 2008). 2−ΔΔCT or the fold change values which are > 1 represents upregulation of miRNA and < 1 indicates downregulation of miRNA (Lutful Kabir et al. 2015). The average of the fold change values from the three experiments was finally taken.
Validation using 5′ RNA ligase-mediated rapid amplification of cDNA ends (RLM-RACE) PCR
In order to validate the predicted target cleavage sites, 5′ RNA Ligase-Mediated Rapid Amplification of cDNA Ends (5′ RLM-RACE) was performed using the FirstChoice RLM-RACE Kit (Ambion, Austin, TX, USA) according to manufacturer’s instructions. Total RNA was extracted from cardamom and RNA oligo adapter was ligated to it. The amplifications were carried out using 5′ RACE outer primer and gene-specific outer primer. After nested PCR with 5′ RACE inner primer and gene-specific inner primer, the 5′ RACE products were purified using the Agarose Gel DNA Purification Kit (TaKaRa Bio), ligated into the pMD19-T vector (TaKaRa Bio), and sequenced. The list of primers used in this study is provided in Supplementary File 2.
Results
Overview of ion torrent sequenced small RNA libraries from cardamom
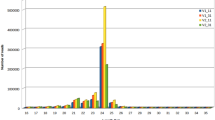
To study miRNA mediated plant response to drought stress in cardamom, two small RNA libraries named as control and treated were prepared from plants raised under well irrigated and drought stressed treatments respectively. The Ion Torrent sequencing of this two small RNA libraries created 3,938,342 (C) and 4,083,181 (T) primary reads under control and treated conditions. 62,915 (1.6%) reads in control library and 63,627 (1.6%) reads in treated library were found to be with 3′ adapters. Following the filtering steps explained in the “Materials and Methods” section, 1,251,632 (C) and 997,432 (T) collapsed sequences were obtained from the two libraries with a size range of 17–27 nt (Table 1). The sequence length distribution of collapsed reads from both control and drought stress small RNA libraries are shown in Fig. 2. Nucleotide sequences of 22, 23 and 24 base lengths are considerably lesser in drought treated library when compared with control library. It may be due to the inhibition of sequences with this type of lengths in drought stressed library (Liu et al. 2015).

Distribution of small RNAs in a control and b drought stress libraries. Nucleotide sequences of 22, 23 and 24 base lengths are considerably lesser in drought treated library when compared with control library. It may be due to the inhibition of sequences with this type of lengths in drought stressed library
Identification of known and novel miRNAs
Cardamom miRNAs which are conserved in other plant species were identified by comparing with miRNAs from miRBase database (Griffiths-Jones et al. 2008). Mapping of filtered reads against miRBase 21.0 identified 1734 (C) and 2078 (T) reads matching against known miRNAs. A total of 150 conserved miRNAs were identified from the two libraries, of which 139 miRNAs belonging to 36 families and 134 miRNAs belonging to 34 families from control and treated libraries respectively (Table 1). miR159, miR396, miR535, miR166b, miR167 and miR396e were the most abundantly expressed miRNAs in both the libraries (Fig. 3). miR156/159/319 family contained 30 members which is the highest among the miRNA families identified while 15 miRNA families (miR3711, miR482, miR5179, miR5250, miR528, miR6478, miR8175, miR8285, miR894, miR9674, miR394, miR8155, miR5810, miR5765 and miR2673) each had only one member (Fig. 4). Known miRNAs are classified into conserved and non-conserved based on the number of different plant families in which they were already reported (Zhang et al. 2006). We identified 24 miRNA families which are conserved and 14 miRNA families which are non-conserved in other plant species (Supplementary File 3).

Count of each known miRNA family. miR159, miR396, miR535, miR166b, miR167 and miR396e were the most abundantly expressed miRNAs in both the libraries

Distribution of number of members in known miRNA families. miR156/159/319 family contained 30 members which is the highest among the miRNA families identified while 15 miRNA families each had only one member
The remaining 1,175,494 (C) and 941,664 (T) reads were retained for novel miRNA prediction. From this 515,716 (C) and 449,475 (T) reads got aligned with the transcriptome sequence of Curcuma longa and were used for the prediction of novel miRNAs. The main step involved was to analyse the precursor sequences of each aligned read and to check the ability to form stem loop secondary structures using RNAfold from the Vienna RNA software package (Hofacker 2003). Using software package miRDeep-P, 9 novel miRNAs from the control and 12 new miRNAs from the treated small RNA libraries were identified. The length of these novel miRNAs varied from 21 to 25 nt, with the majority being 24 nt. The stability of stem loop secondary structure is measured in terms of minimum free energy (MFE), a lower value of MFE indicates greater stability of RNA secondary structure and it is a characteristic of miRNAs (Bonnet et al. 2004). The average MFE for cardamom miRNA precursors was comparatively lower and found to be − 59.28 kcal mol−1 which is in accordance with MFE calculated for miRNA precursors of other plant species like Chick pea (− 57.58 kcal mol−1), Arabidopsis (− 76.2 kcal mol−1), rice (− 71.57 kcal mol−1), soybean (− 56.83 kcal mol−1), Medicago (− 67.73 kcal mol−1) and Sorghum (− 54.29 kcal mol−1) (Jain et al. 2014; Katiyar et al. 2015).
Differentially expressed miRNAs in response to drought in cardamom
Differential expression analysis was performed between the control and treated libraries to identify drought responsive miRNAs in cardamom. Seventeen known miRNAs which belong to twelve miRNA families were identified to be differentially expressed under drought stress (Table 2). Of these, 11 miRNAs were upregulated and 6 miRNAs got downregulated. miR169f and miR172 were the most significantly upregulated (fold change 3.64) and downregulated (fold change − 2.6) miRNAs respectively (Fig. 5; Table 3). 16 conserved and 8 novel miRNAs were discovered only in control library whereas 11 conserved and 11 novel miRNAs were found only in the treated library (Supplementary File 4). These miRNAs from the treated plants might be expressed under the influence of drought stress in cardamom.

Differentially expressed miRNAs under drought stress. Seventeen known miRNAs which belong to 12 miRNA families were identified to be differentially expressed under drought stress. Of these, 11 miRNAs were upregulated and 6 miRNAs got downregulated. miR169f and miR172 were the most significantly upregulated and downregulated miRNAs respectively
Target prediction and functional annotation
Target prediction of known and novel miRNAs is required for annotation of molecular functions (Liu et al. 2015). 1261 unique potential targets were identified for known and novel miRNAs in cardamom using psRNATarget software. Gene ontology enrichment analysis for all the predicted miRNA targets of cardamom was carried out using the tool AgriGO. Target genes were found to be involved in biological process, cellular component and molecular function. Large number of target genes were mainly found to be associated with cellular process (GO:0009987), metabolic process (GO:0008152), biological regulation (GO:0065007), response to stimulus (GO:0050896), developmental process (GO:0032502) and multicellular organismal process (GO:0032501) in the category biological process. For cellular component the main terms are cell part (GO:0044464), cell (GO:0005623), organelle (GO:0043226) and important terms for molecular functions are binding (GO:0005488), catalytic activity (GO:0003824) and transcription regulator activity (GO:0030528) (Fig. 6). Six genes with GO term ‘response to water deprivation’ (GO:0009414), four genes with GO term ‘response to heat’ (GO:0009408), eight genes with GO term ‘response to cold’ (GO:0009409), eight genes with GO term ‘response to salt stress’ (GO:0009651), two genes with GO term ‘cellular response to phosphate starvation’ (GO:0016036) and seven genes with GO term ‘response to oxidative stress’ (GO:0006979) were identified by further analysis of genes related with GO term ‘response to stimulus’ (Figs. 6, 7). These identified GO terms are associated with the major abiotic stress factors which limits the productivity of plants. We also identified four genes with GO term ‘response to abscisic acid stimulus’ (GO:0009737) and three genes with GO term ‘response to peroxidase activity’ (GO:0004601). Abscisic acid (ABA) and peroxidase activity play important roles in drought stress conditions. miRNAs which target the genes involved in ‘response to water deprivation’ is shown in Table 4.

Gene ontology analysis of target genes for all known and novel miRNAs in cardamom. Gene ontology enrichment analysis for all the predicted miRNA targets of cardamom was carried out using the tool AgriGO. Target genes were found to be involved in biological process, cellular component and molecular function

Stress responsive genes associated with GO term ‘response to stimulus’. Six genes with GO term ‘response to water deprivation’ (GO:0009414), four genes with GO term ‘response to heat’ (GO:0009408), eight genes with GO term ‘response to cold’ (GO:0009409), eight genes with GO term ‘response to salt stress’ (GO:0009651), two genes with GO term ‘cellular response to phosphate starvation’ (GO:0016036) and seven genes with GO term ‘response to oxidative stress’ (GO:0006979) were identified by further analysis of genes related with GO term ‘response to stimulus’
qRT PCR validation
Among the eight drought responsive miRNAs selected for qRT PCR study, seven showed similar expression pattern with that of the small RNA sequencing results (Fig. 8). miR156d, miR169f, miR3711 and miR397 were upregulated and miR172, miR166d5p and miR171b were found to be downregulated. In contrast to the high throughput sequencing results, miR477e was downregulated in qRT PCR results. It may be due to the low quality of the primers or low abundance of the miRNAs and more study is needed to confirm this observation. Previous studies conducted in Glycine max, Populus euphratica and rice roots have reported this type of contradiction between the deep sequencing and qRT PCR results (Li et al. 2011a, b; Bakhshi et al. 2016).

Relative expression level of miRNAs evaluated by qRT PCR method. Among the eight drought responsive miRNAs selected for qRT PCR study, seven showed similar expression pattern with that of the small RNA sequencing results. miR156d, miR169f, miR3711 and miR397 were upregulated and miR172, miR166d5p and miR171b were found to be downregulated. In contrast to the high throughput sequencing results, miR477e was downregulated in qRT PCR results
Validation of miRNA targeted cleavage on mRNAs
RLM RACE was done to confirm two unigene sequences as targets for cardamom miRNAs (Fig. 9). Unigene 8377-2_P1263 encoding MYB domain protein was mapped with a cleavage site at the 12th nucleotide of the miR159 from 5′end. Cleavage site at the 9th base of the miR169 binding site on the unigene 8377-1_P1263 which encodes the nuclear factor Y; subunit 1 having a considerable role in drought tolerance was identified.

Mapping of target gene cleavage sites by 5′RLM-RACE. For each cardamom miRNA, miRNA sequence is shown in blue colour at the bottom and the partial target sequence is shown in black and red colour at the top. Arrow indicates the mapped cleavage sites on miRNA aligned position on the target mRNAs and number denotes the fraction of cloned pcr product. (Color figure online)
Discussion
Plants are continuously exposed to both biotic and abiotic stresses which limit crop yields. Among those factors, the negative influence of abiotic stress is increasing worldwide. This pointed out to the fact that unraveling the complex mechanisms underlying stress resistance of plants has profound significance to tackle the situation. Recently, the newly developed sequencing technologies, such as the Illumina Genome Analyzer, the ABI SOLiD system and Ion Torrent sequencing, show advances over traditional methods with improved throughput, speed and reduced cost. Currently, such next generation sequencing technologies offer applications such as identification of miRNAs in control and stress environments, which detect differential expression of those miRNAs and deliver new insights into the role of miRNAs in plant development, and stress related regulation. To date, nothing is known about the functions of miRNAs in abiotic stress responses in cardamom.
High-throughput sequencing of cardamom microRNAs
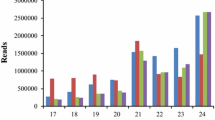
Small RNA libraries were constructed from wild cardamom plants grown under irrigated and drought conditions and Ion torrent sequencing was performed. After the pre-processing steps of the raw reads, sequences with 17–27 nt length were obtained. A total of 150 known and 20 novel miRNAs were identified from both the control and drought treated libraries. Sequences having 21 nt length were abundant among the miRNAs identified for both control and drought stressed small RNA libraries (Fig. 10). miR159, miR396, miR535, miR166b, miR167 and miR396e were the most abundantly expressed miRNAs. miR159 target mRNAs coding for MYB proteins which are known to bind to the promoter of the floral meristem identity gene LEAFY (Reyes and Chua 2007). miR396 target mRNA coding for Growth regulating factor (GRF) transcription factors, Rhodenase like protein and kinesin like protein B (Liu et al. 2009). miR535 mediates the cleavage of an SPL gene, controlling a number of fundamental aspects of plant growth and development, including vegetative phase change, flowering time, branching, and leaf initiation rate. miR166b cleave their target mRNAs of HD-ZIP III genes, play overlapping, distinct and antagonistic roles in key aspects of development that have evolved during land plant evolution (Boualem et al. 2008). miR167 has been implicated in auxin signalling by regulating the expression of certain auxin response factor (ARF) genes to determine the plant developmental process (Ebrahimi Khaksefidi et al. 2015). miR3711 which is a non-conserved miRNA reported only in Norway spruce and Astragalus chrysochlorus targets the hydroxycinnamoyltransferase gene which is known to promote lignin synthesis. Lignin is known to have pest resistance characteristics because of the insolubility and complexity of lignin polymer. There is a proposition that cardamom plants with high lignin content show more resistance to pest (Soumya and Sabu 2014). The targets of novel miRNAs were predicted using psRNAtarget. The functions employed by these targets were analysed by searching against the TAIR database (Supplementary File 5).

Distribution of miRNA sequences obtained from control and drought treated small RNA libraries. Sequences having 21 nt length were abundant among the miRNAs identified for both control and drought stressed small RNA libraries
Drought responsive microRNAs in cardamom
Seventeen miRNAs belonging to 11 families were found to be drought responsive in cardamom. Of these miRNA families, 9 were previously reported to be upregulated or downregulated under drought stress. miR156/159 family members like miR156d and miR159b were found to be induced and miR156l was repressed under drought stress in cardamom. Gene ontology analysis has shown that miR156l cleaves the target mRNA AT5G08620.1 which was identified to be involved in response to water deprivation (Table 4).There are reports that miR156 was up-regulated in A. thaliana (Liu et al. 2008), Triticum dicoccoides (Kantar et al. 2011), Hordeum vulgare (Kantar et al. 2010), Populus euphratica (Li et al. 2009), Prunus persica (Eldem et al. 2012) and was downregulated in Oryza sativa (Zhou et al. 2010). miR159b in cardamom was upregulated like that in Arabidopsis, but was discovered to be repressed in Oryza and Prunus. Both miR166 family members, miR166d-5p and miR166p were identified to be downregulated in this study. miR166 was downregulated in Triticum, but was induced in Glycine max. miR167 was upregulated in Arbidopsis and downregulated in Prunus. In this study miR167f-5p was induced under drought stress. miR169 targets Nuclear factor Y (NF-Y) transcription factor subunit A-5 which have predominant roles in developmental process and response to abiotic stresses in plants (Ding et al. 2013). miR169 was upregulated in tomato in response to drought. With the increased expression of miR169c in tomato, stomatal conductance and water loss become decreased and shows increased tolerance to drought (Li et al. 2008). Induced expression of this miRNA under drought stress was also observed in other plants like Oryza, Glycine max (Li et al. 2011b) and Populus euphratica. We also observed that miR169d and miR169f were upregulaed in cardamom. Using AgriGO software, miR169c in cardamom was found to target AT1G54160.1 mRNA which express in response to water deprivation. In contrast to this, downregulation of this miR169 was found in Arabidopsis, Medicago (Wang et al. 2011) and Prunus. NFYA5 gene is more expressed in guard cells and vascular tissues. In guard cells, NFYA5 controls the opening and closing of stomata and in vascular tissues, NFYA5 regulates the expression of many drought responsive proteins (Li et al. 2008). This drought responsive proteins include dehydrins, vacoular acid invertase, glutathione-S-transferase (GST), helicase, proline, carbohydrates and abscicic acid regulating genes producing proteins like late embryo abundant (LEA), responsive to abscisic acid (RAB), cold regulated (COR) and 5-bisphosphate carboxylase oxygenase (Rubisco) (Close 1996; Pnueli et al. 2002; Trouverie et al. 2003; Anderson and Davis 2004; Nezhadahmadi et al. 2013). Expression of two members of the family miR17l was obtained in a contradictory manner. miR171i was upregulated and miR171b was downregulated in cardamom under drought stress. There are many reports of such differentially expressed miRNA members of the same families in response to water stress condition(Ferdous et al. 2015). In rice miR171 family members showed both induced and repressed expression, miR171b was upregulated and miR171i, miR171a, miR171c and miR171 show downregulation (Zhou et al. 2010). Upregulation of this miRNA family was found in other plant species like Arabidopsis, Prunus and down-regulation was observed in Triticum and Medicago. miR172 was downregulated in cardamom with a significant fold change of − 2.6. It was downregulated in rice also, but an induced expression level was noticed in case of Arabidopsis. miR396 was repressed in most of the plant species like rice, Medicago and Prunus. In cardamom miR396f-5p was upregulated as in Arabidopsis. In this study, Gene ontology has shown that the gene AT2G35930.1 and AT3G45140.1, which functions in response to water stress condition is targeted by miR396 and miR397 respectively. miR397 which showed upregulation in cardamom was found to be induced in Arabidopsis also, but was downregulated in rice and Prunus. miR399 was reported to be upregulated under drought condition in Medicago. In cardamom miR399b was identified to be downregulated. A repression of miR408 was observed in plant species like rice, Prunus and Populus trichocarpa (Shuai et al. 2013) which targets drought responsive genes like early responsive dehydration-related protein (ERD) and polyphenol oxidase (PPO). In cardamom, miR408 shows an upregulation like in Arabidopsis and Medicago. Studies have shown that upregulation of miR408 cleaves the target genes of COX5b, CSD1 and plantacyanin in Medicago (Trindade et al. 2010). miR477e in cardamom was upregulated under water stress. miR477 was found to be significantly differentiated in bread wheat under drought condition. miR3711 which is a non-conserved miRNA discovered as drought regulating in cardamom was not previously reported to be responsive to drought stress. miR3711 was reported to be upregulated in selenium treated tissues of Astragalus chrysochlorus (Cakir et al. 2016). Cross adaptation is a phenomenon in which plants subjected to a stress develop resistance against other stresses (Sabehat et al. 1998). In Medicago, upregulation of miR2089 and miR2118 was observed under drought stress whose target genes are responsive to disease condition (Wang et al. 2011). Upregulated miRNAs during stress condition leads to the inhibition of target genes which negatively affects the stress tolerance and downregulated miRNAs helps in accumulation of target mRNAs which positively contributes towards stress tolerance (Zhang 2015).
Monocot specific miRNAs in cardamom
miR396d and miR396e, reported to be present only in monocots were observed in cardamom which belongs to the monocotyledons family Zingiberaceae (Sunkar and Jagadeeswaran 2008). miR528 which is a monocot specific family detected in rice, sorghum and maize was found in our study (Franke and Green 2015). SsCBP1 was experimentally proven to be target for miR528 (Zanca et al. 2010). miR528 target recognition site in SsCBP1 is present only in monocot genomes and was confirmed that miR528 is monocot specific. Other miRNAs specific to monocots (Katiyar et al. 2015) observed in cardamom are miR156b, miR319a-b, miR395, miR396a and miR529.
Conclusions
This study provides an insight into the drought responsive miRNAs of cardamom by combined small RNA sequencing and bioinformatics analysis. A total of 150 conserved and 20 novel miRNAs were identified from both the control and treated libraries. Discovery of 17 differentially expressed miRNAs under drought stress suggests that these miRNAs might have involved in various biological processes to improve plant tolerance to water stress. Target genes were found to be involved in cellular, metabolic, biological regulation, response to stimulus, developmental and multiorganismal processes. Expression profiles of a group of differentially expressed miRNAs were successfully validated by using qRT PCR. This study would provide valuable contribution towards understanding miRNA-mediated regulatory mechanisms underlying drought response in monocot plants.
References
Agharbaoui Z, Leclercq M, Remita MA et al (2015) An integrative approach to identify hexaploid wheat miRNAome associated with development and tolerance to abiotic stress. BMC Genom 16:1–17. https://doi.org/10.1186/s12864-015-1490-8
Akdogan G, Tufekci ED, Uranbey S, Unver T (2015) miRNA-based drought regulation in wheat. Funct Integr Genom. https://doi.org/10.1007/s10142-015-0452-1
Anderson JV, Davis DG (2004) Abiotic stress alters transcript profiles and activity of glutathione S-transferase, glutathione peroxidase, and glutathione reductase in Euphorbia esula. Physiol Plant 120:421–433. https://doi.org/10.1111/j.0031-9317.2004.00249.x
Bakhshi B, Mohseni Fard E, Nikpay N et al (2016) MicroRNA signatures of drought signaling in rice root. PLoS ONE 11:1–25. https://doi.org/10.1371/journal.pone.0156814
Barrera-Figueroa BE, Gao L, Diop NN et al (2011) Identification and comparative analysis of drought-associated microRNAs in two cowpea genotypes. BMC Plant Biol 11:1–11. https://doi.org/10.1186/1471-2229-11-127
Bej S, Basak J (2014) MicroRNAs: the potential biomarkers in plant stress response. Am J Plant Sci 5:748–759. https://doi.org/10.4236/ajps.2014.55089
Boke H, Ozhuner E, Turktas M et al (2015) Regulation of the alkaloid biosynthesis by miRNA in opium poppy. Plant Biotechnol J 13:409–420. https://doi.org/10.1111/pbi.12346
Bonnet E, Wuyts J, Rouze P, Van de Peer Y (2004) Evidence that microRNA precursors, unlike other non-coding RNAs, have lower folding free energies than random sequences. Bioinformatics 20:2911–2917. https://doi.org/10.1093/bioinformatics/bth374
Boualem A, Laporte P, Jovanovic M et al (2008) MicroRNA166 controls root and nodule development in Medicago truncatula. Plant J Cell Mol Biol 54:876–887. https://doi.org/10.1111/j.1365-313X.2008.03448.x
Brodersen P, Sakvarelidze-Achard L, Bruun-Rasmussen M et al (2008) Widespread translational inhibition by plant miRNAs and siRNAs. Science 320:1185–1190. https://doi.org/10.1126/science.1159151
Cakir O, Candar-Cakir B, Zhang B (2016) Small RNA and degradome sequencing reveals important microRNA function in Astragalus chrysochlorus response to selenium stimuli. Plant Biotechnol J 14:543–556. https://doi.org/10.1111/pbi.12397
Close TJ (1996) Dehydrins: emergence of a biochemical role of a family of plant dehydration proteins. Physiol Plant 97:795–803. https://doi.org/10.1111/j.1399-3054.1996.tb00546.x
Dai X, Zhao PX (2011) psRNATarget: a plant small RNA target analysis server. Nucleic Acids Res 39:W155–W159. https://doi.org/10.1093/nar/gkr319
Ding Y, Tao Y, Zhu C (2013) Emerging roles of microRNAs in the mediation of drought stress response in plants. J Exp Bot 64:3077–3086. https://doi.org/10.1093/jxb/ert164
Ding X, Li J, Zhang H et al (2016) Identification of miRNAs and their targets by high-throughput sequencing and degradome analysis in cytoplasmic male-sterile line NJCMS1A and its maintainer NJCMS1B of soybean. BMC Genom 17:1–16. https://doi.org/10.1186/s12864-015-2352-0
Du Z, Zhou X, Ling Y et al (2010) agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res 38:W64–W70. https://doi.org/10.1093/nar/gkq310
Ebrahimi Khaksefidi R, Mirlohi S, Khalaji F et al (2015) Differential expression of seven conserved microRNAs in response to abiotic stress and their regulatory network in Helianthus annuus. Front Plant Sci 6:741. https://doi.org/10.3389/fpls.2015.00741
Eldem V, Celikkol Akcay U, Ozhuner E, et al (2012) Genome-wide identification of miRNAs responsive to drought in peach Prunus persica by high-throughput deep sequencing. PLoS ONE 7:e50298.
Ferdous J, Hussain SS, Shi B (2015) Role of microRNAs in plant drought tolerance. Plant Biotechnol J 13:293–305. https://doi.org/10.1111/pbi.12318
Franke KR, Green PJ (2015) The microRNAs of Brachypodium, genetics and genomics of Brachypodium. In: Vogel J (ed) Plant genetics and genomics: crop models. Springer, Cham
Griffiths-Jones S, Saini HK, van Dongen S, Enright AJ (2008) miRBase: tools for microRNA genomics. Nucleic Acids Res 36:D154–D158. https://doi.org/10.1093/nar/gkm952
Hajyzadeh M, Turktas M, Khawar KM, Unver T (2015) miR408 overexpression causes increased drought tolerance in chickpea. Gene 555:186–193. https://doi.org/10.1016/j.gene.2014.11.002
Hivrale V, Zheng Y, Puli COR et al (2016) Characterization of drought- and heat-responsive microRNAs in switchgrass. Plant Sci 242:214–223. https://doi.org/10.1016/j.plantsci.2015.07.018
Hofacker IL (2003) Vienna RNA secondary structure server. Nucleic Acids Res 31:3429–3431
Hwang E, Shin S, Kwon H (2011) Identification of microRNAs and their putative targets that respond to drought stress in Solanum tuberosum. J Korean Soc Appl Biol Chem 54:317–324. https://doi.org/10.3839/jksabc.2011.051
Jain M, Chevala VVSN, Garg R (2014) Genome-wide discovery and differential regulation of conserved and novel microRNAs in chickpea via deep sequencing. J Exp Bot 65:5945–5958. https://doi.org/10.1093/jxb/eru333
Jeong D, Green PJ (2013) The role of rice microRNAs in abiotic stress responses. J Plant Biol 56:187–197. https://doi.org/10.1007/s12374-013-0213-4
Kader SMA, Bauomi AA, Abdel-Rahman M et al (2015) Antioxidant potentials of (Elletaria cardamomum) cardamom against uranium hazards. Int J Basic Life Sci 3:2320–2513X
Kantar M, Unver T, Budak H (2010) Regulation of barley miRNAs upon dehydration stress correlated with target gene expression. Funct Integr Genom 10:493–507. https://doi.org/10.1007/s10142-010-0181-4
Kantar M, Lucas SJ, Budak H (2011) miRNA expression patterns of Triticum dicoccoides in response to shock drought stress. Planta 233:471–484. https://doi.org/10.1007/s00425-010-1309-4
Katiyar A, Smita S, Muthusamy SK et al (2015) Identification of novel drought-responsive microRNAs and trans-acting siRNAs from Sorghum bicolor (L.) Moench by high-throughput sequencing analysis. Front Plant Sci 6:506
Kelly H, Downing T, Tuite NL et al (2015) Cross platform standardisation of an experimental pipeline for use in the identification of dysregulated human circulating MiRNAs. PLoS ONE 10:e0137389
Kuriakose G, Sinu PA, Shivanna KR (2009) Domestication of cardamom (Elettaria cardamomum) in Western Ghats, India: divergence in productive traits and a shift in major pollinators. Ann Bot 103:727–733. https://doi.org/10.1093/aob/mcn262
Lander ES (2001) Initial sequencing and analysis of the human genome. Nature 409:860–921
Langmead B, Trapnell C, Pop M, Salzberg SL (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10:R25. https://doi.org/10.1186/gb-2009-10-3-r25
Li W, Oono Y, Zhu J et al (2008) The Arabidopsis NFYA5 transcription factor is regulated transcriptionally and posttranscriptionally to promote drought resistance. Plant Cell 20:2238–2251. https://doi.org/10.1105/tpc.108.059444
Li B, Yin W, Xia X (2009) Identification of microRNAs and their targets from Populus euphratica. Biochem Biophys Res Commun 388:272–277. https://doi.org/10.1016/j.bbrc.2009.07.161
Li B, Qin Y, Duan H et al (2011a) Genome-wide characterization of new and drought stress responsive microRNAs in Populus euphratica. J Exp Bot 62:3765–3779. https://doi.org/10.1093/jxb/err051
Li H, Dong Y, Yin H et al (2011b) Characterization of the stress associated microRNAs in Glycine max by deep sequencing. BMC Plant Biol 11:1–12. https://doi.org/10.1186/1471-2229-11-170
Li J, Fu F, An M et al (2013) Differential expression of microRNAs in response to drought stress in maize. J Integr Agric 12:1414–1422. https://doi.org/10.1016/S2095-3119(13)60311-1
Li W, Wang P, Li Y et al (2015) Identification of microRNAs in response to different day lengths in soybean using high-throughput sequencing and qRT-PCR. PLoS ONE 10:e0132621
Lima JC, Loss-Morais G, Margis R (2012) MicroRNAs play critical roles during plant development and in response to abiotic stresses. Genet Mol Biol 35:1069–1077
Lin S, Chen T, Qin X et al (2014) Identification of microRNA families expressed in sugarcane leaves subjected to drought stress and the targets thereof. Pak J Agric Sci 51:925–934
Liu H, Tian X, Li Y et al (2008) Microarray-based analysis of stress-regulated microRNAs in Arabidopsis thaliana. RNA 14:836–843. https://doi.org/10.1261/rna.895308
Liu D, Song Y, Chen Z, Yu D (2009) Ectopic expression of miR396 suppresses GRF target gene expression and alters leaf growth in Arabidopsis. Physiol Plant 136:223–236. https://doi.org/10.1111/j.1399-3054.2009.01229.x
Liu W, Xu L, Wang Y et al (2015) Transcriptome-wide analysis of chromium-stress responsive microRNAs to explore miRNA-mediated regulatory networks in radish (Raphanus sativus L.). Sci Rep 5:14024. https://doi.org/10.1038/srep14024
Lutful Kabir FM, DeInnocentes P, Bird RC (2015) Altered microRNA expression profiles and regulation of INK4A/CDKN2A tumor suppressor genes in canine breast cancer models. J Cell Biochem 116:2956–2969. https://doi.org/10.1002/jcb.25243
Martin M (2011) Cutadapt removes adapter sequences from highthroughput sequencing reads. EMBnet J. https://doi.org/10.14806/ej.17.1.200
Morozova O, Marra MA (2008) Applications of next-generation sequencing technologies in functional genomics. Genomics 92:255–264. https://doi.org/10.1016/j.ygeno.2008.07.001
Murugan M, Shetty PK, Ravi R et al (2011) Environmental impacts of Intensive cardamom (small) cultivation in Indian cardamom hills: the need for sustainable and efficient practices. Recent Res Sci Technol 3:9–15
Muthusamy M, Uma S, Backiyarani S, Saraswathi MS (2014) Computational prediction, identification, and expression profiling of microRNAs in banana (Musa spp.) during soil moisture deficit stress. J Hortic Sci Biotechnol 89:208–214. https://doi.org/10.1080/14620316.2014.11513070
Nadiya F, Anjali N, Gangaprasad A, Sabu K (2015) High-quality RNA extraction from small cardamom tissues rich in polysaccharides and polyphenols. Anal Biochem 485:25–27
Nezhadahmadi A, Prodhan ZH, Faruq G (2013) Drought tolerance in wheat. Sci World J 2013:610721. https://doi.org/10.1155/2013/610721
Nitasha BGM, Nagendra N, Vinodraj K et al (2015) Comparison of the efficacy of cardamom (Elettaria cardamomum) with pioglitazone on dexamethasone-induced hepatic steatosis, dyslipidemia, and hyperglycemia in albino rats. J Adv Pharm Technol Res. https://doi.org/10.4103/2231-4040.157981
Patel RK, Jain M (2012) NGS QC toolkit: a toolkit for quality control of next generation sequencing data. PLoS ONE 7:e30619
Pnueli L, Hallak-Herr E, Rozenberg M et al (2002) Molecular and biochemical mechanisms associated with dormancy and drought tolerance in the desert legume Retama raetam. Plant J Cell Mol Biol 31:319–330
Qiblawi S, Dhanarasu S, Faris MA (2015) Chemopreventive effect of cardamom (Elettaria cardamomum L.) against benzo(alpha)pyrene-induced forestomach papillomagenesis in Swiss albino mice. J Environ Pathol Toxicol Oncol 34:95–104
Ravindran PN, Madhusoodanan KJ (2002) Cardamom, the genus Elettaria. Taylor and Francis, London
Reyes JL, Chua N (2007) ABA induction of miR159 controls transcript levels of two MYB factors during Arabidopsis seed germination. Plant J Cell Mol Biol 49:592–606. https://doi.org/10.1111/j.1365-313X.2006.02980.x
Sabehat A, Weiss D, Lurie S (1998) Heat-shock proteins and cross-tolerance in plants. Physiol Plant 103:437–441. https://doi.org/10.1034/j.1399-3054.1998.1030317.x
Schmittgen TD, Lee EJ, Jiang J et al (2008) Real-time PCR quantification of precursor and mature microRNA. Methods 44:31–38. https://doi.org/10.1016/j.ymeth.2007.09.006
Shuai P, Liang D, Zhang Z et al (2013) Identification of drought-responsive and novel Populus trichocarpa microRNAs by high-throughput sequencing and their targets using degradome analysis. BMC Genom 14:233
Shweta B, Tamara B, Martin HA et al (2015) Detecting miRNA mentions and relations in biomedical literature [version 3; referees: 2 approved, 1 approved with reservations]
Song C, Wang C, Zhang C et al (2010) Deep sequencing discovery of novel and conserved microRNAs in trifoliate orange (Citrus trifoliata). BMC Genom 11:431
Song JB, Gao S, Sun D et al (2013) miR394 and LCR are involved in Arabidopsis salt and drought stress responses in an abscisic acid-dependent manner. BMC Plant Biol 13:210. https://doi.org/10.1186/1471-2229-13-210
Song A, Wang L, Chen S et al (2015) Identification of nitrogen starvation-responsive microRNAs in Chrysanthemum nankingense. Plant Physiol Biochem 91:41–48. https://doi.org/10.1016/j.plaphy.2015.04.003
Soumya SD, Sabu KK (2014) Development and use of SSR markers for analysis of genetic diversity and correlation with lignin content in cardamom (Elettaria cardamomum Maton). Vellayani Department of Plant Biotechnology, College of Agriculture, Trivandrum, p 67
Sun F, Guo G, Du J et al (2014) Whole-genome discovery of miRNAs and their targets in wheat (Triticum aestivum L.). BMC Plant Biol 14:1–17. https://doi.org/10.1186/1471-2229-14-142
Sunkar R, Jagadeeswaran G (2008) In silico identification of conserved microRNAs in large number of diverse plant species. BMC Plant Biol 8:37. https://doi.org/10.1186/1471-2229-8-37
Tang C, Yang M, Wu F et al (2015) Identification of miRNAs and their targets in transgenic Brassica napus and its acceptor (Westar) by high-throughput sequencing and degradome analysis. RSC Adv 5:85383–85394. https://doi.org/10.1039/C5RA14672K
Trindade I, Capitao C, Dalmay T et al (2010) miR398 and miR408 are up-regulated in response to water deficit in Medicago truncatula. Planta 231:705–716. https://doi.org/10.1007/s00425-009-1078-0
Trivedi UH, Cezard T, Bridgett S et al (2014) Quality control of next-generation sequencing data without a reference. Front Genet 5:111. https://doi.org/10.3389/fgene.2014.00111
Trouverie J, Thevenot C, Rocher J et al (2003) The role of abscisic acid in the response of a specific vacuolar invertase to water stress in the adult maize leaf. J Exp Bot 54:2177–2186. https://doi.org/10.1093/jxb/erg234
Voelkerding KV, Dames SA, Durtschi JD (2009) Next-generation sequencing: from basic research to diagnostics. Clin Chem 55:641–658. https://doi.org/10.1373/clinchem.2008.112789
Wang T, Chen L, Zhao M et al (2011) Identification of drought-responsive microRNAs in Medicago truncatula by genome-wide high-throughput sequencing. BMC Genom 12:367. https://doi.org/10.1186/1471-2164-12-367
Wang Z, Jiang D, Zhang C et al (2015) Genome-wide identification of turnip mosaic virus-responsive microRNAs in non-heading Chinese cabbage by high-throughput sequencing. Gene 571:178–187. https://doi.org/10.1016/j.gene.2015.06.047
Xia K, Wang R, Ou X et al (2012) OsTIR1 and OsAFB2 downregulation via OsmiR393 overexpression leads to more tillers, early flowering and less tolerance to salt and drought in rice. PLoS ONE 7:e30039
Yang X, Li L (2011) miRDeep-P: a computational tool for analyzing the microRNA transcriptome in plants. Bioinformatics 27:2614–2615. https://doi.org/10.1093/bioinformatics/btr430
Yang J, Zhang N, Zhou X et al (2015a) Identification of four novel stu-miR169s and their target genes in Solanum tuberosum and expression profiles response to drought stress. Plant Syst Evol 302:55–66. https://doi.org/10.1007/s00606-015-1242-x
Yang T, Fang L, Zhang X et al (2015b) High-throughput development of SSR markers from pea (Pisum sativum L.) based on next generation sequencing of a purified Chinese commercial variety. PLoS ONE 10:e0139775
Yi F, Chen J, Yu J (2015) Global analysis of uncapped mRNA changes under drought stress and microRNA-dependent endonucleolytic cleavages in foxtail millet. BMC Plant Biol 15:241. https://doi.org/10.1186/s12870-015-0632-0
Yin F, Qin C, Gao J et al (2015) Genome-wide identification and analysis of drought-responsive genes and microRNAs in tobacco. Int J Mol Sci 16:5714–5740. https://doi.org/10.3390/ijms16035714
Zanca AS, Vicentini R, Ortiz-Morea FA et al (2010) Identification and expression analysis of microRNAs and targets in the biofuel crop sugarcane. BMC Plant Biol 10:1–13. https://doi.org/10.1186/1471-2229-10-260
Zandkarimi H, Bedre R, Solis J et al (2015) Sequencing and expression analysis of salt-responsive miRNAs and target genes in the halophyte smooth cordgrass (Spartina alternifolia Loisel). Mol Biol Rep 42:1341–1350. https://doi.org/10.1007/s11033-015-3880-z
Zhang B (2015) MicroRNA: a new target for improving plant tolerance to abiotic stress. J Exp Bot 66:1749–1761. https://doi.org/10.1093/jxb/erv013
Zhang B, Pan X, Cannon CH et al (2006) Conservation and divergence of plant microRNA genes. Plant J 46:243–259. https://doi.org/10.1111/j.1365-313X.2006.02697.x
Zhang Y, Zhu X, Chen X et al (2014) Identification and characterization of cold-responsive microRNAs in tea plant (Camellia sinensis) and their targets using high-throughput sequencing and degradome analysis. BMC Plant Biol 14:271. https://doi.org/10.1186/s12870-014-0271-x
Zhao B, Liang R, Ge L et al (2007) Identification of drought-induced microRNAs in rice. Biochem Biophys Res Commun 354:585–590. https://doi.org/10.1016/j.bbrc.2007.01.022
Zhou L, Liu Y, Liu Z et al (2010) Genome-wide identification and analysis of drought-responsive microRNAs in Oryza sativa. J Exp Bot 61:4157–4168. https://doi.org/10.1093/jxb/erq237
Acknowledgements
The authors thank the Director of the Jawaharlal Nehru Tropical Botanic Garden & Research Institute (JNTBGRI) for providing the necessary facilities to carry out this research work. We acknowledge the permission granted by Kerala Forest Department for collecting the plant samples. Thanks are due to Vishnu, J. S. and Shefeek, S. for various assistances rendered for the study. We are grateful to the staff at the Centre for Cellular and Molecular Platforms (C-CAMP, Bangalore, India) and Origin Labs, Kerala for performing high throughput sequencing and qRT-PCR respectively. The research was supported by Kerala State Council for Science, Technology, and Environment (KSCSTE), Trivandrum by granting Research Fellowship to N. Anjali (010-31/FSHP/10/CSTE).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Anjali, N., Nadiya, F., Thomas, J. et al. Identification and characterization of drought responsive microRNAs and their target genes in cardamom (Elettaria cardamomum Maton). Plant Growth Regul 87, 201–216 (2019). https://doi.org/10.1007/s10725-018-0462-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10725-018-0462-9