
Abstract
Plant microRNAs are important endogenous gene regulators which regulate gene expression at post-transcriptional level. Previous studies have identified that miR169 family members regulated the NF-YA transcription factors which have been implicated in plant development and stress responses. At present, reported potato genome sequence data offered an opportunity for global insights into the molecular mechanisms of the miR169/NF-YA modules in potato. In this work, 4 novel stu-miR169 family members were predicted in potato based on potato genome sequence data. miRNA target prediction showed that mature stu-miR169 sequences have a bite sit on the 5 of StNF-YA genes in potato, and three of them were validated by RNA ligase-mediated 5′RACE (5′ RLM-RACE) assay. The result from investigation of the expression patterns of mature stu-miR169 and their predicted target genes also showed that mature stu-miR169 was down-regulated in response to the drought stress. There were some targeted StNF-YA genes that exhibited a negative expression pattern with mature stu-miR169 during the different periods of drought-treated samples. Taken together, the decreased expression of stu-miR169 might drive over-expression of NF-YA family members, they are related to resistances against drought stress.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
MicroRNAs (miRNAs) are a class of noncoding small RNAs (sRNAs) which have been ubiquitously demonstrated to be critical regulators of gene expression either through degradation of the transcript or translational attenuation at post-transcriptional level (Voinnet 2009). miRNA genes are transcribed by RNA polymerase II and termed pri-microRNAs (pri-miRNAs). Mature miRNAs are derived from longer noncoding pri-miRNAs, and being processed from these transcripts by multiple cleavage steps process involving many complicated enzymes (Kurihara and Watanabe 2004; Bartel 2004; Parizotto et al. 2004). The mature miRNAs are usually 21–24 nucleotides (nt) in length and can more easily combine with RNA-induced silencing complex (RISC) to regulate the expression of genes either through degradation of the transcript or translational attenuation, which mainly depend on the complementarities between miRNA and targeted mRNA (Kidner and Martienssen 2005).
In plants, most miRNAs have perfect or near perfect complementarity to their mRNA targets and cleave them (Rhoades et al. 2002; Bartel 2004; Chen 2005; Du and Zamore 2005; Zhang et al. 2006a). Since the first plant miRNAs were reported in Arabidopsis (Llave et al. 2002; Park et al. 2002), increasing amounts of researches have shown that miRNA-regulated targets are involved in plant multiple developmental pathways and biological processes. A significant fraction has been shown to play crucial roles in plant responses to a variety of abiotic and biotic stresses, including disease stress (Chapman et al. 2004), mechanical stress (Lu et al. 2005, 2008), nutrient homeostasis (Pant et al. 2009; Liang et al. 2010), drought (Zhou et al. 2010; Li et al. 2011; Zhang et al. 2014), salinity (Macovei and Tuteja 2012; Li et al. 2013), cold (Zhang et al. 2009a; Barakat et al. 2012), heat (Chen et al. 2012a; Yu et al. 2012), oxidative stress (Sunkar et al. 2006), and heavy metal stress (Ding et al. 2011; Zhou et al. 2012; Chen et al. 2012b; Zeng et al. 2012). These studies were mainly focused on model plant species, such as Arabidopsis thaliana, Oryza sativa and Zea mays. Potato (Solanum tuberosum) is an important crop around the world, accounting for a significant amount of human food consumption. But the growth and productivity of potato are severely affected by drought stress. In previous studies, many potato conserved miRNAs were identified based on bioinformatics analysis using potato EST, GSS and nr databases (Zhang et al. 2009b; Xie et al. 2010; Yang et al. 2010; Kim et al. 2011). Recently, 28 conserved and 120 potato-specific miRNA families were also identified by deep sequencing from potato leaf and stolon (Zhang et al. 2013). However, a few of miRNAs and regulated target genes have been identified responses to drought stress and other abiotic stresses in potato (Hwang et al. 2011a, b; Zhang et al. 2014).
The miR169 family is one of the most conserved miRNA families in plants and has been shown to be involved in plant responses to abiotic stress (Li et al. 2008; Zhou et al. 2008; Lee et al. 2010; Zhao et al. 2011). Some of miR169 families were also identified by deep sequencing in potato (Zhang et al. 2013). However, the function of those miR169 families remains largely unknown in potato. Computational prediction and experimental analysis suggested that miR169-targeted members targeted to NF-YA gene family members (Jones-Rhoades et al. 2006; Zhao et al. 2009; Ni et al. 2013), which was an evolutionarily conserved transcription factor presented in nearly all eukaryotes. NF-YA was encoded by a single gene in yeast and animals, but in plants, which was encoded by multigene family. At least 10 NF-YA genes were present in rice, 36 potential NF-YA genes in maize and 38 potential NF-YA genes in potato in the plant transcription factor database (http://planttfdb.cbi.pku.edu.cn/). The CCAAT box was one of the most common cis-elements in plant promoters. It was located 80 to 100 base pairs (bp) upstream of the transcription start site (TSS) and presented in 30 % of all eukaryotic gene promoters (Maity and de Crombrugghe 1998; Mantovani 1999). NF-YA as a CCAAT-binding protein complex has high affinity and sequence specificity for the extensive CCAAT box in the eukaryotic genome, which plays significant roles in the mediation of diverse genes and involves in regulating plant development and various stress responses including plant nodule development (Combier et al. 2006), chloroplast biogenesis (Petroni et al. 2012), light signaling (Laloum et al. 2012), nitrogen nutrition (Warpeha and Kaufman 2007; Xu et al. 2011), and ABA response (Laloum et al. 2012). In addition, miR169/NF-YA module was involved in drought tolerance and stress-induced early flowering in Arabidopsis (Li et al. 2008; Xu et al. 2013). However, the miR169/NF-YA modules respond to drought or other abiotic stress have not been reported in potato.
To understand the regulating mechanism of miR169/NF-YA module respond to drought stress in potato, we explored 4 novel stu-miR169 family members based on bioinformatics analysis. We predicted and confirmed that StNF-YA family members were regulated by stu-miR169 family members and helped potato responsible for drought stress.
Materials and methods
Plant material and drought treatments
Potato variety ‘Longshu 3’ was grown in pot in the controlled environment chamber at 24 °C with 16-h day-length and supplemental high-intensity sodium lighting. When the plant was 20 cm long, drought conditions were simulated by withholding watering on the treated pots while the other pots were grown under normal condition as controls. Fresh leaves were collected from treated potato plants every 5 days, and lasted for 5 times. Entire plant leaves were immediately transferred into liquid nitrogen and were stored at −80 °C for further use.
Identification of miR169 family members and their precursors in potato by homology search
To genome-wide identification of potential miR169 family members in potato, a total of 427 previously known plant miR169 mature sequences were obtained from 35 species (miRBase Release 20.0, June, 2013; http://microrna.sanger.ac.uk/). To avoid the redundant or overlapping mature sequences of miR169s, mature miRNAs which were carefully screened as non-redundant sequences were used as nucleotide reference set for homology search for miR169 and its precursor in potato. The procedure and general strategy used to search for potential miRNAs in potato are shown in Fig. 1. Based on mature miRNAs likely to be conserved among species, screened non-redundant plant miR169 mature sequences were used against the potato EST, GSS, and nr databases. Sequences with less than two mismatches with known plant mature sequences of miR169s were used further against the protein database (http://www.ncbi.nlm.nih.gov/) to remove coding sequences.

Bioinformatic prediction procedure of potential miR169 family members in potato
The remaining candidate sequences were then carefully predicted and generated by the secondary structures using the web-based software UNAFold which was publicly available at the website (http://www.bioinfo.rpi.edu/applications/mfold/old/rna/). The parameters were used the default parameters except the folding temperature was fixed at 37 °C. The following criteria were then further screened the potential miR169s or pre-miR169s: (1) pre-miRNA sequence can fold into an standard stem-loop hairpin secondary structure; (2) the mature candidate miRNA sequence was located in one arm of the hairpin structure and had less than six mismatches with the opposite miRNA* sequence in the other arm; (3) no loop or break in mature miRNA sequences was allowed; (4) the secondary structures of predicted pre-miRNA had high negative minimal free energies (MFEs) and minimal free energy index (MFEIs); MFEI more than 0.85 and greater than 60 nt with a minimum free energy (MFE) of lower than −15 kcal/mol was accepted as being potential miRNA precursor sequences (Zhang et al. 2006a, b, 2007); (5) an A + U content filter and a loop-length filter were applied to the hairpin structures obtained only hairpins with 20–22 mature nucleotide sequences with a C + G content of 50 % or less were analyzed further. If one sequence has met all of these criteria, we considered it as a precursor of miR169 family members in potato.
Prediction and identification of miR169-regulated NF-YA family members in potato
Previous studies have showed that plant miRNAs regulate gene expression by binding to target mRNA sequences in a perfect or near perfect complementary site (Rhoades et al. 2002; Chen 2005). This makes it possible to predict plant miRNA targets using a homology search. In this study, the putative target genes for miR169s were predicted from potato genome 3.4 transcripts (Solanum tuberosum group phureja DM1-3) using the plant miRNA target prediction online software psRNATarget (http://plantgrn.noble.org/psRNATarget/) (Dai and Zhao 2011) with default parameters. To identify function of these predicted mRNA sequences, they were used to blast against the no-redundant protein sequences (nr) database (http://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastx&PAGE_TYPE=BlastSearch&LINK_LOC=blasthome) with Blastx in NCBI. Blastp and Tblaxtn (basic local alignment search tool: http://blast.ncbi.nlm.nih.gov) were used with the e value cutoff set to 1e-003 (Altschul et al. 1990). Protein sequences derived from the candidate genes were examined using the domain analysis programs Pfam database (protein family: http://pfam.Sanger.ac.uk/) with the default cutoff parameters (Bateman et al. 2002).
Bioinformatic analysis of potential targets
Potential FN-YA genes were selected and used to further bioinformatic analysis. The chromosomal locations and gene structures were retrieved from the potato genome data which were downloaded from the PGSC database (http://potatogenome.net/index.php/Main_Page). The multiple alignments of protein sequences were conducted using the Clustalx1.83 program. MEGA 5.0 software was used to generate the phylogenetic tree analysis using the neighbor-joining (NJ) method and reliability values at each branch representing bootstrap samples (1000 replicates) (Tamura et al. 2011). The pI/Mw and physicochemical property of the deduced polypeptide were predicted using the programs listed in Expasy (http://www.EXPASY.org). The genomic sequences were identified from Potato Genome Sequencing Consortium database (http://potatogenome.net/index.php/Main_Page). The gene structures (exon and intron) were analyzed with splign (http://www.ncbi.nlm.nih.gov/sutils/splign) (Kapustin et al. 2008).
RNA extraction
Potato leaves harvested in liquid nitrogen were used to extract the RNA immediately. Total RNA without genome DNA was extracted with TRIzol Total RNA Reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s protocol. RNA concentration was quantified using NanoPhotometer (Implen) and then stored at −80 °C for further use.
Quantitative real-time PCR analysis of mature miRNAs
Expression of mature stu-miR169 sequences was analyzed using quantitative real-time PCR (qRT-PCR). Reverse transcription was carried out in 20-μl reaction mixture. 1 μg of total RNA of each sample was treated with DNase and reverse transcribed using One Step PrimeScript® miRNA cDNA Synthesis Kit (TaKaRa) according to the manufacturer’s instructions. The reverse transcription product was amplified using a miRNA-specific forward primer and a universal reverse primer. The PCR amplification was set at 40 cycles of denaturation at 95 °C for 10 s, annealing at 60 °C for 30 s, and extension at 72 °C for 1 min (Feng et al. 2009). 1 mol of each Uni-miR qPCR primer and miRNA-specific primer were used for each PCR reaction. Uni-miR qPCR primers were provided with the kit, and the miRNA-specific primers were as following Table 1. The expression of miRNA was normalized with St18sRNA.
qRT-PCR analysis of target genes
qRT-PCR analysis was performed for miR169-regulated NF-YAs expression analysis. Briefly, 20-ml PCR reaction contained about 100 ng cDNA, 10 ml SYBR® Premix Ex Taq™ II (TaKaRa), 0.8 μl each primer. The reactions were mixed gently and incubated at 94 °C for 2 min, followed by 40 cycles of 94 °C for 30 s, 60 °C for 34 s and 72 °C for 30 s. ef1a was used for each sample as an internal control. All samples were performed on an optical 96-well plate with the ABI3000 (Applied Biosystems 3000 Real-Time PCR System) with 3 biological replicates and 2 technical replicates. The relative expression level of target genes was detected using the 2−∆∆Ct method. The standard deviations of the data were obtained from the three independent replicates.
Experimental validation miRNA-directed cleavage site using the 5′ RLM-RACE
To validate the NF-YA mRNA cleavage sites caused by the miR169 family members in potato, a modified RNA ligase-mediated 5′RACE (5′ RLM-RACE) method was used. Total RNAs from potato leaves were ligated to the 5′-RNA adapter by T4 RNA ligase 1 (NEB), and reversely transcribed to yield cDNAs. The 5′ ends of the miR169 cleavage products were amplified by PCR using the primers 5′-adapter and the gene-specific primer of stNF-YAs (Table 2). The different PCR fragments were then cloned and sequenced to determine the cleavage sites.
Results and discussion
Prediction of potential novel miR169 family members in potato
According to miRBase version 20 (http://www.mirbase.org/) and published data, 427 mature sequences of miR169 families have been discovered in 35 plant species, which including monocots and dicots as well as some ancient Coniferophytas (Fig. 2). Eight miR169s were discovered in potato. In this research, we searched the potato GSS and EST databases by BLAST based on the mature sequences of all miR169s previously identified in plants. We identified 4 novel miR169 family members in potato, which can be folded to the typical secondary structure of the miRNA family and named as stu-miR169i, stu-miR169j, stu-miR169k and stu-miR169l. The detailed information of 4 predicted novel miR169 family members is listed in Table 3.

All of known plant miR169 family members in different species. Aly (Arabidopsis lyrata); Aqc (Aquilegia caerulea); Ath (Arabidopsis thaliana); Ata (Aegilops tauschii); Atr (Amborella trichopoda); Bdi (Brachypodium distachyon); Bna (Brassica napus); Cca (Cynara cardunculus); Cme (Cucumis melo); Cpa (Carica papaya); Csi (Citrus sinensis); Far (Festuca arundinacea); Ghb (Gossypium herbaceum); Ghr (Gossypium hirsutum); Gma (Glycine max); Hvu (Hordeum vulgare); Lus (Linum usitatissimum); Mdm (Malus domestica); Mes (Manihot esculenta); Mtr (Medicago truncatula); Nta (Nicotiana tabacum); Osa (Oryza sativa); Pde (Pinus densata); Ppe (Prunus persica); Ptc (Populus trichocarpa); Rco (Ricinus communis); Sbi (Sorghum bicolor); Sly (Solanum lycopersicum); Ssl (Salvia sclarea); Ssp (Saccharum sp); Stu (Solanum tuberosum); Tcc (Theobroma cacao); Vun (Vigna unguiculata); Vvi (Vitis vinifera); Zma (Zea mays)
All of them were identified in GSS database and contained the mature miRNA sequence within one arm of the hairpin structure (Fig. 3). Among them, 3 mature miRNA sequences were located at the 5′ end of the miRNA precursor sequence, and only one was located at the 3′ end. The length of mature miRNA sequences was 21 or 22 nucleotides and had less six mismatches with the opposite miRNA* sequences in the other arm, which were similar to the typical length of plant mature miRNAs (21–24 nt) and no more than 6 nt mismatches (Griffiths-Jones et al. 2006); no loop or break in mature miRNA sequences of hairpin structure was found. The length of miRNA precursors varied from 80 to 119 nucleotides (Online Resource 1), which was also similar to the length of plant miRNA precursors range from 60 to more than 400 nucleotides (Zhang et al. 2006c). The sequences of the mature miRNA sequences had C + G content ranging from 32.8 to 45.7 %, which were also in agreement with the notion that miRNA precursors and mature miRNAs contained more A + U nucleotides than G + C (Guddeti et al. 2005). Pre-miRNAs had high minimal folding free energy index (MFEI) and the average MFEI of miRNA precursors was 0.97 in previously known plant pre-miRNAs (Zhang et al. 2006d). More importantly, the MFEI of over 90 % of miRNA precursors is greater than 0.85 and no other RNAs higher than 0.85. From this prediction, the novel identified 4 pre-miR169s had MFEI values ranging from 0.935 to 1.895, which greatly larger than 0.85, were most likely to be the miRNAs. To make certain that the miRNA candidates were real existent in potato, qRT-PCR was used to detect and quantify the predicted miRNAs of differently treated samples. The result showed that mature sequences of novel predicted stu-miR169 were successfully detected.

Predicted hairpin secondary structures of 4 novel stu-miR169 family members in this research. Mature miRNA sequences are in red and underlined
Potential target genes of stu-miR169 in potato
Gene regulation under drought stress is mediated by multiple transcription factors (Zhu 2002; Yamaguchi-Shinozaki and Shinozaki 2006). NF-YAs act as a DNA sequence-seeking component of the trimeric transcription factor and are expected to be localized to the nucleus where they can combine with the NF-YB/C dimer to create the active transcription factor (Nardini et al. 2013). Plant NF-YA genes are known to have function in plant nodule development (Combier et al. 2006), N deficiency (Xu et al. 2011), and ABA response (Warpeha and Kaufman 2007). Previous studies have identified that miR169/NF-YA modules were important regulators for development and stress responses in Arabidopsis thaliana, Oryza sativa and Glycine max (Li et al. 2008; Mantovani 1999; Zhao et al. 2009). In this research, NF-YA mRNAs were predicted to be the targets of stu-miR169s based on analysis using psRNA target online software (Table 4). All of five StNF-YAs had conserved domain of NF-YB/C interaction and DNA binding domain and the typical StNF-YAs proteins (Fig. 4). The full-length of cDNA sequences of them varied from 1076 to 3182 nucleotides, and the length of CDS sequence varied from 591 to 933 nucleotides. The detailed information of isoelectric point (pI), molecular mass and the length of predicted protein is shown in Table 5. The peptide length and the molecular weight of these StNF-YA genes were extremely close to the plant typical NF-YA family members that had been reported in Arabidopsis, rice, maize and tomato.

Alignment analysis of the full-length amino acid sequences of 5 identified potato NF-YA genes using Clustalx1.83. The domain of NF-YA gene protein sequences was predicted using Pfam
Phylogenetic and gene structure analysis of miR169-regulated StNF-YAs
Orthology predictions are important for transferring functional information between different species (Gabaldon 2008). To evaluate the evolutionary and functional relationships of miR169-regulated StNF-YA genes with tomato and Arabidopsis NF-YAs, multiple alignments were developed using full-length amino acid sequences of all known potato, tomato and Arabidopsis NF-YA genes (Dryad: http://datadryad.org/, DOI:10.5061/dryad.3s057). They were subjected to a multiple sequence alignment using the neighbor-joining (NJ) method of the MEGA 5.0 program. The results showed that NF-YA genes were highly conserved in potato, tomato and Arabidopsis. For five StNF-YA genes, StNF-YA3, StNF-YA4 and StNF-YA6 had highly significant orthology matches to AtNF-YA9, AtNF-YA8 and AtNF-YA3, respectively (Fig. 5). Regulation of these three AtNF-YAs had been implicated in drought tolerance (Warpeha and Kaufman 2007). Ath-miR169a/AtNF-YA module was also involved in drought tolerance (Li et al. 2008), therefore, StNF-YA3, StNF-YA4 and StNF-YA6 represented the most likely candidates for similar functions in potato.

A phylogenetic tree constructed with Clustalx1.83 using the full-length amino acid sequences of all known potato, tomato and Arabidopsis NF-YA genes. The bootstrap analysis was performed using 1000 replicates in MEGA 5.0 to evaluate the reliability of different phylogenetic groups
The patterns of exon–intron structure can provide important insights into the evolution of gene families. Comparison between the genomic and cDNA sequences indicated that StNF-YA1 had 4 exons; StNF-YA3 and StNF-YA6 had 5 exons; StNF-YA4 and StNF-YA5 had 6 exons (Fig. 6). StNF-YA1 and StNF-YA5 had a conserved exon with the length of 86 bp. StNF-YA3 and StNF-YA4 had highly conserved exon with the length of 165 bp. StNF-YA5 and StNF-YA6 had highly conserved exon with the length of 72 bp. This conserved exon numbers and intron phase among species supported their close evolutionary relationship and the introduced classification of subgroups.

Gene structures of five NF-YA genes in potato. Introns and exons were represented by lines and filled boxes, respectively. The numbers above the exons indicate the length (bp) of the exons
stu-miR169 family members contribute to the regulation of StNF-YA family members under drought stress
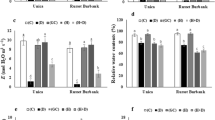
Expressional products of induced genes have been reported to function both in the initial stress response and in establishing stress tolerance in plants (Kasuga et al. 1999). The expression of these genes was controlled by a complex regulatory cascade. miRNAs can act as switches in such cascades negatively affecting gene expression at the post-transcription (Voinnet 2009). The miR169 family was one of the largest and most conserved miRNA families in plants. Previous studies have identified that miR169 family regulated the conserved NF-YA transcription factors. To understand the roles of stu-miR169 family members and their target genes in potato response to drought, RLM-5′RACE assay was used to validate the predicted targets, and the cleavage sites of three stu-miR169 target genes were validated by RLM-5′RACE assay (Fig. 7). qRT-PCR was also carried out to validate and detect the relative expression levels of stu-miR169 family members and their accordingly predicted target genes. The results showed that the relative expression levels of mature stu-miR169 sequence had a negative expression pattern with their target genes StNF-YA1 and StNF-YA5 during the different period of drought stress. However, StNF-YA4 was not increased too much. StNF-YA3 and StNF-YA6 were expressed at low levels in all six period of drought stress (Fig. 8). The accumulation of StNF-YAs under drought stress was highly diversified in potato, and these proteins may mediate a different metabolic process under normal growing conditions, and play distinct roles in response to adverse environments. Taking together, the result suggested that stu-miR169 may regulate over-expression of StNF-YA family members under drought stress in potato.

Mapping of StNF-YAs cleavage sites generated by stu-miR169

Expression pattern of stu-miR169 (a) and their potential target genes (b). Relative expression levels of mature stu-miR169 sequence and their potential target genes (StNF-YA1, StNF-YA3, StNF-YA4, StNF-YA5 and StNF-YA6) were measured by qRT-PCR in 5 periods under drought treatment. The X axis was the time of days after plant experiencing drought stress. The expression level of each gene in 0 day was set as 1, and following states were quantified relative to it using the 2−∆∆Ct method. St18sRNA and ef1a were chosen as endogenous controls for stu-miR169 and StNF-YA genes, respectively. The error bars indicated the standard deviations obtained from three independent replicates
In conclusion, we identified 4 novel miR169 family members in potato and found they were down-regulated along with drought stress, and 5 StNF-YA genes of potato were predicted as the target of stu-miR169s. The result suggested that decreased expression of stu-miR169s may drive over-expression of StNF-YA genes in potato, which can help potato to adapt to drought stress.
References
Altschul SF, Gish W, Miller W, Myers EW, Lipman DJF (1990) Basic local alignment search tool. J Molec Biol 215:403–410
Barakat A, Sriram A, Park J, Zhebentyayeva T, Main D, Abbott A (2012) Genome wide identification of chilling responsive microRNAs in Prunus persica. BMC Genom 13:481
Bartel DP (2004) MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297
Bateman A, Birney E, Cerruti L, Durbin R, Etwiller L, Eddy SR, Griffiths-Jones S, Howe KL, Marshall M, Sonnhammer EL (2002) The pfam protein families database. Nucl Acids Res 30:276–280
Chapman EJ, Prokhnevsky AI, Gopinath K, Dolja VV, Carrington JC (2004) Viral RNA silencing suppressors inhibit the microRNA pathway at an intermediate step. Gene Dev 18:1179–1186
Chen X (2005) MicroRNA biogenesis and function in plants. FEBS Lett 579:5923–5931
Chen L, Ren YY, Zhang YY, Xu JC, Sun FS, Zhang ZY, Wang YW (2012a) Genome-wide identification and expression analysis of heat responsive and novel microRNAs in Populus tomentosa. Gene 504:160–165
Chen L, Wang TZ, Zhao MG, Tian QY, Zhang WH (2012b) Identification of aluminum-responsive microRNAs in Medicago truncatula by genome-wide high-throughput sequencing. Planta 235:375–386
Combier JP, Frugier F, de Billy F, Boualem A, El-Yahyaoui F, Moreau S, Vernié T, Ott T, Gamas P, Crespi M, Niebel A (2006) MtHAP2-1 is a key transcriptional regulator of symbiotic nodule development regulated by microRNA169 in Medicago truncatula. Genes Dev 20:3084–3088
Dai X, Zhao PX (2011) psRNATarget: a plant small RNA target analysis server. Nucl Acids Res 39:W155–W159
Ding YF, Chen Z, Zhu C (2011) Microarray-based analysis of cadmium-responsive microRNAs in rice (Oryza sativa). J Exp Bot 62:3563–3573
Du T, Zamore PD (2005) microPrimer: the biogenesis and function of microRNA. Development 132:4645–4652
Feng J, Wang K, Liu X, Chen S, Chen J (2009) The quantification of tomato microRNAs response to viral infection by stem-loop real-time RT-PCR. Gene 437:14–21
Gabaldon T (2008) Large-scale assignment of orthology: back to phylogenetics? Genome Biol 9:235
Griffiths-Jones S, Grocock RJ, van Dongen S, Bateman A, Enright AJ (2006) miRBase: microRNA sequences, targets and gene nomenclature. Nucl Acids Res 34:140–144
Guddeti S, Zhang DC, Li AL, Leseberg CH, Kang H, Li XG, Zhai WX, Johns MA, Mao L (2005) Molecular evolution of the rice miR395 gene family. Cell Res 15:631–638
Hwang EW, Shin SJ, Park SC, Jeong MJ, Kwon HB (2011a) Identification of miR172 family members and their putative targets responding to drought stress in Solanum tuberosum. Genes Genom 33:105–110
Hwang EW, Shin SJ, Yu BK, Byun MO, Kwon HB (2011b) miR171 family members are involved in drought response in Solanum tuberosum. J Pl Biol 54:43–48
Jones-Rhoades MW, Bartel DP, Bartel B (2006) MicroRNAs and their regulatory roles in plants. Annual Rev Pl Biol 57:19–53
Kapustin Y, Souvorov A, Tatusova T, Lipman D (2008) Splign: algorithms for computing spliced alignments with identification of paralogs. Biol Direct 3:1–13
Kasuga M, Liu Q, Miura S, Yamaguchi-Shinozaki K, Shinozaki K (1999) Improving plant drought, salt, and freezing tolerance by gene transfer of a single stress-inducible transcription factor. Nat Biotechnol 17:287–291
Kidner CA, Martienssen RA (2005) The developmental role of microRNA in plants. Curr Opin Pl Biol 8:38–44
Kim HJ, Baek KH, Lee BW, Choi D, Hur CG (2011) In silico identification and characterization of microRNAs and their putative target genes in Solanaceae plants. Genome 54(2):91–98
Kurihara Y, Watanabe Y (2004) Arabidopsis micro-RNA biogenesis through Dicer-like 1 protein functions. Proc Natl Acad Sci USA 101:12753–12758
Laloum T, De MS, Gamas P, Baudin M, Niebel A (2012) CCAAT-box binding transcription factors in plants: y so many? Trends Pl Sci 18:157–166
Lee H, Yoo SJ, Lee JH, Kim W, Yoo SK, Fitzgerald H, Carrington JC, Ahn JH (2010) Genetic framework for flowering-time regulation by ambient temperature-responsive miRNAs in Arabidopsis. Nucl Acids Res 38:3081–3093
Li WX, Oono Y, Zhu J, He XJ, Wu JM, Iida K, Lu XY, Cui X, Jin H, Zhu JK (2008) The Arabidopsis NFYA5 transcription factor is regulated transcriptionally and posttranscriptionally to promote drought resistance. Pl Cell 20:2238–2251
Li BS, Qin YR, Duan H, Yin WL, Xia XL (2011) Genome-wide characterization of new and drought stress responsive microRNAs in Populus euphratica. J Exp Bot 62:3765–3779
Li BS, Duan H, Li JG, Deng XW, Yin WL, Xia XL (2013) Global identification of miRNAs and targets in Populus euphratica under salt stress. Pl Molec Biol 81:525–539
Liang G, Yang F, Yu D (2010) MicroRNA395 mediates regulation of sulfate accumulation and allocation in Arabidopsis thaliana. Plant J 62:1046–1057
Llave C, Kasschau KD, Rector MA, Carrington JC (2002) Endogenous and silencing- associated small RNAs in plants. Pl Cell 14:1605–1619
Lu S, Sun YH, Shi R, Clark C, Li L, Chiang VL (2005) Novel and mechanical stress-responsive microRNAs in Populus trichocarpa that are absent from Arabidopsis. Pl Cell 17:2186–2203
Lu S, Sun YH, Chiang VL (2008) Stress-responsive microRNAs in Populus. Pl J 55:131–151
Macovei A, Tuteja N (2012) microRNAs targeting DEAD-box helicases are involved in salinity stress response in rice (Oryza sativa L.). BMC Pl Biol 12:183
Maity SN, de Crombrugghe B (1998) Role of the CCAAT-binding protein CBF/NF-Y in transcription. Trends Biochem Sci 23:174–178
Mantovani R (1999) The molecular biology of the CCAAT-binding factor NF-Y. Gene 239:15–27
Nardini M, Gnesutta N, Donati G, Gatta R, Forni C, Fossati A, Vonrhein C, Moras D, Romier C, Bolognesi M, Mantovani R (2013) Sequence specific transcription factor NF-Y displays histone-like DNA binding and H2B-like ubiquitination. Cell 152:132–143
Ni Z, Hu Z, Jiang Q, Zhang H (2013) GmNFYA3, a target gene of miR169, is a positive regulator of plant tolerance to drought stress. Pl Molec Biol 82:113–129
Pant BD, Musialak-Lange M, Nuc P, May P, Buhtz A, Kehr J, Walther D, Scheible WR (2009) Identification of nutrient-responsive Arabidopsis and rapeseed microRNAs by comprehensive real-time polymerase chain reaction profiling and small RNA sequencing. Pl Physiol 150:1541–1555
Parizotto EA, Dunoyer P, Rahm N, Himber C, Voinnet O (2004) In vivo investigation of the transcription, processing, endonucleolytic activity, and functional relevance of the spatial distribution of a plant miRNA. Genes Dev 18:2237–2242
Park W, Li J, Song R, Messing J, Chen X (2002) CARPEL FACTORY, a Dicer homolog, and HEN1, a novel protein, act in microRNA metabolism in Arabidopsis thaliana. Curr Biol 12:1484–1495
Petroni K, Kumimoto RW, Gnesutta N, Calvenzani V, Fornari M, Tonelli C, Holt BF, Mantovani R (2012) The promiscuous life of plant NUCLEAR FACTOR Y transcription factors. Pl Cell 24:4777–4792
Rhoades MW, Reinhart BJ, Lim LP, Burge CB, Bartel B, Bartel DP (2002) Prediction of plant microRNA targets. Cell 110:513–520
Sunkar R, Kapoor A, Zhu JK (2006) Posttranscriptional induction of two Cu/Zn superoxide dismutase genes in Arabidopsis is mediated by downregulation of miR398 and important for oxidative stress tolerance. Pl Cell 18:2051–2065
Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molec Biol Evol 28:2731–2739
Voinnet O (2009) Origin, biogenesis, and activity of plant microRNAs. Cell 136:669–687
Warpeha KM, Kaufman LS (2007) Opposite ends of the spectrum: plant and animal g-protein signaling. Pl Signal Behav 2:480–482
Xie F, Frazier TP, Zhang B (2010) Identification, characterization and expression analysis of microRNAs and their targets in the potato (Solanum tuberosum). Gene 473:8–22
Xu Z, Zhong S, Li X, Li W, Rothstein SJ, Zhang S, Bi Y, Xie C (2011) Genome-wide identification of microRNAs in response to low nitrate availability in maize leaves and roots. PLoS ONE 6:e28009
Xu MY, Li WW, Hu XL, Wang MB, Fan YL, Zhang CY, Wang L (2013) Stress-induced early flowering is mediated by miR169 in Arabidopsis thaliana. J Exp Bot 65:89–101
Yamaguchi-Shinozaki K, Shinozaki K (2006) Transcriptional regulatory networks in cellular responses and tolerance to dehydration and cold stresses. Annual Rev Pl Biol 57:781–803
Yang W, Liu X, Zhang J, Feng J, Li C, Chen J (2010) Prediction and validation of conservative microRNAs of Solanum tuberosum L. Molec Biol Rep 37:3081–3087
Yu X, Wang H, Lu YZ, Ruiter M, Cariaso M, Prins M, Tunen A, He YK (2012) Identification of conserved and novel microRNAs that are responsive to heat stress in Brassica rapa. J Exp Bot 63:1025–1038
Zeng QY, Yang CY, Ma QB, Li XP, Dong WW, Nian H (2012) Identification of wild soybean miRNAs and their target genes responsive to aluminum stress. BMC Pl Biol 12:182
Zhang B, Pan X, Cobb GP, Anderson TA (2006a) Plant microRNA: a small regulatory molecule with big impact. Dev Biol 289:3–16
Zhang B, Pan X, Anderson TA (2006b) Identification of 188 conserved maize microRNAs and their targets. FEBS Lett 580:3753–3762
Zhang B, Pan X, Cannon CH, Cobb GP, Anderson TA (2006c) Conservation and divergence of plant microRNA genes. Pl J 46:243–259
Zhang BH, Pan XP, Cox SB, Cobb GP, Anderson TA (2006d) Evidence that miRNAs are different from other RNAs. Cell Molec Life Sci 63:246–254
Zhang B, Wang Q, Wang K, Pan X, Liu F, Guo T, Cobb GP, Anderson TA (2007) Identification of cotton microRNAs and their targets. Gene 397:26–37
Zhang J, Xu Y, Huan Q, Chong K (2009a) Deep sequencing of Brachypodium small RNAs at the global genome level identifies microRNAs involved in cold stress response. BMC Genom 10:449
Zhang W, Luo Y, Gong X, Zeng W, Li S (2009b) Computational identification of 48 potato microRNAs and their targets. Comput Biol Chem 33:84–93
Zhang R, Marshall D, Bryan GJ, Hornyik C (2013) Identification and characterization of miRNA transcriptome in potato by high-throughput sequencing. PLoS ONE 8:e57233
Zhang N, Yang J, Wang Z, Wen Y, Wang J, He W, Liu B, Si H, Wang D (2014) Identification of novel and conserved microRNAs related to drought stress in potato by deep sequencing. PLoS ONE 9:e95489
Zhao B, Ge L, Liang R, Li W, Ruan K, Lin H, Jin Y (2009) Members of miR-169 family are induced by high salinity and transiently inhibit the NF-YA transcription factor. BMC Molec Biol 10:29
Zhao M, Ding H, Zhu JK, Zhang F, Li WX (2011) Involvement of miR169 in the nitrogen-starvation responses in Arabidopsis. New Phytol 190:906–915
Zhou X, Wang G, Sutoh K, Zhu JK, Zhang W (2008) Identification of cold inducible microRNAs in plants by transcriptome analysis. Biochim Biophys Acta 1779:780–788
Zhou L, Liu Y, Liu Z, Kong D, Duan M, Luo L (2010) Genome-wide identification and analysis of drought-responsive microRNAs in Oryza sativa. J Exp Bot 61:4157–4168
Zhou ZS, Song JB, Yang ZM (2012) Genome-wide identification of Brassica napus microRNAs and their targets in response to cadmium. J Exp Bot 63:4597–4613
Zhu JK (2002) Salt and drought stress signal transduction in plants. Annual Rev Pl Biol 53:247–273
Acknowledgments
This research program was sponsored by the National Natural Science Foundation of China (No. 31460370), the Specialized Research Fund for the Doctoral Program of Higher Education of China (No. 20106202120003), the Fundamental Research Funds for the Universities of Gansu Province (2014), and International Science and Technology Cooperation Program of China (No. 0102014DFG31570).
Author information
Authors and Affiliations
Corresponding author
Additional information
Handling editor: Jorg Fuchs.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Yang, J., Zhang, N., Zhou, X. et al. Identification of four novel stu-miR169s and their target genes in Solanum tuberosum and expression profiles response to drought stress. Plant Syst Evol 302, 55–66 (2016). https://doi.org/10.1007/s00606-015-1242-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00606-015-1242-x