
Abstract
Ceriops is a viviparous mangrove with widespread species Ceriops decandra and C. tagal, and an endemic species C. australis. Genetic diversity of the three species was screened in 30 populations collected from 23 locations in the Indo West Pacific (IWP) using Inter-simple sequence repeats (ISSR) and sequences of partial nuclear gene (G3pdh) and chloroplast DNA (trnV-trnM). At the species level, the total gene diversity (Ht) revealed by ISSRs was 0.270, 0.118, and 0.089 in C. decandra, C. tagal, and C. australis, respectively. A total of six haplotypes of G3pdh and five haplotypes of trnV-trnM were recognized among the three species. Only C. decandra was detected containing more than one haplotype from each sequence data set (four G3pdh haplotypes and three trnV-trnM haplotypes). At the population level, genetic diversity of Ceriops was relatively low inferred from ISSRs (He = 0.028, 0.023, and 0.053 in C. decandra, C. tagal, and C. australis, respectively). No haplotype diversity within population was detected from any of the three species. Cluster analysis based on ISSRs identified three major geographical groups in correspond to the East Indian Ocean (EIO), South China Sea (SCS), and North Australia (NA) in both C. decandra and C. tagal. The cladogram from DNA sequences also detected the same three geographical groups in C. decandra. Analysis of molecular variance (AMOVA) revealed that most of the total variation was accounted for by differentiation between the three major geographical regions of both C. decandra and C. tagal. The significant genetic structure may result from the geological events in these regions during the recent Pleistocene glaciations. This study also provided insights into the phylogenetics of Ceriops.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Mangrove forests are the most dominant intertidal ecosystems along the tropical and subtropical coastlines. Ceriops Arnold (Rhizophoraceae), the “Yellow Mangrove,” is one of the typical constituents of the inner mangrove, characterized by vivipary (i.e., the development of the progeny when still attached to the maternal parent) (Tomlinson 1986). This genus comprises three species, Ceriops decandra, C. tagal, and C. australis (Ballment et al. 1988; Duke 1992). Indeed, C. australis was previously treated as a variety of C. tagal, C. tagal var. australis, but more recently, a reclassification of C. tagal var. australis as C. australis was reported based on allozyme analysis (Ballment et al. 1988). Geographically, C. australis has a rather limited distribution area endemic in the littoral zone of Australasia (i.e., Australia, New Zealand, and New Guinea). C. tagal is widespread in the most extensive range throughout the tropics of the Indo West Pacific (IWP) region, whereas C. decandra is native to a narrower range in this region (including Australasia and Indo-Malesia, but not extensive to East Africa) (Duke 1992).
The IWP is one of the major biogeographic global regions of mangrove distribution, which encompasses two potential discontinuities: the Malay Peninsula, and the Indonesia archipelago (including New Guinea), which lie on a shallow continental shelf that has been exposed repeatedly to create land connections in relatively recent geological history (Benzie 1998; Voris 2000). It was reported that past climatic and geomorphological events may have shaped population genetic structure through their effects on species distributions and levels of gene flow among populations (Benzie 1998; Dodd et al. 2002). Mangrove species whose propagules are disseminated in the oceans provide an excellent model for studying the effects of historical biogeographical events on population genetic structure. C. decandra and C. tagal, with their wide geographical distributions within the IWP region, are a good model affording the opportunity to investigate genetic differentiation at different spatial scales that integrate various historical biogeographical events.
Molecular markers, such as allozymes, restriction fragment length polymorphism (RFLP), random amplified polymorphic DNA (RAPD), amplified fragment length polymorphism (AFLP), microsatellites (SSR), Inter-simple sequence repeats (ISSR), and DNA sequence, have proven to be a very efficient means to investigate population genetics of mangrove species (Dodd et al. 2002; Maguire et al. 2000; Jian et al. 2004; Sun et al. 1998; Chiang et al. 2001; Tang et al. 2003; Takayama et al. 2006). A study on C. tagal using ISSR from Thailand and South China presented low-genetic variation at the population level (He = 0.008); however, a relatively high genetic variation (S. Huang, personal communication) within population was found using isozyme analyses (He = 0.195); more recently, cpDNA also revealed a high average haplotype diversity (Hd = 0.549) of total populations in C. tagal from Malay Peninsula and Borneo (Liao et al. 2006).
Here, a comparative study was conducted on the population genetic structure of the three species of Ceriops within the IWP region using different molecular markers. We selected DNA sequence fragments, the trnV-trnM and G3pdh, and ISSR as genetic markers. The trnV-trnM is a cpDNA intergenic spacer as described by Taberlet et al. (1991), whereas the G3pdh is a portion of a single-copy nuclear gene encoding glyceraldehyde 3-phosphate dehydrogenase (Strand et al. 1997). Both of the two fragments have been applied in some plant population genetic and phylogeographic studies (e.g., Huang et al. 2002; Hwang et al. 2000; Olsen and Schaal 1999; Olsen 2002). ISSR (Inter-simple sequence repeat) analysis from selective amplification of genomic DNA was also used in this study due to the high polymorphism usually found at ISSR loci in genetic diversity studies (Zietkiewicz et al. 1994; Gupta et al. 1994). We tried to address the following questions: (1) what is the genetic diversity and population structure in the three species of Ceriops and how efficient are ISSR and DNA sequence markers in studying these species? (2) Has the historical biogeographical events in the IWP had an important influence on population genetic structure of the widespread species C. decandra and C. tagal? and (3) what can be inferred from these data that has implications for the phylogenetics of this genus?
Materials and methods
Sample collection and DNA extraction
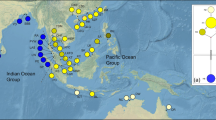
A total of 385 individuals of leaf samples across three Ceriops species were collected from 30 natural populations in the IWP, including three geographical regions, i.e., the East Indian Ocean (EIO), the South China Sea (SCS), and North Australia (NA) (Table 1; Fig. 1). The distance of sampled trees is at least 30 m. Total samples for ISSR analysis consisted of 189 individuals from 13 populations of C. decandra, 152 from 14 populations of C. tagal, and 44 from 3 populations of C. australis. The sample number for DNA sequencing was 107, 117, and 28 from C. decandra, C. tagal, and C. australis, respectively. Genomic DNA of each individual was extracted using the CTAB method (Doyle and Doyle 1987).

Map showing locations of Ceriops populations sampled. Filled squares are sampled locations of C. decandra; asterisks are sampled locations of C. tagal; filled triangles are sampled locations of C. australis. Population names are 1. Bangkok; 2. Khanom; 3. Lu-un; 4. Phang-Nya; 5. Ranong; 6. Kukup; 7. Tanjung Piai; 8. Pasir Ris; 9. Daintree River (A); 10. Daintree River (B); 11. Cairns airport; 12. Channel Island; 13. Channel Island Driveway; 14. Tanmen; 15. Yalongwan; 16. Linggao; 17. Dongge; 18. Dongzhaigang; 19. Godavari delta; 20. Neganbe; 21. Pambala; 22. Kuching; 23. Tuaran
PCR amplification of G3pdh and trnV-trnM, and DNA sequencing
The G3pdh region was amplified with primers GPDX7F and GPDX9R following Olsen (2002). The same primers were used for sequencing. For trnV-trnM spacer, PCR was performed following Huang et al. (2002) with the forward primer trnV and the reverse primer trnM. In addition to the two external primers, two internal primers were used in sequencing: trnV-2 (5′ TGGGCTCTTTCAATAACT 3′) and trnM-2 (5′ TTGATTACATGATATACTCCT 3′), forward and reverse, respectively. PCR products were purified using agarose gel purification Kit (QIAGEN, Hilden, Germany) following the protocol provided by the manufacturers. Sequences were analyzed with the ABI PRISM 3700 sequencer.
ISSR amplification
One hundred ISSR primers from the Biotechnology Laboratory, at the University of British Columbia (UBC set no. 9) were initially screened and eleven of the primers that produced clear and reproducible fragments were used: 807 (AG)8T, 808 (AG)8C, 810 (GA)8T, 825 (AC)8T, 834 (AG)8YT, 835 (AG)8YC, 841 (GA)8YC, 842 (GA)8YG, 857 (AC)8YG, 889 DBD(AC)7, and 891 HVH(TG)7. PCR amplifications were performed in 10 mM Tris–HCl, 50 mM KCl, 1.5 mM MgCl2, 0.1 mM dNTPs, 0.1% Triton X-100, 2% formamide, 200 nM primer, 1 U of Taq polymerase, and 20 ng of genomic DNA per 10 μL reaction. Amplification was performed in a PTC-200 thermocycler under the following cycle profile: initial denaturation at 94°C for 5 min, followed by 45 s at 94°C, 45 s annealing at 52–56°C, and 1.5 min extension at 72°C for 45 cycles, and 7 min at 72°C for a final extension. The PCR products were separated on 1.5% agarose gels buffered with 0.5× TBE, and detected by staining with ethidiumbromide.
Nucleotide sequences data analysis
DNA sequences were assembled using DNASTAR software (DNASTAR 1994) and aligned using ClustalX (Thompson et al. 1997). All sequences were submitted to GenBank. For each data set, maximum parsimony (MP) analysis was performed using a branch-and-bound search using PAUP* 4.0b5 (Swofford 1998) to construct phylogenic trees. According to the phylogenetic study of Rhizophoraceae (Zhong et al. 2000; Shi et al. 2002), we selected Bruguiera gymnorrhiza as outgroup in this study. Strict consensus trees were constructed from all most-parsimonious trees. Bootstrap analyses were carried out with 1,000 replicates using TBR branch-swapping of the heuristic search with random taxon addition (Felsenstein 1985). Characters were assigned equal weights at all nucleotide positions (Fitch 1971). Gaps were treated as missing data. Sequence divergences were estimated using P distance (Saito and Nei 1987).
ISSR data analysis
ISSR bands were scored as present (1) or absent (0) for each DNA sample excluding the smeared and weak ones. The banding patterns were analyzed as phenetic data, or as genetic data for tests of population genetic diversity and genetic architecture. As genetic data, the binary data matrix was input into POPGENE (Yeh et al. 1997), assuming Hardy–Weinberg equilibrium. The following indices were estimated: the percentage of polymorphic loci (P), the expected heterozygosity (He) (Nei 1973), total heterozygosity (Ht), heterozygosity value within populations (Hs), and the proportion of genetic diversity between populations (G ST). Bootstrap analysis (1,000 duplicates) and Neighbor-Joining diagrams were constructed on genetic distances (Nei 1978) among populations using RAPDDIST (Black 1995) and the NEIGHBOR program in PHYLIP 3.5C (Felsenstein 1993). Gene diversity statistics were further calculated for the three geographical regional groups identified by the cluster analysis, i.e., EIO, SCS, and the coast of NA.
For phenetic analyses, analysis of molecular variance (AMOVA) was performed using AMOVA Version 1.55 (Excoffier 1993) to determine whether genetic variation was distributed according to current boundaries within Ceriops, or, alternatively, whether genetic variance is partitioned geographically as might be the case if morphospecies are not monophyletic groups. If the results support the morphospecies are independent evolutionary lineages, most genetic variance within our samples should be partitioned among groups of populations defined by morphology (see Nice et al. 2005). A second AMOVA were arranged to calculate the genetic variation distribution among regions, among populations within a region, and within populations. In order to test for the correlation between genetic and geographical distances among populations, a Mantel test was performed using NTSYSpc V2.02j (Rohlf 1998) by computing 5,000 permutations.
Results
DNA sequences
The total length of G3pdh of all Ceriops individuals was 862 bp (GenBank accession no. EF423375, EF423376, EF423383, EF423384, EF423385, and EF423386). Excluding a 18-bp deletion occurring only on the haplotype of C. tagal, a total of 41 polymorphic sites (4.8% of the total length) were detected, which consist of 38 point mutations and 3 indels. Of the 812-bp trnV-trnM aligned sequences of Ceriops, four long fragments of indels (7, 19, 20, and 21 bp, respectively) were detected. A total of 28 trnV-trnM sites were polymorphic (3.4% of the total sequence) with 20 point mutations and 8 indel sites (excluding the long indel fragments) (GenBank accession no. EF423377, EF423378, EF423379, F423380, and EF423381). Among species, the lowest nucleotide divergence was between C. tagal and C. australis (estimates of pairwise divergence between them is 13, and 1, in G3pdh and in trnV-trnM, respectively, with the exception of indel fragments).
A total of six haplotyes of G3pdh and five haplotyes of trnV-trnM were recognized among the three species. No haplotype diversity was found within populations. Only C. decandra was detected having more than one haplotype from each DNA sequence data set. In C. decandra, a total of four G3pdh haplotypes (the estimates of pairwise distances among them varied from 2 to 15) and three trnV-trnM haplotypes (the value of pairwise divergence among them ranged from 3 to 16) were detected. Both G3pdh and trnV-trnM data of C. decandra identified three groups in correspond to the geographical region EIO, SCS, and NA (Fig. 2a, b).

Trees of Ceriops constructed by molecular data. (a) The most parsimony tree inferred from sequences of G3pdh; (b) the strict consensus tree inferred from sequences of trnV-trnM. NA North Australia, SCS South China Sea, EIO East Indian Ocean, WM West Malay Peninsula, IS India/Sri Lanka area; (c) Neighbor-Joining tree obtained with 1,000 bootstraps on Nei’s genetic distance (1978) (see Table 1 and Fig. 1 for population locations). Bootstrap values are reported as percentages
ISSRs
Genetic diversity and phylogenetic analysis of the genus
A total of 179 loci and 201 genotypes were presented from the 11 ISSR primers for the three species across 385 individuals of 30 populations. The size of the ISSR fragments varied from 0.2 to 1.4 kb. Of the 179 loci surveyed, 159 were polymorphic in at least one of the sampled populations. The number of unique loci of each species detected in C. decandra, C. tagal, and C. australis was 21, 4, and 2, respectively. The total ISSR diversity (Ht) of all individuals from the three species is 0.308 (data not shown). The expected heterozygosity of each population (He) was presented in Table 1. Analyses of variance (ANOVA) indicated the value of expected heterozygosity was independent of population size (P > 0.05, data not shown).
The Neighbor-Joining dendrogram revealed three distinct groups in correspond to C. decandra, C. tagal, and C. australis (Fig. 2c). AMOVA revealed that most of the variation (56.24%) was held between species (Table 2). All components of molecular variance were highly significant (P < 0.001).
Genetic differences and population structure within species
Of the species C. decandra, 11 ISSR primers produced a total of 164 loci and 78 ISSR genotypes across the 189 individuals from 13 populations. At the species level, the percentage of polymorphic loci (P) was 79% (Table 3). Ht was 0.270 compared with the heterozygosity value of 0.028 within populations (Hs), and the proportion of genetic diversity between populations (G ST) was 0.897.
In the case of C. tagal, a total of 136 loci and 88 ISSR genotypes were presented across all 152 individuals of 14 populations. At the species level, the value of P was 54%. The estimate of Ht in C. tagal was 0.118, whereas the heterozygosity within populations (Hs) was 0.023, with a G ST value of 0.802 (Table 3).
For the three populations of C. australis, a total of 117 loci and 35 ISSR genotypes were detected across all 44 individuals of the three populations. The percentage of polymorphic loci was 29% at the species level. The Ht, Hs, and G ST of C. australis is 0.089, 0.053, and 0.402, respectively (Table 3).
Three major groups in correspond to the geographical regions SCS, EIO, and NA were revealed by cluster analysis in C. decandra and C. tagal with the bootstrap values varied from 57.1 to 100% (Fig. 2c). Mantel test indicated no statistically significant correlation between pairwise genetic distance and corresponding geographic distance among populations within species (data not shown).
AMOVA revealed that significant population differentiation for the two widespread species, C. decandra and C. tagal (PHIST = 0.898, and 0.790, respectively) (Table 2). Most of genetic variation (75.83% for C. decandra, and 46.25% for C. tagal) was partitioned among regions (SCS, EIO, and NA), with a small part of genetic variation accounting for among populations within regions and among individuals within populations. For the endemic species C. australis, 43.24% of the total variation was accounted for among populations, with a variation component of 56.76% among individuals within a population. All components of molecular variance were highly significant (P < 0.001).
Discussion
DNA sequence and ISSR markers
The detection of high levels of polymorphism (P = 79, 54, and 29% in C. decandra, C. tagal, and C. australis, respectively) makes ISSR analysis a powerful tool for assessing genetic diversity within species. Although only C. decandra was detected containing more than one haplotype from DNA sequences, there were still some highlights contributed to this study. The geographical groups of the chloroplast haplotypes within C. decandra is correlated to that of nucleotide haplotypes. The spatial pattern of genetic structure inferred from nuclear sequences (G3pdh) of C. decandra was as well consistent with that revealed by the nuclear genome data (ISSRs).
Phylogenetics of the genus
White (1926) observed that in Australia and Papua New Guinea there was a form of C. tagal whose propagules had smooth, terete hypocotyls rather than the angled or ribbed hypocotyls typical of C. tagal, and described the atypical form as a variety of C. tagal, C. tagal (Perr.) C. B. Robinson var. australis C. T. White. However, in a more recent systematic revision, the evidence of distinct allozyme phenotypes and the reproductive isolation between C. tagal and C. tagal var. australis confirmed that the variety should be recognized as a species distinct from C. tagal (Ballment et al. 1988). In our study, two monophyletic clades corresponding to the C. tagal and C. australis were supported by Neighbor-Joining dendrogram inferred from the ISSR data (Fig. 2c). AMOVA analysis also revealed that most of the variances of these two species were accounted for between species (Table 2). In addition, no haplotype was shared by C. tagal and C. australis (Fig. 2a, b). All of these results in our study supported C. tagal and C. australis should be treated as separate groups.
Genetic diversity and population structure within species
At the population level, the expected heterozygosity (He) of Ceriops was low inferred from ISSRs (Table 3), and from DNA sequences as well (no haplotype diversity and no nucleotide diversity within populations were detected). Such low population genetic diversity was also presented in the previous study of C. tagal from South China and Thailand (Ge and Sun 2001), and in some other mangrove species, e.g., Aegiceras corniculatum, B. gymnorrhiza, Avicennia marina (Ge and Sun 1999; Ge 2001), using the ISSR technology. In this study, the expected heterozygosity varied widely among populations of each species, but was not related to any obvious characteristics of the populations, such as their isolation at the margins of the range of the species or sample size (Table 1). Although species of Ceriops show self compatibility, their pollination may favor outcrossing (Tomlinson 1986). Such a mixed mating model has existed in some other mangrove species of Rhizophoraceae (Lowenfeld and Klekowski 1992; Klekowski et al. 1994; Sun et al. 1998; Ge 2001). Variation between populations in outcrossing rates has commonly been revealed in mixed mating species (Schoen 1982). Low-gene diversity in this study may also arise from population substructure or founder events, in which high level of inbreeding have led to demes of genetically similar individuals (see Tan et al. 2005). The higher genetic variation inferred from isozyme (S. Huang, personal communication) and high cpDNA haplotype diversity (Liao et al. 2006) in C. tagal may be due to different molecular markers being used and different sampling methods.
At the species level, both of the ISSR data and DNA sequences revealed the greatest diversity within C. decandra of the genus. As Hamrick et al. (1991) suggested, several factors are important in determining levels of genetic diversity. Geographic range is strongly associated with the level of variation maintained at the species level. Generally endemic species have lower genetic diversity than widespread species. In contrast to the restricted sampling of the endemic species C. australis, C. decandra, and C. tagal were both sampled covering a wide geographic range throughout the IWP in this study. Other factors such as breeding systems, vegetative reproduction, dispersal pattern, sample size, etc., also significantly influence the genetic diversity of a species. Considering the geographic sampling range (C. tagal have a wider sampling range than C. decandra, see Fig. 1), the sample size (152 individuals from 14 population in C. tagal versus 189 from 13 population in C. decandra), and the similar life history and biological characteristics, it is interesting that C. tagal exhibited much lower genetic diversity than C. decandra. DNA sequences may give some highlight on time scale. No haplotype diversity was detected in C. tagal, whereas four G3pdh haplotypes and three trnV-trnM haplotypes (the diversity between these haplotypes is also high) were found in C. decandra. The species C. tagal which seems to be able to survive in a wider range of climate may not have been splitted as much as C. decandra in different refugia, and thus the separation of the different C. tagal groups might be more recent.
Significant population genetic structure was detected in both C. decandra and C. tagal. Both genetic data and phenetic data (AMOVA) indicated significant inter-population variation in genetic structure. The fixation index (G ST = 0.897, and 0.802 in C. decandra, and C. tagal, respectively) was close to the partition of genetic variance (PHIST) by AMOVA (Table 2), based on similarities of ISSR fingerprints. A dendrogram based on genetic distances of ISSR revealed three major clades corresponding to three well-delineated geographical regions (i.e., the EIO, SCS, and NA) within the distribution of C. decandra and C. tagal. Similarly, the G3pdh MP tree and the trnV-trnM strict consensus tree displayed the same three geographical groups in C. decandra. Furthermore, hierarchical AMOVA revealed that most of the total variation (76% in C. decandra, and 46% in C. tagal) was accounted for by differentiation between regions. The separation of the SCS populations from the EIO populations in this study is consistent with the result of previous studies on C. tagal within the Malay Peninsula using cpDNA sequences and ISSRs (Liao et al. 2006; Ge and Sun 2001). Generally, the present distribution of genetic structure is influenced by both natural history and evolutionary history (Hamrick and Godt 1996). For plants whose seeds are passively dispersed, the seed dispersal ability of a species is one of the major determinants accounting for population differentiation (Dodd et al. 2002). Ceriops are viviparous mangroves, whose propagules are about 10–20 cm long (Tomlinson 1986). Their viviparous seedlings are buoyant, presumably capable of long-distance dispersal by ocean currents. However, field experiments suggested their propagule dispersal is apparently limited by surface currents, the presence of suitable environmental conditions, and longevity of propagules during dispersal (Clarke et al. 2001; see Ge and Sun 2001). In addition, a field experiment has revealed very limited dispersal capability on C. australis propagules (only 9% of marked propagules were found more than 3 m from the parent trees) (McGuinness 1997). Thus, the long-distance dispersal of propagules is rather limited in Ceriops species. The geographic history is another factor that may have influenced genetic differentiation in this area (Ge and Sun 2001). During the Pleistocene, sea levels fluctuated repeatedly, dropping as low 200 m below, and occasionally rising above, present levels. The genetic differentiation was thought to occur when gene flow was restricted, probably at times of lowered sea level when land connected much of South East Asia, New Guinea and Australia, almost closing the sea connecting between the Indian and Pacific Oceans (Benzie 1998). Even today, the Indian Ocean is connected with SCS only through the Strait of Malacca, which make it an effective barrier to some mangrove species between the Pacific and Indian Oceans (Ge and Sun 2001; Tan et al 2005). Within-region differentiation seems to be high in the EIO and SCS, but these areas need further study. With more extensive sampling, especially in Australia and Indonesia, and a more efficient DNA sequence marker will provide more information to infer the phylogeographic pattern of the Ceriops species.
References
Ballment ER, Smith TJ, Stoddart JA (1988) Sibling species in the mangrove genus Ceriops (Rhizophoraceae), detected using biochemical genetics. Aust Syst Bot 1:391–397
Benzie JAH (1998) Genetic structure of marine organisms and SE Asian biogeography. In: Hall R, Holloway JD (eds) Biogeography and geological evolution of SE. Asia. Backhuys Publishers, The Netherlands, pp 197–209
Black WC IV (1995) FORTRAN programs for the analysis of RAPD-PCR markers in populations. Colorado State University, Ft. Collins, CO 80523
Chiang TY, Chiang YC, Chen YJ, Chou CH, Havanond S, Hong TN, Huang S (2001) Phylogeography of Kandelia candel in East Asiatic mangroves based on nucleotide variation of chloroplast and mitochondrial DNAs. Mol Ecol 10:2697–2710
Clarke PJ, Kerrigan RA, Westphal CJ (2001) Dispersal potential and early growth in 14 tropical mangroves: do early life history traits correlate with patterns of adult distribution? J Ecol 89:648–659
DNASTAR (1994) Lasergene. DNASTAR Inc., Madison, Wis
Dodd RS, Afzal-Rafii Z, Kashani N, Budrick J (2002) Land barriers and open oceans: effects on gene diversity and population structure in Avicennia germinans L. (Avicenniaceae). Mol Ecol 11:1327–1338
Doyle JJ, Doyle JL (1987) A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem Bull 19:11–15
Duke NC (1992) Mangrove floristics and biogeography. In: Robertson AI, Alongi DM (eds) Tropical mangrove ecosystems. American Geophysical Union, Washington, DC, pp 63–100
Excoffier L (1993) Analysis of molecular variance. Version 1.55. Genetics and Biometry Laboratory, University of Geneva
Felsenstein J (1985) Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol 17:368–376
Felsenstein J (1993) Phylogeny inference package (PHYLIP), Version 3.5. University of Washington, Seattle
Fitch WM (1971) Toward defining the course of evolution: minimum change for a specific tree topology. Syst Zool 20:406–416
Ge XJ (2001) Reproductive biology and conservation genetics of mangroves in south China and Hong Kong. Dissertation, University of Hong Kong
Ge XJ, Sun M (1999) Reproductive biology and genetic diversity of a cryptoviviparous mangrove Aegiceras corniculatum (Myrsinaceae) using allozyme and inter-simple sequence repeat (ISSR) analysis. Mol Ecol 8:2061–2069
Ge XJ, Sun M (2001) Population genetic structure of Ceriops tagal (Rhizophoraceae) in Thailand and China. Wetlands Ecol Manage 9:203–209
Gupta M, Chyi YS, Romero-Severson J, Owen JL (1994) Amplification of DNA markers from evolutionarily diverse genomes using single primers of simple-sequence repeats. Theor Appl Genet 89:998–1006
Hamrick JL, Godt MJW (1996) Conservation genetics of endemic plant species. In: Avise JC, Hamrick JL (eds) Conservation genetics. Chapman & Hall, New York, pp 281–304
Hamrick JL, Godt MJW, Murawski DA, Loveless MD (1991) Correlations between species and allozyme diversity: implications for conservation biology. In: Falk DA, Holsinger KE (eds) Genetics and conservation of rare plants. Oxford University Press, New York, pp 75–86
Hwang LH, Hwang SY, Lin TP (2000) Low chloroplast DNA variation and population differentiation of Chamaecypaeis formosensis and Chamaecyparis taiwanensis. Taiwan J For Sci 15:229–236
Huang SF, Hwang SH, Lin T (2002) Spatial pattern of chloroplast DNA variation of Cyclobalanopsis glauca in Taiwan and East Asia. Mol Ecol 11:2349–2358
Jian SG, Tang T, Zhong Y, Shi SH (2004)Variation in inter-simple sequence repeat (ISSR) in mangrove and non-mangrove populations of Heritiera littoralis (Sterculiaceae) from China and Australia. Aquat Bot 79:75–86
Klekowski EJJ, Lowenfeld R, Hepler PK (1994) Mangrove genetics. II. Outcrossing and lower spontaneous mutation rates in Puerto Rican Rhizophora. Int J Plant Sci 155:373–381
Liao PC, Havanond S, Huang S (2006) Phylogeography of Ceriops tagal (Rhizophoraceae) in Southeast Asia: the land barrier of the Malay Peninsula has caused population differentiation between the Indian Ocean and South China Sea. Conserv Genet 8:89–98
Lowenfeld R, Klekowski EJJ (1992) Mangrove genetics. I. Mating system and mutation rates of Rhizophora mangle in Florida and San Salvador Island. Int J Plant Sci 153:394–399
Maguire TL, Saenger P, Baverstock P, Henry R (2000) Microsatellite analysis of genetic structure in the mangrove species Avicennia marina (Forsk.) Vierh. (Aveicenniaceae). Mol Ecol 9:1853–1862
McGuinness KA (1997) Dispersal, establishment and survival of Ceriops tagal propagules in a north Australian mangrove forest. Oecologia (Berlin) 109:80–87
Nei M (1973) Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci USA 70:3321–3323
Nei M (1978) Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89:583–590
Nice CC, Anthony N, Gelembiuk G et al (2005) The history and geography of diversification within the butterfly genus Lycaeides in North America. Mol Ecol 14:1741–1754
Olsen KM (2002) Population history of Manihot esculenta (Euphorbiaceae) inferred from nuclear DNA sequences. Mol Ecol 11:901–911
Olsen KM, Schaal BA (1999) Evidence on the origin of cassava: phylogeography of Manihot esculenta. Proc Natl Acad Sci USA 96:5586–5591
Rohlf FJ (1998) NTSYS-pc, numerical taxonomy and multivari- ate analysis system, Version 2.02a. Exeter Software. Setauket, New York
Saitou N, Nei M (1987) The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol Biol Evol 4:406–425
Schoen DJ (1982) The breeding system of Gilia achilleifolia: variation in floral characteristics and outcrossing rate. Evolution 36:596–613
Shi S, Zhong Y, Huang Y, Du Y, Qiu X, Chang H (2002) Phylogenetic relationships of the Rhizophoraceae in China based on sequences of the chloroplast gene matK and the internal transcribed spacer regions if nuclear ribosomal DNA and combined data set. Biochem Syst Ecol 30:309–319
Strand AE, Leebens-Mack J, Milligan BG (1997) Nuclear DNA-based markers for plant evolutionary biology. Mol Ecol 6:113–118
Sun M, Wong KC, Lee JSY (1998) Reproductive biology and population genetic structure of Kandelia candel (Rhizophoraceae), a viviparous mangrove species. Am J Bot 85:1631–1637
Swofford DL (1998) PAUP*. Phylogenetic analysis using parsimony and other methods, Version 4. Sinauer Associates, Sunderland, MA
Taberlet P, Gielly L, Pautou G, Bouvet J (1991) Universal primers for amplification of three non-coding regions of chloroplast DNA. Plant Mol Biol 17:1105–1109
Takayama K, Kajita T, Murata J, Tateishi Y (2006) Phylogeography and genetic structure of Hibiscus tiliaceus—speciation of a pantropical plant with sea-drifted seeds. Mol Ecol 15:2871–2881
Tan F, Huang Y, Ge X, Su G, Ni X, Shi S (2005) Population genetic structure and conservation implications of Ceriops decandra in Malay Peninsula and North Australia. Aquat Bot 81:175–188
Tang T, Zhong Y, Jian S, Shi S (2003) Genetic diversity of Hibiscus tiliaceus (Malvaceae) in China assessed suing AFLP markers. Ann Bot 92:409–414
Thompson JD, Gibson TJ, Plewniak F (1997) The Clustal X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876–4882
Tomlinson PB (1986) The botany of mangroves. Cambridge University Press, Cambridge, pp 374–381
Voris HK (2000) Maps of Pleistocene sea levels in Southeast Asia: shorelines, river systems, and time durations. J Biogeogr 27:1153–1167
White CT (1926) A variety of Ceriops tagal C. B. Rob. (C. candolleana W. & A.). J Bot 64:220–221
Yeh FC, Yang RC, Boyle TBJ, Ye ZH, Mao JX (1997) POPGENE, the user-friendly shareware for population genetic analysis. Mol. Biol. Biotech. Center, University of Alberta, Canada
Zhong Y, Shi S, Tang X, Huang Y, Tan F, Zhang X (2000) Testing relative evolutionary rates and estimating divergence time among six genera of Rhizophoraceae using cpDNA and nrDNA sequences. Chin Sci Bull 45:1011–1015
Zietkiewicz E, Rafalski A, Labuda D (1994) Genome fingerprinting by simple sequence repeat (SSR)-anchored polymerase chain reaction amplification. Genomics 20:176–183
Acknowledgments
We thank Dr. Norman Duke, Dr. Xuejun Ge, Dr. Sonjai Havanond, and Dr. Peichun Liao for their assistance in collecting or providing some plant materials, and Mr. Miles Tracy for reading the manuscript. We also appreciate the helpful comments on this manuscript from three anonymous referees. This study was supported by grants from the National Natural Science Foundation of China (30300033, 30500049, 30230030, 30470119, U0633002), the Natural Science Foundation of Guangdong Province (500316), the Ministry of Education Special Foundation (20010558013), the Chang Hungta Science Foundation of Sun Yat-Sen University, the Qiu Shi Science and Technology Foundation and the National Key Project for Basic Research (973) grant 2003CB715904.
Author information
Authors and Affiliations
Corresponding author
Additional information
Yelin Huang and Fengxiao Tan contributed equally to this work.
Rights and permissions
About this article
Cite this article
Huang, Y., Tan, F., Su, G. et al. Population genetic structure of three tree species in the mangrove genus Ceriops (Rhizophoraceae) from the Indo West Pacific. Genetica 133, 47–56 (2008). https://doi.org/10.1007/s10709-007-9182-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10709-007-9182-1