
Abstract
Acanthus ilicifolius is a widespread mangrove species in the Indo-West Pacific (IWP) with high ecological value. In this study, we analyzed the patterns of genetic variation in the species across its distribution range and assessed factors that may have influenced its genetic structure across the IWP using data from four chloroplast DNA fragments. Our results demonstrated high genetic diversity at the species level with little variation within populations, indicating restricted gene flow among A. ilicifolius populations. Strong genetic divergence was found between populations in the Indian Ocean and populations in the Pacific Ocean, likely due to the land barrier effect of the Malay Peninsula (or in the past, the Sundaland). Ecological niche modeling revealed no significant niche differentiation between the two groups, suggesting that ecological divergence was less responsible for the divergence of A. ilicifolius. Despite the ability for long-distance dispersal, genetic barriers identified within the Pacific Ocean suggested ocean currents to be a cryptic barrier for gene flow in this region. This study provides new insights into the historical population dynamics of mangrove species in the IWP region and is valuable for the long-term conservation of A. ilicifolius.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Mangroves are one of the dominant intertidal ecosystems along the tropical and subtropical coastlines (Tomlinson, 2016). They are of great ecological and economic importance for balancing the coastal ecosystem, sequestering carbon, purifying water, providing habitats for marine life and protecting coastlines (Barbier et al., 2011). However, mangrove resources are severely depleted due to overexploitation and natural ecological events in many parts of the tropics (Umali et al., 1987; Duke et al., 2007), and proper management of mangrove resources have become an urgent task (Minobe et al., 2010). Understanding the patterns and factors affecting the distribution and differentiation of mangroves species is important for their conservation.
As widespread tropical and subtropical intertidal plants, mangroves are dispersed mainly by ocean currents and their propagules (or seeds) can usually float for an extended period of time in saline, brackish, and freshwater (Clarke et al., 2001; Tomlinson, 2016). Such an adaptive feature allowed for long-distance dispersal through ocean currents and strongly influenced the present-day distribution of mangroves as previously revealed in several studies (Duke, 1995; Clarke et al., 2001; Gallaher et al., 2017; Tomlinson, 2016; Guo et al., 2018b). High potential for long-distance dispersal can weaken the influence of geographic distance and lead to potentially higher genetic connectivity across populations (Wee et al., 2014; Guo et al., 2018b). For a species with less capability for long-distance dispersal, gene flow among populations can occur in a stepping-stone manner along its dispersal route, generating genetic differentiation that increases with physical distance (i.e., isolation-by-distance, IBD). Since dispersal capability varies among mangrove species (Clarke et al., 2001; Drexler, 2001; Duke et al., 2002), it is important to conduct targeted studies on how and to what extent the capability of dispersal has influenced the distribution and genetic differentiation of individual species. Besides, as inconsistent evidence weighs in on the significance of IBD, the role of geographical distance in shaping the genetic structure of mangrove species is still unsure (Wee et al., 2014; Mori et al., 2015; Ngeve et al., 2016). Other than dispersal capability and geographic distance, the genetic connectivity of mangrove species can also be influenced by ocean circulation patterns (Pil et al., 2011; Wee et al., 2014), as observed in other passively dispersed marine species (e.g., Thiel & Gutow, 2005; Galindo et al., 2006); ocean currents can be both facilitator and barrier for long-distance dispersal (Ayre & Dufty, 1994; Waters, 2008; Gallaher et al., 2017).
Another major barrier to gene flow is vicariance, caused by physical barriers that result in distinct genetic discontinuities across a landscape. Genetic discontinuities associated with land barriers, usually related to sea-level fluctuations during the Quaternary climatic oscillations, have often been reported in studies on mangrove species. Among them, the Malay Peninsula (or the Sundaland, in the past) is often reported as the land barrier responsible for genetic differentiation between mangrove populations in the Indian Ocean (referring to the coasts located on the west of the peninsula) and populations in the Pacific Ocean (referring to the coasts located on the east of the peninsula) (Su et al., 2007; Huang et al., 2008; Li et al., 2016; Yang et al., 2016; Guo et al., 2018a, b). Similar implications have been made in studies on marine fauna in the region (Benzie, 1999; Barber et al., 2000; Gaither et al., 2011). Other barriers implicated in causing significant genetic differentiation in marine and mangrove species include Wallace’s line (Wee et al., 2015; Guo et al., 2016, 2018b) and the Central American Isthmus (CAI) (Dodd et al., 2002; Lessios, 2008). However, these genetic barriers do not affect all mangrove species similarly. For example, while an east–west division across the Malay Peninsula was widely found in many mangrove species, exceptions were observed, e.g., in Rhizophora mucronata (Inomata et al., 2009; Wee et al., 2014). Another factor often ignored by studies on mangrove species is the differentiation of ecological conditions among regions, which together with adaptation and other evolutionary processes, can be a powerful driving force for genetic divergence (Zhou et al., 2012). The additional estimation of niche overlap between regions of high genetic divergence is thus necessary to achieve a holistic understanding of factors influencing the genetic structure of a species.
This study attempts to characterize patterns of genetic variation in the Holly mangrove Acanthus ilicifolius Lam., the predominant mangrove species in the genus (McDade et al., 2005) to elucidate the genetic structure and evolutionary history of the species. In addition to protecting the coast, the species is also valued for its aesthetic and medicinal values (Babu et al., 2002; Wu et al., 2003). This species commonly grows in landward edges of the mangroves just above the high tide mark, but also occurs in inner mangroves as the understory. As a non-viviparous mangrove species, A. ilicifolius primarily disperses via explosively dehiscent capsules, with secondary seed dispersal through water currents which may allow for long-distance dispersal of the species (McDade et al., 2005; Tomlinson, 2016). The species has an extremely wide distribution from India to the Western Pacific (New Caledonia), tropical Australia, and China (Duke, 2013; Tomlinson, 2016). The genetic structure and underlying factors responsible for its present-day distribution, however, remains mostly unexplored. Up until now, the only molecular analysis on populations of this species is by Lakshmi et al. (1997) who studied several populations of A. ilicifolius along the Indian coastline using mostly dominant genetic markers. In this study, we characterized the phylogeographic pattern of A. ilicifolius across its distribution range using maternally inherited chloroplast DNA (cpDNA) markers, which are especially informative in the reconstruction of a species’ historical range shifts and recolonization routes (Triest 2008). With special focus on 1) the genetic diversity and differentiation of A. ilicifolius across the distributional range, 2) population structure and demography of A. ilicifolius, and 3) the influences of biological features (e.g., dispersal capability), ecological factors (e.g., niche overlap) and phylogeographic forces (e.g., geographical distance, land barrier and oceanography) on the present distribution of the species, we envision that outcome from this study would provide solid information for the better management of A. ilicifolius populations.
Materials and methods
Sample collection and molecular experiments
A total of 399 samples from 41 natural populations of A. ilicifolius were collected across its entire distribution range in the IWP, with sample sizes varying from six to 13 individuals per population (Table 1). The sampled individuals within each population were located at least 10 m apart. Young leaves were collected from individual plants and stored with silica gel until DNA isolation.
Genomic DNA of each individual was extracted using the CTAB method (Doyle & Doyle, 1990). Among an initial list of commonly used cpDNA primer pairs, which were screened on a subset of samples, four primer pairs that amplified polymorphic fragments within or among populations, including the cpDNA regions of trnS-trnG, trnV-trnM, trnL-trnF, and 5′rps12-rpl20 (Taberlet et al., 1991; Hamilton, 1999; Cheng et al., 2005), were used for PCR amplification in all samples. DNA amplification was carried out in 30 μl PCR reaction mixtures containing approximately 10–15 ng of total DNA, 5 pmol of each primer, 10 mM of Tris–HCl (pH 8.4), 1.5 mM of MgCl2, 0.1 mM of dNTP, and 2 U of Taq polymerase (Shengong Inc., Shanghai, China). PCR reactions were performed under the following cycle profile: initial denaturation at 94°C for 4 min, followed by 1 min at 94°C, 45 s at 53°C, and 1 min at 72°C for 35 cycles, and 10 min at 72°C for final extension. The PCR products were separated on 1.0% agarose gel, stained with ethidium bromide and viewed under UV light. Purified DNA fragments were then sequenced from both directions on an ABI 3730XL DNA Analyzer (Applied Biosystems).
Phylogeographic and phylogenetic analysis
The cpDNA sequences were assembled and manually edited using SeqManTM (DNASTAR). The sequences of A. ilicifolius generated in this study have been deposited in GenBank under the accession numbers MT264980-MT264992. The geographic distribution of cpDNA haplotypes was plotted on a map using GenGis v2.11 (Parks et al., 2009), and the relationships among haplotypes were inferred using the median-joining method implemented in NETWORK v4.6.1.2 (Bandelt et al., 1999). Continuous indels were treated as single mutational events in the analysis. A principal coordinate analysis (PCoA) was performed using GenAlEx v6.5 (Peakall & Smouse, 2012) based on the pairwise Kimura-2-parameter genetic distances of all populations computed using MEGA v6 (Tamura et al., 2013). Population groups were further determined using the spatial analysis of molecular variance method in SAMOVA v2.0 (Dupanloup et al., 2002). We considered models with putative numbers of populations (K) ranging from 1 to 10, and for each K we used 1000 simulations of the annealing process for each of the 100 repeated runs. Pairwise Kimura-2-parameter genetic distances for all populations were also used to identify biogeographical boundaries or areas exhibiting the largest genetic discontinuities between population pairs using the Monmonier’s maximum difference algorithm as implemented in BARRIER v2.2 (Manni et al., 2004). The robustness of the identified barriers was assessed by 100 bootstrap replicates.
Haplotype diversity (Hd) and nucleotide diversity (π) were calculated using DnaSP v5.10.1 (Librado & Rozas, 2009). Average gene diversity within populations (HS), total gene diversity (HT) and two coefficients of genetic variation (GST and NST) were estimated using PERMUT (Pons & Petit, 1996). GST and NST were further compared through a test with 1000 permutations to examine the presence of phylogeographic structure. Compared with GST which is dependent of haplotype frequencies, NST takes into account both haplotype frequencies and their sequence similarities (Pons & Petit, 1996), thus a significantly higher NST than GST could be an indication of the presence of phylogeographic structure. To further characterize the population structure and genetic variation, multiple hierarchical analyses of molecular variance (AMOVA) were performed using ARLEQUIN v3.5 (Excoffier & Lischer, 2010) with the statistical significance determined by 1000 permutations. Isolation-by-distance (IBD) was examined by testing the relationship between pairwise FST and natural log-transformed (Ln-transformed) geographical distances between the populations using the Mantel test implemented in GenAlEx with 1000 permutations.
To test the presence of recent population expansion in A. ilicifolius, mismatch distribution analysis was conducted using ARLEQUIN in which the distribution of the number of pairwise differences between haplotypes was compared with their theoretical distribution expected under a sudden demographic expansion model. Goodness-of-fit was tested with the sum-of-squared deviations (SSD) between observed and expected mismatch distributions, and the raggedness index of Harpending (HRag; Harpending, 1994) was estimated using 1000 parametric bootstrap replicates. Neutrality tests, including Tajima’s D test (Tajima, 1989) and Fu’s Fs test (Fu, 1997), were also conducted on DnaSP to test for possible recent demographic expansions. Significant D values and large negative Fs values suggest rapid demographic expansions (Hudson, 1990). In cases where the hypothesis of rapid expansion was not rejected, the parameter value for the mode of the mismatch distribution (s) and its 95% confidence interval (CI) were converted into estimates of time since expansion (t) using the formula: t = s/2u (Rogers & Harpending, 1992). The value u was calculated as u = 2 μkg, where μ is the substitution rate, k is the average sequence length used for analysis and g is the generation time in years. For μ, we assumed the same minimum and maximum values of 1.0 and 8.24 × 10−9 substitutions per site/year, respectively, based on the lower and upper ranges for previously estimated (synonymous) substitution rates for cpDNA in angiosperms (Wolfe et al., 1987; Richardson et al., 2001). For g, 10 years were used as an approximation based on the description by Liao (2009).
Ecological niche modeling
To reconstruct the paleoclimate geographic distributions of A. ilicifolius, species distribution model under present-day were generated using the maximum entropy model implemented in MAXENT v3.4.1 (Phillips et al., 2006; Phillips & Dudı´k, 2008) and projected to those of the Last Glacial Maximum (LGM) and the Last Inter Glacial (LIG) periods. A total of 60 occurrence records were included in the niche modeling, including our 41 sampling locations and 19 other locations from our field investigations. Among 19 bioclimatic variables downloaded from the WORLDCLIM database (http://www.worldclim.org/) (Hijmans et al., 2005), seven bioclimatic variables that exhibited pairwise Pearson correlation coefficients r < 0.7 were chosen for final analyses, namely, mean diurnal range (BIO2), max temperature of warmest month (BIO5), min temperature of coldest month (BIO6), mean temperature of wettest quarter (BIO8), precipitation of wettest quarter (BIO16), precipitation of driest quarter (BIO17), and precipitation of warmest quarter (BIO18). For LGM prediction, the Community Climate System Model 4 (CCSM4) was used. For LIG prediction, the climatic model from Otto-Bliesner et al. (2006) was used. These variables were at a resolution of 2.5 m.
To test the ecological divergence between different population groups of A. ilicifolius, ecological niche models (ENMs) were generated for each group using the seven environmental variables described above and the maximum entropy model implemented in the MAXENT v3.4.1. ENMTOOLS v1.3 (Warren et al., 2010) was used to estimate niche overlap between different population groups assessed by Schoener’s D and Warren’s I similarity statistics, with 100 pseudo-replicates. Values of both D and I may range from 0 (no niche overlap) to 1 (identical niches). Information of 31 and 10 sampling sites investigated in this study was used as occurrence records for the Pacific Ocean group (i.e., the eastern lineage) and the Indian Ocean group (i.e., the western lineage) (see results), respectively. All analysis performed on MAXENT v3.4.1 was run using default program conditions with the machine-learning algorithm being configured to use 75% of species records for training and 25% for testing the model. Area Under Curve (AUC) of the Receiver Operating Characteristic (ROC) plot was used for model evaluation. DIVA-GIS v7.5 (Hijmans et al. 2005) was used to draw the range of suitable distributions.
Results
Chloroplast DNA variation and haplotype distribution
The alignment lengths of the four cpDNA fragments trnL-trnF, 5′rps12- rpl20, trnV-trnM and trnS-trnG were 866, 790, 830 and 729 bp, respectively. A total of 23 polymorphic sites were present in the total concatenated length of 3215 bp, producing seven haplotypes (H1-H7; Table 1) in the 399 samples analyzed. Based on a genealogical network reflecting the relationship among populations and frequency of each haplotype (Fig. 1a), the seven haplotypes formed two haplotype groups. Haplotype group I comprised a frequently observed haplotype (H1) at the center and other less frequent haplotypes (H2-H5) arranged in a star-shape fashion. However, H1 is not likely to be the ancestral haplotype in group I since it is not directly connected to group II haplotypes. Haplotype group II comprised a dominant haplotype (H6) and a rare haplotype (H7), with the former one being closer to the group I haplotypes, suggesting H6 is the more ancient haplotype in this group. The haplotypes within the group I (H1-H5) and group II (H6-H7) showed a close relationship with each other, with a maximum of one mutational step within each group. On the other hand, a minimum of 18 mutational steps between haplotype group I and group II (i.e., between an unidentified haplotype mv1 and H6; Fig. 1a) can be observed, indicating substantial differentiation between the two haplotype groups.

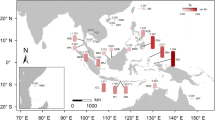
Map of sampling sites and the geographical distribution of cpDNA haplotypes in A. ilicifolius. a Median-joining network for the seven haplotypes detected. b Sampling region and distribution of all inferred haplotypes. In the median-joining network, the size of the circle is proportional to the frequency of each sampled haplotype. The small red circle indicates median vectors (i.e., unsampled or extinct haplotypes). The black line on the branches indicates the number of steps separating adjacent haplotypes. Two hypothetical haplotype groups are indicated as Group I and Group II
Geographic distribution of these haplotypes showed distinct distribution ranges of the two haplotype groups, with the group I haplotypes being more dominant among the studied populations (Fig. 1b). Groups I haplotypes were widely distributed across the Pacific Ocean (PO) region: H1 was observed in populations from southern China, east coast of Thailand and the Malay Peninsula, Borneo, Sumatra, and Java (BL); H2 was observed in populations from Java (ICT), northern Australia, and Vanuatu; H3 was restricted to a single population (HK) in southern China; H4 was observed in populations along the east coast of Thailand (TDM), Borneo (KC), and the Philippines (SA); while H5 was found to be private to a population in the Philippines (IBJ). In contrast, group II haplotypes were restricted to the Indian Ocean (IO) region: H6 was widely distributed in populations from southern India, Sri Lanka, Bay of Bengal, and the west coasts of Thailand and the Malay Peninsula; while H7 was only observed in a single population in Sri Lanka (CLB). Notably, populations in/near the south of the Malay Peninsula (SJW, TP, SBW, KS), which are situated at/close to the contact regions between the east and west coasts of the peninsula were all group I haplotypes (H1 and H4).
Population structure and genetic diversity
The PCoA (Fig. 2) showed a population grouping pattern consistent with the above haplotype aggregation analysis. The first axis (X-axis) explained most of the variation (93.83%), along which two population groups were observed. Geographically, population group I comprised mainly populations from the PO region, including those around the South China Sea, Sumatra, Java, north Australia and Vanuatu. Population group II comprised mainly populations from the IO region, including those around the Bay of Bengal and the Andaman Sea (including the west coasts of Thailand and the Malay Peninsula). The SAMOVA divided the 41 populations into two genetic groups matching exactly the two population groups with high FCT value (0.972) when K = 2. Further investigation based on the Monmonier’s algorithm identified a potential geographical barrier (barrier A) associated with the genetic abruption between PO and IO region populations with bootstrap support of 100% (Fig. 3), reflecting significant genetic isolation between them. Although populations within the PO region were found to be close along the X-axis in the PCoA analysis, potential geographical barriers were also identified between populations SA and IBJ in the Philippines (barrier B; bootstrap support of 85%), and between populations BL and ICT in Indonesia (barrier C; bootstrap support of 55%), suggesting genetic isolation (barriers of gene flow) between these populations.

Principal coordinate analysis (PCoA) for all sampled populations of A. ilicifolius from the Indo-West Pacific (IWP). The 41 populations were clustered into two groups (i.e., I and II), generally consistent with the two haplotype groups indicated in Fig. 1

Spatial separation and gene flow barriers of A. ilicifolius populations. Barriers (thick red/blue lines) were detected and numbered A–D in the map. The thickness of the red/blue line indicates the support of the barriers based on 100 permutations. Black dots with ID numbers indicate sampled populations (see Table 1)
Hierarchical AMOVA revealed that a high level of variation could be attributed to the differentiation between IO and PO population groups (97.25%), while little variation resides among populations within groups (2.36%) and within populations (0.39%) (Table 2), supporting the geographical divergence of A. ilicifolius between the two groups. When performing the analysis on each group independently, most (87.12%) of the genetic variation was found among populations (FST = 0.871; P < 0.001) in the PO. Conversely, only 8.31% of the genetic variation reside among populations (FST = 0.083; P > 0.05) while most (91.69%) of the genetic variation was found within populations in IO. Mantel tests revealed weak but significant correlation between population differentiation (FST) and geographical distance for all sampled populations (R2 = 0.0923, P < 0.05) and PO populations (R2 = 0.143, P < 0.05), while no significant correlation was found for IO populations (R2 = 0.0911, P = 0.05).
High levels of haplotype (Hd = 0.707) and nucleotide (π = 2.40 × 10−3) diversities were observed across the distribution range of A. ilicifolius (Table 3). The haplotype diversity ranged from 0.000 to 0.509 and the nucleotide diversity ranged from 0.000 to 0.16 × 10−3. Total gene diversity (HT = 0.715) across all sampled populations was much higher than the average intra-population gene diversity (HS = 0.007), suggesting that the majority of cpDNA diversity is distributed among populations. Population differentiation was high (GST = 0.894; NST = 0.990) across the distribution range of A. ilicifolius. Similar results were produced by AMOVA, with the among population component of variation up to 99.00% (Table 2). Genetic variation that takes into account haplotype similarities (NST) is higher than that considers all haplotypes equally divergent (GST), although the differences between the two estimates was not significant (P > 0.05) (Table 3). When population groups were considered separately, the genetic diversity was much higher in the PO populations (Hd = 0.587; π = 0.00022 ; HT = 0.589) than in the IO populations (Hd = 0.042; π = 0.00001; HT = 0.033). Population differentiation and number of substitution types were also remarkably higher in PO (GST = 0.848; NST = 0.870) than in IO (GST = 0.100; NST = 0.100). Average intra-population gene diversity was low in both groups, with PO populations (HS = 0.077) slightly higher than IO populations (HS = 0.033).
Population demography and environmental niche modeling
To infer the population demography of the two genetic clusters identified above, the existence of recent population expansion was examined by mismatch distribution analysis and neutrality tests. The distributions of pairwise differences for the samples from PO and IO were both unimodal (Fig. 4). Further analyses using the non-significant variance and raggedness index tests (SSD = 1.85 × 10−6, P = 0.14; HRag = 0.8420, P = 0.87) suggested that the observed distribution for IO populations did not differ significantly from those expected under a sudden expansion model (Table 4). The negative (although not significant) values of Tajima’s D (− 0.9112, P > 0.1) and Fu’s Fs (− 1.443) were also consistent with the range expansion inference for the IO group. Based on the corresponding τ value, and assuming minimum and maximum mutation rates of 1.0 × 10−9 and 8.24 × 10−9 substitutions per site/year, this expansion event was dated at 46,875 and 5690 years before present (BP), respectively. Taking into account the 95% confidence interval of τ, much broader estimates of 11,015–46,875 and 1335–5690 years BP, respectively, were generated (Table 4). For PO populations, however, the pattern significantly rejected the expansion model in terms of sum-of-squared deviations (SSD = 0.0180, P = 0.01) and raggedness index (HRag = 0.1634, P = 0.00). Neutrality tests (Tajima’s D = 0.1397, P > 0.1; Fu’s Fs = 0.136) also provided no support for the hypothesis of sudden population expansion in PO (Table 4). Contrasting results were also obtained when analyzing all populations from both groups together. While mismatch distribution analysis displayed a bimodal graph, the expansion model statistically fit well to the non-unimodal distribution (SSD = 0.0826, P = 0.23; HRag = 0.1025, P = 0.41), which suggested no significant rejection of the demographic expansion model. However, positive Tajima’s D (3.0527, P < 0.01) and Fu’s Fs (21.527) values indicated no evidence of recent range expansion of the total populations across the IWP.

Mismatch distribution analysis for a A. ilicifolius across IWP, b populations from PO group, and c populations from IO group, respectively. The solid lines show observed distributions of pairwise differences among cpDNA haplotypes and the dashed lines represent the distributions expected for an expanding population
Ecological niche modeling (ENM) showed that the environmental variable that contributed most to model prediction was the mean temperature of the wettest quarter for A. ilicifolius. A high AUC value was obtained from the current potential distribution (AUC > 0.97), indicating good predictive model performance. The current niche-predicted distribution of this species was similar to its actual distribution in the IWP (Fig. 5a). When projecting the current niche into historical climatic conditions, the suitable climate space generally stay in situ during the LIG period, but contracted greatly during the LGM period and was mainly located, under the CCSM4 model, in segregated refugia in the IWP (e.g., the Andaman Sea, the South China Sea, and the northern edge of the Sahul Shelf) (Fig. 5a). For ENMs built for the PO and IO groups, all models had AUC > 0.95, implying that the results greatly differed from random prediction. Multiple niche overlap values and identity tests revealed that empirically observed values for both I and D (I = 0.670, D = 0.549; Fig. 5b) were not remarkably different from those expected from the pseudo-replicated data sets, indicating no significant ecological differentiation between the two groups.

Results of ecological niche modeling of A. ilicifolius. a Predicted distributions of A. ilicifolius in IWP at the present time, the LGM (Last Glacial Maximum) and LIG (Last Inter Glacial). Colors represent the probability of a species occurring in an area, from highest (red) to lowest (blue). b The results of niche identity tests (I and D) between PO and IO groups. Blue bars show the simulated identity values generated from 100 randomizations, arrows indicate actual values from MAXENT modeling
Discussion
Genetic diversity and population genetic structure in A. ilicifolius
Characterization of genetic diversity is a prerequisite for efficient conservation and utilization of genetic resources. Different mangrove species are likely to display various degrees of polymorphism depending on their different genetic architecture and edaphic preferences and adaptations to the constantly stressful growing conditions (Lakshmi et al., 1997). The high genetic variation of A. ilicifolius across its distribution range (HT = 0.715) was comparable to other mangrove species such as Excoecaria agallocha (HT = 0.740) and Avicennia germinans (HT = 0.87) using cpDNA markers (Nettel & Dodd, 2007; Guo et al., 2018a). At the population level, however, the genetic variation was extremely low (HS = 0.007) in A. ilicifolius, implying that the high level of total genetic diversity was mainly contributed by the genetic divergence between populations. A. ilicifolius populations exhibited high levels of cytoplasmic structure (GST/NST = 0.894/0.990) across its distributional range. However, the non-significance (P > 0.05) of the phylogeographic structure of cpDNA haplotype distributions observed in A. ilicifolius indicated that historical disruptions of gene flow might have influenced the lineage composition of contemporary populations in the IWP (Avise 2000; Fan et al., 2013).
Long-distance dispersal in A. ilicifolius
Continental drift (e.g., the break-up of the Gondwanaland) and life history characteristics (e.g., long-distance dispersal) are two major factors that have contributed to the pantropical distribution of mangroves today (Duke, 1995; Clarke et al., 2001; Guo et al., 2018a, b). Given the estimated clade ages as discussed below, the wide-range distribution (India to Australia and the Philippines) of A. ilicifolius is not likely explained by continental drift. The break-up of Gondwanaland began more than 100 million years ago (MYA), and the relevant landmasses were widely separated by ca. 50 MYA (Scotese, 2003); the present forms of the Malay Peninsula, Sumatra, and Borneo has been formed at least 60 MYA (Lee & Lawver, 1995). Although the primary dispersal of A. ilicifolius via explosively dehiscent capsules can only happen up to about 2 m, with suitable conditions (e.g., ocean currents), secondary dispersal via water could provide further opportunity for seeds to disperse over long distances (Tomlinson, 2016), which could explain the extremely wide range of this species (McDade et al., 2005). The sharing of haplotypes among distant populations, like the H6 haplotype being shared among populations from the Andaman Sea and the Bay of Bengal with the absence of IBD in this area (Fig. 1; Table 3), indicated the existence of long-distance dispersal among transoceanic populations.
Major lineage divergence in A. ilicifolius
A major finding of this study is the presence of two lineages of A. ilicifolius—PO and IO—as inferred from the haplotype network analysis. Such strong genetic differentiation between the populations around the Pacific and Indian Oceans (or simply the genealogical break across the Malay Peninsula) has been reported previously for several mangrove species, including R. apiculata (Inomata et al., 2009), Bruguiera gymnorrhiza (Urashi et al., 2013), Ceriops decandra (Huang et al., 2008), C. tagal (Liao et al., 2007), Lumnitzera littorea (Su et al., 2007) and other marine organisms (Benzie, 1999; Alfaro et al., 2004; Antoro et al., 2006). We utilized the average synonymous substitution rate of cpDNA in angiosperms (1.0 to 8.24 × 10−9 per site/year; Wolfe et al. 1987; Richardson et al. 2001) as the approximation to estimate the divergence time between the IO and PO lineages, which dated their divergence to about 0.36–2.97 MYA. Given the absence of fossil calibration and species-specific synonymous substitution rates, this estimation is likely to be underestimated.
In mangroves, genetic divergence across the Malay Peninsula has often been attributed to the founding history of populations in this region; during the LGM in the Pleistocene (ca. 20,000 years ago), Sumatra and Borneo were connected with the Eurasian continent as one landmass (i.e., the Sunda Shelf) due to the lowering of sea levels, and relict mangrove habitats were likely to have been restricted to two major refugia at the outer margins of the shelf—one in the present South China Sea and another in the present Andaman Sea, with the landmass becoming a barrier impeding genetic exchange between the two regions (Cannon et al., 2009). During deglaciation in the Holocene (ca. 10,000 years ago), populations from the South China Sea and the Andaman Sea refugia most likely founded the mangrove populations on the east and west coasts of the Malay Peninsula, respectively (Cannon et al., 2009; Wee et al., 2014). Such a hypothesis may also apply to A. ilicifolius as indicated by the paleoclimate niche modeling in this study. Our niche modeling results indicated that the potential range for A. ilicifolius is generally similar to its current distribution during the LIG period (0.14–0.12 MYA), but contracted to small areas in the Andaman Sea and the South China Sea separated by the Sunda Shelf during the LGM (0.021–0.018 MYA). Populations in the isolated refugia would have drifted apart genetically due to reduced gene flow. In addition, our investigation based on Monmonier’s algorithm identified a significant barrier along the Thai-Malay Peninsula, separating the PO and IO populations of A. ilicifolius. Taken together, these two groups may have been isolated a long time ago, and their genetic divergence was related to, if not driven by, the range fragmentation across the present Malay Peninsula (or the Sunda Shelf, in the past) during the LGM, as observed in other mangrove species.
Aside from geographical factors (i.e., land barrier) impeding gene flow between populations, ecological factors (i.e., habitat differentiation) could also serve as a major force that drives lineage divergence given that highly divergent haplotypes may also reflect local adaptation to selective pressures during a species’ range expansion into ecologically divergent environments (Fan et al., 2013). Estimating the niche overlap between the two population groups, we found that the ecological conditions were not significantly different, indicating that the Malay Peninsula as a land barrier, instead of differential adaptation, would better explain the genetic break.
Demographic histories of IO and PO populations
Given the different geographic characteristics of the IO and PO regions, populations at the east and west of the Malay Peninsula might have experienced different demographic histories following their initial divergence. The western lineage (i.e., the IO group) was fixed for an ancestral haplotype (H6) except for a single population in Sri Lanka (CLB) which additionally harbored a minor haplotype (H7) derived from H6. While distinct genetic differences between PO and IO populations indicated a much longer history of isolation between the two lineages, the high genetic identities among populations within IO region indicated recent coancestry of these populations, suggesting that the present distribution pattern of A. ilicifolius in IO region was established rather recently by the rapid expansion of a small number of founders in this region. This is consistent with previous findings that mangrove populations in the Indian Ocean were derived from populations in one of the major refugia, the Andaman Sea, after the LGM (Flenley, 1998; Cannon et al., 2009). The sudden range expansion of IO populations during the glacial period was supported by the mismatch analysis which showed a unimodal profile and a good statistical fit of the expansion model as indicated by the non-significant SSD and HRag values. The inferred population expansion was dated to about 5690 (1335–5690) or 46,875 (11,015–46,875) years ago, depending on the mutation rates used, probably corresponding to the last glacial periods even though these estimates should be treated with caution given their wide confidence intervals. Population reductions and expansions caused by recent fluctuations in sea level might have caused bottlenecks and founder effects leading to the reduced genetic diversity of individual populations. Similar scenarios of population contraction and range expansion from a refugium after glaciations have also been frequently proposed or detected in other mangrove and non-mangrove species (Saenger & Bellan, 1995; Maguire et al., 2000; Hewitt, 2000; Nettel & Dodd, 2007; Kennedy et al., 2016).
The demographic history appeared to be more complex in the PO group (i.e., the eastern lineage), for which five haplotypes formed a star-like evolutionary unit in the genealogical network centered around the H1 haplotype. However, instead of by haplotype H1, the eastern lineage was first connected to the western lineage by H4 and H5 through a missing haplotype mv1, suggesting that the most recent ancestor of the eastern lineage is either missing during the evolution or unsampled in this study because of its rarity. Furthermore, the eastern lineage exhibited significant SSD and HRag values which, despite the unimodal mismatch distribution profile, could be interpreted as reflectance of past demographic stability. High levels of cytoplasmic substructure (GST/NST = 0.848/0.870) along with significant IBD of PO populations (Table 3) is further evidence of restricted seed dispersal in this region; note that the IBD was probably not simply a result of physical distance among populations but also a reflection of the resistance posed by barriers to gene flow for mangroves like A. ilicifolius (Meirmans, 2012; Guo et al., 2018a). Long-term persistence of populations in the east may have allowed for maintenance of higher total cpDNA haplotypic diversity (HT = 0.589). The emergence of young haplotypes (e.g., H2 and H3), on the other hand, could be ascribed to other factors such as adaptability and the microclimatic conditions where they inhabit. These findings are indicative that A. ilicifolius might exist in multiple independent glacial refugia within the Pacific Ocean region, consistent with a scenario of ‘refugia within refugia’ as observed in other plants from different geographic regions such as the Hengduan Mountains in China (Fan et al., 2013) and the Iberian Peninsula in Europe (Gómez & Lunt, 2007). They also lend support to the earlier interpretation that mangrove populations in the Pacific Ocean were mainly derived from the South China Sea refugium after the LGM (Flenley, 1998; Cannon et al., 2009).
In this context, populations in China could have been brought from the nearby refugia in the South China Sea with the aid of suitable ocean currents. A. ilicifolius fruits from April to May (Ragavan et al., 2015); in summer (from May to September), the surface current of the tropical Indian Ocean flows northward into the South China Sea and then towards the Pacific Ocean through the Bashi Strait (Fang et al., 2012). Besides, another northeastward current that originates from the southeast of Hainan Island flows along the Guangdong coast and extends to the Taiwan Strait (Qu et al., 2009). These two currents provide the opportunity for gene flow of A. ilicifolius populations from the south of the South China Sea to southern China. Notably, although there were both H1 and H4 in the east coast of Malay Peninsula and Borneo, only H1 haplotype as observed in southern China, probably indicating better adaptability of H1 in the colonized area or smaller chance of H4 dispersion due to its lower frequency. Similarly, the observation that populations located in Java (BL) and Sumatra (PD) fixed for haplotype H1 should have resulted from gene flow out of the Sunda Shelf (Guo et al., 2018b). On the other hand, BARRIER analysis identified genetic barriers between two localities in the Philippines (barrier B), between two localities in Indonesia (barrier C) as well as between Java and north Australia (barrier D). In the absence of any landmass in these regions and the relatively close geographic distance between these populations indicated the presence of cryptic barriers (i.e., ocean currents) in this region. The genetic discontinuity detected between ICT-RC-DW and other populations in Indonesia, in particular, was probably influenced by the Indonesian throughflow that finally flows to the Timor Sea and turns west to the Indian Ocean. Such pattern of the genetic discontinuity between populations incongruent with ocean current patterns was also observed in other mangroves such as R. mucronata, the major genetic discontinuity of which was found to be explained by the prevailing ocean currents at the Malacca Strait and Andaman Sea (Wee et al., 2014). Further analysis of ocean circulation simulation could provide more powerful evidence on which and how ocean currents have shapen the genetic structure of the species. These results indicated that although ocean currents can serve as a carrier for seeds enabling genetic exchange across a long distance, it can also act as a cryptic barrier impeding genetic exchange between geographically close populations. However, the restrictions may vary across species depending on the features of the contemporary ocean currents and the mobility and survivability of the propagules (Duke et al., 2002).
Conclusions
This is the first report of chloroplast DNA sequence variation within and among natural populations of A. ilicifolius across most of its distribution range. Our analysis identified two population groups (genetic clusters) corresponding to populations in the Indian Ocean and the Pacific Ocean regions. Such genealogical break was related to the repeated fragmentation following sea-level fluctuations during the Pleistocene glaciation, in which the Malay Peninsula (or the Sundaland, in the past) acting as a land barrier has at least maintained the genetic differentiation in A. ilicifolius. In contrast, ecological factors (i.e., niche conditions of IO and PO populations) are likely to play a relatively inconsequential role in this process. Populations from west and east of the Malay Peninsula probably have experienced different demographic histories. Ocean currents could either serve as carrier enabling long-distance dispersal or be a barrier impeding gene flow of A. ilicifolius in different regions.
The two genetic clusters we identified here were geographically discrete, suggesting prolonged genetic and physical isolation, which provide a basis for the definition of evolutionary significant units (ESUs) in A. ilicifolius, even though the exact levels of molecular phylogenetic distinctiveness required for the definition of ESUs are still debatable (Moritz, 1994). We recommend that these genetic clusters should be managed separately, and care should be taken to avoid artificial transplantation of individuals from a different cluster. Besides, genetic differentiation has also been observed within each population group, especially within the PO group, suggesting that the collection of samples from these genetically distinct populations, instead of selection within a particular population, would be a better option for conserving the diverse gene pools of this species. Our findings are complementary for better understanding of the genetic diversity and evolution of mangroves and provide new insights into the conservation of mangrove plants and for future restoration efforts.
References
Alfaro, M. E., D. R. Karns, H. K. Voris, E. Abernathy & S. L. Sellins, 2004. Phylogeny of Cerberus (Serpentes: Homalopsinae) and phylogeography of Cerberus rynchops: diversification of a coastal marine snake in Southeast Asia. Journal of Biogeography 31: 1277–1292.
Antoro, S., U. Na-Nakorn & W. Koedprang, 2006. Study of genetic diversity of orange-spotted grouper, Epinephelus coioides, from Thailand and Indonesia using microsatellite markers. Marine Biotechnology 8: 17–26.
Avise, J. C., 2000. Phylogeography: the history and formation of species. Harvard University Press, Cambridge, MA.
Ayre, D. J. & S. Dufty, 1994. Evidence for restricted gene flow in the viviparous coral Seriatopora hystrix on Australia’s Great Barrier Reef. Evolution 48: 1183–1201.
Babu, B. H., B. S. Shylesh & J. Padikkala, 2002. Tumour reducing and anticarcinogenic activity of Acanthus ilicifolius in mice. Journal of Ethnopharmacology 79: 27–33.
Bandelt, H.-J., P. Forster & A. Röhl, 1999. Median-joining networks for inferring intraspecific phylogenies. Molecular Biology and Evolution 16: 37–48.
Barber, P. H., S. R. Palumbi, M. V. Erdmann & M. K. Moosa, 2000. A marine Wallace’s line? Nature 406: 692–693.
Barbier, E. B., S. D. Hacker, C. Kennedy, E. W. Kock, A. C. Stier & B. R. Sillman, 2011. The value of estuarine and coastal ecosystem services. Ecological Monographs 81: 169–193.
Benzie, J. A., 1999. Major genetic differences between crown-of-thorns starfish (Acanthaster planci) populations in the Indian and Pacific Oceans. Evolution 53: 1782–1795.
Cannon, C. H., R. J. Morley & A. B. G. Bush, 2009. The current refugial rainforests of Sundaland are unrepresentative of their biogeographic past and highly vulnerable to disturbance. Proceedings of the National Academy of Sciences, USA 106: 11188.
Cheng, Y. P., S. Y. Hwang & T. P. Lin, 2005. Potential refugia in Taiwan revealed by the phylogeographical study of Castanopsis carlesii Hayata (Fagaceae). Molecular Ecology 14: 2075–2085.
Clarke, P. J., R. A. Kerrigan & C. J. Westphal, 2001. Dispersal potential and early growth in 14 tropical mangroves: do early life history traits correlate with patterns of adult distribution? Journal of Ecology 89: 648–659.
Dodd, R., Z. Afzal-Rafii, N. Kashani & J. Budrick, 2002. Land barriers and open oceans: effects on gene diversity and population structure in Avicennia germinans L. (Avicenniaceae). Molecular Ecology 11: 1327–1338.
Doyle, J. J. & J. L. Doyle, 1990. A rapid total DNA preparation procedure for fresh plant tissue. Focus 12: 13–15.
Drexler, J. Z., 2001. Maximum longevities of Rhizophora apiculata and R. mucronata propagules. Pacific Science 55: 17–22.
Duke, N. C., 1995. Genetic diversity, distributional barriers and rafting continents? More thoughts on the evolution of mangroves. Hydrobiologia 295: 167–181.
Duke, N. C., 2013. World mangrove ID: expert information at your fingertips. MangroveWatch Publications, Brisbane.
Duke, N. C., E. Lo & M. Sun, 2002. Global distribution and genetic discontinuities of mangroves–emerging patterns in the evolution of Rhizophora. Trees 16: 65–79.
Duke, N. C., J. O. Meynecke, S. Dittmann, A. M. Ellison, K. Anger, U. Berger, S. Cannicci, K. Diele, K. C. Ewel, C. D. Field, N. Koedam, S. Y. Lee, C. Marchand, I. Nordhaus & F. Dahdouh-Guebas, 2007. A world without mangroves? Science 317: 41–43.
Dupanloup, I., S. Schneider & L. Excoffier, 2002. A simulated annealing approach to define the genetic structure of populations. Molecular Ecology 11: 2571–2581.
Excoffier, L. & H. E. Lischer, 2010. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Molecular Ecology Resources 10: 564–567.
Fan, D. M., J. P. Yue, Z. L. Nie, Z. M. Li, H. P. Comes & H. Sun, 2013. Phylogeography of Sophora davidii (Leguminosae) across the ‘Tanaka-Kaiyong Line’, an important phytogeographic boundary in Southwest China. Molecular Ecology 22: 4270–4288.
Fang G., G. Wang, Y. Fang & W. Fang, 2012. A review on the South China Sea western boundary current. Acta Oceanologica Sinica 31: 1–10.
Flenley, J. R., 1998. Tropical forests under the climates of the last 30,000 years. In Markham, A. (ed.), Potential impacts of climate change on tropical forest ecosystems. Springer, Netherlands, Dordrecht: 37–57.
Fu, Y.-X. & W.-H. Li, 1993. Statistical tests of neutrality of mutations. Genetics 133: 693–709.
Fu, Y. X., 1997. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147: 915–925.
Gaither, M. R., B. W. Bowen, T.-R. Bordenave, L. A. Rocha, S. J. Newman, J. A. Gomez, L. van Herwerden & M. T. Craig, 2011. Phylogeography of the reef fish Cephalopholis argus (Epinephelidae) indicates Pleistocene isolation across the indo-pacific barrier with contemporary overlap in the coral triangle. BMC Evolutionary Biology 11: 189.
Galindo, H. M., D. B. Olson & S. R. Palumbi, 2006. Seascape genetics: a coupled oceanographic-genetic model predicts population structure of Caribbean corals. Current Biology 16: 1622–1626.
Gallaher, T., M. W. Callmander, S. Buerki, S. Setsuko & S. C. Keeley, 2017. Navigating the ‘broad freeway’: ocean currents and inland isolation drive diversification in the Pandanus tectorius complex (Pandanaceae). Journal of Biogeography 44: 1598–1611.
Gómez, A. & D. H. Lunt, 2007. Refugia within refugia: patterns of phylogeographic concordance in the Iberian Peninsula. In Weiss, S. & N. Ferrand (eds), Phylogeography of Southern European Refugia. Springer, Dordrecht: 155–188.
Guo, Z., Y. Huang, Y. Chen, N. C. Duke, C. Zhong & S. Shi, 2016. Genetic discontinuities in a dominant mangrove Rhizophora apiculata (Rhizophoraceae) in the Indo-Malesian region. Journal of Biogeography 43: 1856–1868.
Guo, W., W. L. Ng, H. Wu, W. Li, L. Zhang, S. Qiao, X. Yang, X. Shi & Y. Huang, 2018a. Chloroplast phylogeography of a widely distributed mangrove species, Excoecaria agallocha, in the Indo-West Pacific region. Hydrobiologia 807: 333–347.
Guo, Z., W. Guo, H. Wu, X. Fang, W. L. Ng, X. Shi, Y. Liu, Z. Huang, W. Li & L. Gan, 2018b. Differing phylogeographic patterns within the Indo-West Pacific mangrove genus Xylocarpus (Meliaceae). Journal of Biogeography 45: 676–689.
Hamilton, M. B., 1999. Four primer pairs for the amplification of chloroplast intergenic regions with intraspecific variation. Molecular Ecology 8: 521–523.
Harpending, H. C., 1994. Signature of ancient population growth in a low-resolution mitochondrial DNA mismatch distribution. Human Biology 66: 591–600.
Hewitt, G., 2000. The genetic legacy of the Quaternary ice ages. Nature 405: 907–913.
Hijmans, R. J., S. E. Cameron, J. L. Parra, P. G. Jones & A. Jarvis, 2005. Very high resolution interpolated climate surfaces for global land areas. International Journal of Climatology: A Journal of the Royal Meteorological Society 25: 1965–1978.
Huang, Y., F. Tan, G. Su, S. Deng, H. He & S. Shi, 2008. Population genetic structure of three tree species in the mangrove genus Ceriops (Rhizophoraceae) from the Indo West Pacific. Genetica 133: 47–56.
Hudson, R. R., 1990. Gene genealogies and the coalescent process. Oxford surveys in evolutionary biology 7: 44.
Inomata, N., X.-R. Wang, S. Changtragoon & A. E. Szmidt, 2009. Levels and patterns of DNA variation in two sympatric mangrove species, Rhizophora apiculata and R. mucronata from Thailand. Genes & Genetic Systems 84: 277–286.
Kennedy, J. P., M. W. Pil, C. E. Proffitt, W. A. Boeger, A. M. Stanford & D. J. Devlin, 2016. Postglacial expansion pathways of red mangrove, Rhizophora mangle, in the Caribbean Basin and Florida. American Journal of Botany 103: 260–726.
Lakshmi, M., S. Rajalakshmi, M. Parani, C. S. Anuratha & A. Parida, 1997. Molecular phylogeny of mangroves I. Use of molecular markers in assessing the intraspecific genetic variability in the mangrove species Acanthus ilicifolius Linn. (Acanthaceae). Theoretical and Applied Genetics 94: 1121–1127.
Lee, T.-Y. & L. A. Lawver, 1995. Cenozoic plate reconstruction of Southeast Asia. Tectonophysics 251: 85–138.
Lessios, H. A., 2008. The great American schism: divergence of marine organisms after the rise of the Central American Isthmus. Annual Review of Ecology, Evolution, Systematics 39: 63–91.
Li, J., Y. Yang, Q. Chen, L. Fang, Z. He, W. Guo, S. Qiao, Z. Wang, M. Guo, C. Zhong, R. Zhou & S. Shi, 2016. Pronounced genetic differentiation and recent secondary contact in the mangrove tree Lumnitzera racemosa revealed by population genomic analyses. Scientific Reports 6: 29486.
Liao, B. W., 2009. Studies on Dongzhai harbor mangrove wetland ecosystem on Hainan island in China. China Ocean University Press, Qingdao.
Liao, P.-C., S. Havanond & S. Huang, 2007. Phylogeography of Ceriops tagal (Rhizophoraceae) in Southeast Asia: the land barrier of the Malay Peninsula has caused population differentiation between the Indian Ocean and South China Sea. Conservation Genetics 8: 89–98.
Librado, P. & J. Rozas, 2009. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25: 1451–1452.
Maguire, T. L., P. Saenger, P. Baverstock & R. Henry, 2000. Microsatellite analysis of genetic structure in the mangrove species Avicennia marina (Forsk.) Vierh. (Avicenniaceae). Molecular Ecology 9: 1853–1862.
Manni, F., E. Guerard & E. Heyer, 2004. Geographic patterns of (genetic, morphologic, linguistic) variation: how barriers can be detected by using Monmonier’s algorithm. Human Biology 76: 173–190.
McDade, L. A., T. F. Daniel, C. A. Kiel & K. Vollesen, 2005. Phylogenetic relationships among Acantheae (Acanthaceae): major lineages present contrasting patterns of molecular evolution and morphological differentiation. Systematic Botany 30: 834–862.
Meirmans, P. G., 2012. The trouble with isolation by distance. Molecular Ecology 21: 2839–2846.
Minobe, S., S. Fukui, R. Saiki, T. Kajita, S. Changtragoon, N.A. Ab Shukor, A. Latiff, B. R. Ramesh, O. Koizumi & T. Yamazaki, 2010. Highly differentiated population structure of a Mangrove species, Bruguiera gymnorhiza (Rhizophoraceae) revealed by one nuclear GapCp and one chloroplast intergenic spacer trnF-trnL. Conservation Genetics 11: 301–310.
Mori, G. M., M. I. Zucchi & A. P. Souza, 2015. Multiple-geographic-scale genetic structure of two mangrove tree species: the roles of mating system, hybridization, limited dispersal and extrinsic factors. PLoS One 10: e0118710.
Moritz, C., 1994. Defining ‘Evolutionarily Significant Units’ for conservation. Trends in Ecology & Evolution 9: 373–375.
Nettel, A. & R. S. Dodd, 2007. Drifting propagules and receding swamps: genetic footprints of mangrove recolonization and dispersal along tropical coasts. Evolution 61: 958–971.
Ngeve, M. N., T. Van der Stocken, D. Menemenlis, N. Koedam & L. Triest, 2016. Contrasting effects of historical sea level rise and contemporary ocean currents on regional gene flow of Rhizophora racemosa in eastern Atlantic mangroves. PLoS One 11: e0150950.
Otto-Bliesner, B. L., S. J. Marsha, J. T. Overpeck, G. H. Miller, A. X. Hu & C. L. I. P. Mem, 2006. Simulating Arctic climate warmth and icefield retreat in the last interglaciation. Science 311: 1751–1753.
Parks, D. H., M. Porter, S. Churcher, S. Wang, C. Blouin, J. Whalley, S. Brooks & R. G. Beiko, 2009. GenGIS: a geospatial information system for genomic data. Genome Research 19: 1896–1904.
Peakall, R. & P. E. Smouse, 2012. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research–an update. Bioinformatics 28: 2537–2539.
Phillips, S. J. & M. Dudík, 2008. Modeling of species distributions with Maxent: new extensions and a comprehensive evaluation. Ecography 31: 161–175.
Phillips, S. J., R. P. Anderson & R. E. Schapire, 2006. Maximum entropy modeling of species geographic distributions. Ecological Modelling 190: 231–259.
Pil, M. W., M. R. Boeger, V. C. Muschner, M. R. Pie, A. Ostrensky & W. A. Boeger, 2011. Postglacial north-south expansion of populations of Rhizophora mangle (Rhizophoraceae) along the Brazilian coast revealed by microsatellite analysis. American Journal of Botany 98: 1031–1039.
Pons, O. & R. J. Petit, 1996. Measuring and testing genetic differentiation with ordered versus unordered alleles. Genetics 144: 1237–1245.
Qu, T., Y. T. Song & T. Yamagata, 2009. An introduction to the South China Sea throughflow: its dynamics, variability, and application for climate. Dynamics of Atmospheres and Oceans 47: 3–14.
Ragavan, P., A. Saxena, P. M. Mohan, R. S. Jayaraj & K. Ravichandran, 2015. Taxonomy and distribution of species of the genus Acanthus (Acanthaceae) in mangroves of the Andaman and Nicobar Islands, India. Biodiversitas 16: 225–237.
Richardson, J. E., R. T. Pennington, T. D. Pennington & P. M. Hollingsworth, 2001. Rapid diversification of a species-rich genus of Neotropical rain forest trees. Science 293: 2242–2245.
Rogers, A. R. & H. Harpending, 1992. Population growth makes waves in the distribution of pairwise genetic differences. Molecular Biology and Evolution 9: 552–569.
Saenger, P. & M. F. Bellan, 1995. The mangrove vegetation of the Atlantic coast of Africa: a review. Université de Toulouse, Toulouse.
Scotese, C. R., 2003. Paleogeographic maps, PALEOMAP project: www.scotese.com.
Su, G., Y. Huang, F. Tan, X. Ni, T. Tang & S. Shi, 2007. Conservation genetics of Lumnitzera littorea (Combretaceae), an endangered mangrove, from the Indo-West Pacific. Marine Biology 150: 321–328.
Taberlet, P., L. Gielly, G. Pautou & J. Bouvet, 1991. Universal primers for amplification of three non-coding regions of chloroplast DNA. Plant Molecular Biology 17: 1105–1109.
Tajima, F., 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595.
Tamura, K., G. Stecher, D. Peterson, A. Filipski & S. Kumar, 2013. MEGA6: molecular Evolutionary Genetics Analysis version 6.0. Molecular Biology and Evolution 30: 2725–2729.
Thiel, M. & L. Gutow, 2005. The ecology of rafting in the marine environment. II. The rafting organisms and community Oceanography and Marine Biology. CRC Press, 289-428.
Tomlinson, P. B., 2016. The botany of mangroves. Cambridge University Press, Cambridge.
Triest, L., 2008. Molecular ecology and biogeography of mangrove trees towards conceptual insights on gene flow and barriers: a review. Aquatic Botany 89: 138–154.
Umali, R., P. Zamora, R. Gatera, R. Jara, A. CAMA-CHO & M. Vannucci, 1987. Mangroves of Asia and the Pacific: Status and management. Technical Report of the UNDP/UNESCO Research and Training Pilot Programme on mangrove ecosystems in Asia and the Pacific.
Urashi, C., K. M. Teshima, S. Minobe, O. Koizumi & N. Inomata, 2013. Inferences of evolutionary history of a widely distributed mangrove species, Bruguiera gymnorrhiza, in the Indo-West Pacific region. Ecology and Evolution 3: 2251–2261.
Warren, D. L., R. E. Glor & M. Turelli, 2010. ENMTools: a toolbox for comparative studies of environmental niche models. Ecography 33: 607–611.
Waters, J. M., 2008. Marine biogeographical disjunction in temperate Australia: historical landbridge, contemporary currents, or both? Diversity and Distributions 14: 692–700.
Wee, A. K., K. Takayama, T. Asakawa, B. Thompson, S. Sungkaew, N. X. Tung, M. Nazre, K. K. Soe, H. T. Tan & Y. Watano, 2014. Oceanic currents, not land masses, maintain the genetic structure of the mangrove Rhizophora mucronata Lam. (Rhizophoraceae) in Southeast Asia. Journal of biogeography 41: 954–964.
Wee, A. K., K. Takayama, J. L. Chua, T. Asakawa, S. H. Meenakshisundaram, Onrizal, B. Adjie, E. R. Ardli, S. Sungkaew, N. B. Malekal, N. X. Tung, S. G. Salmo, 3rd, O. B. Yllano, M. N. Saleh, K. K. Soe, Y. Tateishi, Y. Watano, S. Baba, E. L. Webb & T. Kajita, 2015. Genetic differentiation and phylogeography of partially sympatric species complex Rhizophora mucronata Lam. and R. stylosa Griff. using SSR markers. BMC Evolutionary Biology 15:57
Wolfe, K. H., W. H. Li & P. M. Sharp, 1987. Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proceedings of the National Academy of Sciences, USA 84: 9054–9058
Wolfe, K. H., W. H. Li & P. M. Sharp, 2003. Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proceedings of the National Academy of Sciences, USA 84: 9054–9058.
Wu, J., S. Zhang, Q. Xiao, Q. Li, J. Huang, L. Long & L. Huang, 2003. Phenylethanoid and aliphatic alcohol glycosides from Acanthus ilicifolius. Phytochemistry 63: 491–495.
Yang, Y., N. C. Duke, F. Peng, J. Li, S. Yang, C. Zhong, R. Zhou & S. Shi, 2016. Ancient geographical barriers drive differentiation among Sonneratia caseolaris populations and recent divergence from S. lanceolata. Frontiers in Plant Science 7:1618.
Zhou, W., Y. Wen, J. Fu, Y. Xu, J. Jin, L. Ding, M. Min, J. Che & Y. Zhang, 2012. Speciation in the Rana chensinensis species complex and its relationship to the uplift of the Qinghai-Tibetan Plateau. Molecular Ecology 21: 960–973.
Acknowledgments
This study is supported by grants from the National Natural Science Foundation of China (Grant No. 31700178 and 41776166), the Natural Science Foundation of Guangdong Province (Grant No. 2017A030313159), Guangdong Basic and Applied Basic Research Foundation (Grant No. 2019A15150122211), and Chang Hungta Science Foundation of Sun Yat-sen University. The authors wish to thank Drs. Suhua Shi, Ying Liu, Renchao Zhou, and Tian Tang (Sun Yat-sen University, China), Dr. Sonjai Havanond (Department of National Parks, Wildlife and Plant Conservation, Thailand), Mr. Chanop Jaengjai (Department of Marine and Coastal Resources, Thailand), and Dr. Albert Williams (Department of Environmental Protection and Conservation, Vanuatu) for assistance in collecting and providing plant material. We would especially like to thank the Associate Editor and the reviewer for their thoughtful and constructive comments towards improving the quality of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Handling editor: Emily M. Dangremond
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Guo, W., Banerjee, A.K., Ng, W.L. et al. Chloroplast DNA phylogeography of the Holly mangrove Acanthus ilicifolius in the Indo-West Pacific. Hydrobiologia 847, 3591–3608 (2020). https://doi.org/10.1007/s10750-020-04372-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10750-020-04372-1