
Abstract
Excoecaria agallocha is one of the predominant mangrove species in the Indo-West Pacific (IWP) region with an extensive range of distribution. To infer the current geographical patterns of genetic variation and provide new insights on the historical population dynamics of mangrove species in the IWP region, we sampled E. agallocha across its distribution range and investigated the phylogeography of this species using four chloroplast DNA (cpDNA) fragments. Our results showed that E. agallocha possessed a high degree of species-level genetic diversity, while the average genetic diversity within populations was much lower. The presence of population genetic structure was supported by the estimates of genetic differentiation and the analysis of molecular variance (AMOVA). Of the ten haplotypes identified, no haplotypes were shared between the East Indian Ocean (EIO), the West Pacific Ocean (WPO), and the North Australian (NA) regions. Genealogy analysis, haplotype distribution patterns, and the principal coordinate analysis (PCoA) consistently suggested the existence of three haplotype groups distributed in distinct geographical locations. The genealogical breaks observed and further analysis of geographic/genetic barriers indicated that both land barriers and oceanic currents may have played important roles in the divergence and demography of E. agallocha.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Mangroves constitute among the most dominant intertidal ecosystem along the tropical and subtropical coastlines. The distribution of mangrove populations is considered to be largely shaped by their responses to colder climate and arid conditions at the limit of their ranges, and have been expanding their ranges along changing coastal zones since the last glacial period (Duke et al., 1998; Saenger, 1998; Dodd et al., 2002; Saintilan et al., 2014). Most mangrove species have viviparous propagules that are buoyant and can be dispersed by ocean currents. These make mangrove species good candidates for the study of population genetic structures influenced by founder effects resulting from frequent local extinction, recolonization, and long-distance dispersal (LDD) of propagules by water currents along coastlines of different forms. Moreover, the accurate assessment of genetic diversity and population genetic structure of mangroves is essential for the preservation and management of this unique ecosystem, and can also provide us a perspective of evolutionary mechanisms that shaped the current diversity and adaptability of coastal species (Urashi et al., 2013).
The geographical distribution of a species is a key determinant of population subdivision and microevolution. The spatial configuration of populations, combined with physical features of the landscape, can either restrict or facilitate gene flow by affecting the movement of individuals or gametes (i.e., pollen) (Slatkin, 1987). For mangrove species, population subdivision across a distribution range resulting from lack of gene flow can be explained by the effects of three factors: geographic distance, land barriers, and ocean currents. First, gene flow through propagule and pollen dispersal could be geographically restricted and genetic exchanges would be limited to neighboring populations, leading to genetic differentiation increasing with physical distance, a process termed isolation-by-distance (IBD; Kimura & Weiss, 1964). Inconsistent evidence on the presence of IBD has been observed in mangrove species (e.g., Dodd et al., 2002; Mori et al., 2015; Ngeve et al., 2016). Since species or region-specific characteristics could exist and resolving the impact of geographic distance on population subdivision in mangrove species will not likely result in a one-size-fits-all conclusion, extended efforts are needed to further explore the impacts of geographical distance on shaping the genetic structure of various mangrove populations. Second, as plants with sea-drifted seeds cannot migrate through land, gene flow among populations of a mangrove species can be blocked by land barriers (Wiley, 1988; Taberlet et al., 1998). For example, genetic discontinuities across the landscape have been examined for mangroves in the Indo-West Pacific (IWP) in several previous studies (e.g., Tan et al., 2005; Su et al., 2006, 2007; Liao et al., 2007), indicating that both the Malay Peninsula and Indonesian archipelago may have acted as land barriers that prevented gene flow among the East Indian Ocean (EIO), West Pacific Ocean (WPO), and North Australia (NA). Third, ocean currents may also be an important force promoting population subdivision thus affecting genetic structure of the extant mangrove populations. For example, ocean currents may act as a barrier to gene flow among geographically proximate marine populations (Ayre & Dufty, 1994; Waters, 2008; Wee et al., 2014). As genetic isolation of populations may result from a complex combination of multiple mechanisms, an explicit interpretation of phylogeography and genetic structure is necessary to clarify the evolutionary forces and historical population dynamics that a species is subjected to.
Excoecaria agallocha L. (Euphorbiaceae), also known as the “milk mangrove,” is the most predominant mangrove species in the genus. This species grows on sandy soil or sandy mud near the terrestrial fringes of mangrove vegetation, and is widely distributed from east Africa to Samoa (Tomlinson, 1986). Besides being an important part of the specific costal ecosystem of mangroves, E. agallocha is also important for its latex, which contains powerful chemicals being used on sores and to treat marine stings. The leaves are also used for fishing, and the leaf sap is currently being tested for its medicinal properties (Das et al., 2011). E. agallocha is dioecious and produces small and buoyant seeds, which are about 3 mM in diameter, with an air space in the seed coat. Although the exact dispersal ability of these seeds (i.e., the dispersal unit in E. agallocha) has not been experimentally tested, their characteristics (i.e., small size and buoyant) are thought to help them float on water and, with the help of surface currents, disperse over long distances (Zhang et al., 2008; Das et al., 2011). Unlike most other mangrove species however, E. agallocha is non-viviparous, and thus represents a suitable model for studying the phylogeographic patterns and population dynamics of non-viviparous mangrove populations in the IWP. Although many studies (e.g., Tan et al., 2005; Su et al., 2006, 2007; Liao et al., 2007) have been conducted on the genetic variation, population structure, and demography of various mangrove species, to our knowledge, only three reports (i.e., Lakshmi et al., 2000; Zhang et al., 2008; Das et al., 2011) exist for E. agallocha. All these studies used dominant markers (i.e., RAPD and RFLP in Lakshmi et al., 2000; ISSR in Zhang et al., 2008; RAPD and ISSR in Das et al., 2011) and were restricted in terms of the sampling regime. Although dominant markers allow the interpretation of genetic diversities and genetic structuring of populations, the inference of spatial haplotype distribution is not possible. Maternally inherited genetic markers, i.e., chloroplast (cp) DNA, are considered especially relevant to phylogeographic surveys (Triest et al., 2008). Most phylogeographic studies of plants have been based on cpDNA and have revealed genetic heterogeneity throughout the range of a species and allowed an inference of historical range shifts and recolonization routes (Bai et al., 2010). Besides, to draw a general picture of genetic structure and evolutionary history, a survey of populations covering most, if not all, areas of a species’ distribution is necessary.
In the present study, we examined the phylogeographic patterns of E. agallocha through cpDNA sequence data and an extensive sampling across the entire range of the species. Fifty-six cpDNA fragments were initially screened, and four loci that showed polymorphism were further sequenced and analyzed for 419 individuals from 47 populations. Our major objectives were to (1) characterize the levels of genetic variation across the geographical range of E. agallocha; (2) determine the genetic structure among these populations; and (3) trace the historical range shift of E. agallocha, and interpret how historical, geographical, and ecological factors (i.e., climate oscillations, land barriers, and oceanic current barriers) may have influenced the present distribution pattern of this species.
Materials and methods
Sample collection
A total of 419 leaf samples from 47 natural populations of E. agallocha were collected across its entire distribution range in the IWP region, including 13 populations from the East Indian Ocean (EIO) region, 32 populations from the West Pacific Ocean (WPO) region, and two populations from the North Australian (NA) region. The sampling details are listed in Table 1. The sampled individuals within each population were located at least 5 m apart. Given that E. agallocha populations usually do not occur as monospecific stands and that the usual distance criterion for mangrove sampling is 3–10 m (Cerón-Souza et al. 2012; Ruan et al., 2013; Dasgupta et al., 2015; Ngeve et al., 2016), the minimum spacing of 5 m used here is adequate and familial sampling could largely be avoided under such a sampling scheme. Plant materials were stored with silica gel in zip-lock plastic bags until DNA isolation.
DNA extraction and PCR amplification
Genomic DNA of each individual was extracted using the CTAB method (Doyle & Doyle, 1990). A total of 56 published primer pairs for different cpDNA regions (Taberlet et al., 1991; Small et al., 1998; Hamilton, 1999; Shaw et al., 2005) were initially screened on a subset of five individuals per population randomly collected from six populations (i.e., ADR, STC, SSB, TPN, HBR, and NSL) that were the farthest apart. Only four fragments that showed polymorphism within or among populations (i.e., trnV-trnM, petB-petD, rpoC1-rpoC2, and trnC-rpoB) were subsequently analyzed for all samples.
DNA amplification was carried out in 30μl PCR reaction mixtures containing approximately 1 μg of total DNA, 5 pmol of each primer, 10 mM of Tris–HCl (pH 8.4), 1.5 mM of MgCl2, 0.1 mM of dNTP, and 2 units of Taq polymerase (Shengong Inc., Shanghai, China). PCR reactions were performed under the following cycle profile: initial denaturation at 94°C for 5 min, followed by 45 s at 94°C, 45 s at 53°C, and 1.5 min at 72°C for 30 cycles, and 10 min at 72°C for final extension. The PCR products were separated on 1.0% agarose gel, stained with ethidium bromide and viewed under UV light. Purified DNA fragments were then sequenced for both strands on an ABI 3730XL DNA Analyzer (Applied Biosystems, Inc.).
Data analysis
The chloroplast DNA sequences were assembled and manually edited using SeqMan™ (DNASTAR). The sequences of E. agallocha generated in this study have been deposited in GenBank under the accession numbers MF381193–MF381206. The geographic distribution of cpDNA haplotypes was plotted on a map using GenGis v2.11 (Parks et al., 2009), and the relationship among haplotypes were inferred using the median-joining method implemented in NETWORK v4.6.1.2 (Bandelt et al., 1999). Continuous indels were treated as single mutational events in the analysis.
Haplotype diversity (Hd) and nucleotide diversity (π) were calculated using DnaSP v5.10.1 (Librado & Rozas, 2009). Average gene diversity within populations (H S) and total gene diversity (H T) were estimated for all populations and for each region using PERMUT (Pons & Petit, 1996). Two parameters of population differentiation, G ST and N ST, were also calculated and compared through a permutation test with 1000 permutations to examine the presence of phylogeographic structure. While G ST considers all haplotypes equally divergent and is dependent of haplotype frequencies, N ST takes into account both haplotype frequencies and their sequence similarities (Pons & Petit, 1996). A significantly higher N ST than G ST could be an indication of the presence of phylogeographic structure.
To further characterize the population structure and genetic variation, multiple hierarchical analyses of molecular variance (AMOVAs) were performed with significance tests of variance components based on 1000 permutations using ARLEQUIN (Excoffier & Lischer, 2010), including three with a region-specific focus, one with an among-region focus, and one that considers all populations without partitioning. Pairwise F ST values for all populations were also calculated to measure the levels of genetic differentiation between populations. Isolation-by-distance (IBD) was examined both across all regions combined and within each region separately by testing the relationship between pairwise F ST and natural-log-transformed (Ln-transformed) geographical distances (Rousset, 1997) using the Mantel test implemented in GenAlEx v6.5 (Peakall & Smouse, 2012), with 1,000 random permutations. A principal coordinate analysis (PCoA) on the Kimura-2-parameter genetic distance of all populations was also performed using GenAlEx.
BARRIER v2.2 (Manni et al., 2004) was used to implement the Monmonier’s maximum difference algorithm to identify biogeographical boundaries or areas exhibiting the largest genetic discontinuities between population pairs using the F ST matrix as the input. The robustness of each barrier was assessed by bootstrapping over loci to generate 100 matrices of genetic differentiation and then tabulating the number of bootstraps that supported the barrier.
Mismatch distribution analysis was conducted using ARLEQUIN to test whether E. agallocha had undergone recent population expansion. An expected distribution of the number of differences between haplotypes under a model of sudden demographic expansion was generated. Goodness-of-fit was tested with the sum-of-squared deviations (SSD) between observed and expected mismatch distributions, and the raggedness index of Harpending (H Rag; Harpending, 1994). We also performed the Tajima’s D test (Tajima, 1989) and Fu’s Fs test (Fu, 1997) to investigate the recent demographic expansions. Significant D values and large negative Fs values generally suggest rapid demographic expansions (Hudson, 1990).
Results
Chloroplast DNA variation and haplotype distribution
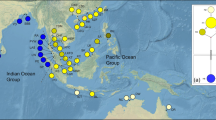
The low proportion (four out of 56 loci; 7.14%) of polymorphic cpDNA regions suggested that the chloroplast genome of E. agallocha is highly conserved. The alignment lengths of the four cpDNA fragments, trnV-trnM, petB-petD, rpoC1-rpoC2, and trnC-rpoB, were 843, 1,626, 1,793, and 1,159 bp, respectively. A total of 10 polymorphic sites were present in the total concatenated length of 5,421 bp, corresponding to 10 haplotypes (C1-C10; Table 1) in the 419 samples analyzed. Of these, two haplotypes (C1 and C2), five haplotypes (C3, C4, C5, C6, and C8), and three haplotypes (C7, C9, and C10) were confined to populations located in EIO, WPO, and NA, respectively, indicating substantial differentiation among these three regions at least during the recent past (Fig. 1). Despite the differences in geographic distribution, these haplotypes showed close relationship with each other, with a maximum of three mutation steps (i.e., between haplotypes C6 and C10).

Map of the sampling sites and the geographical distribution of cpDNA haplotypes in E. agallocha. (a) Sampling region and distribution of all inferred haplotypes; (b) median-joining network for the ten haplotypes detected. In the median-joining network, the size of the circle is proportional to the frequency of each sampled haplotype. The small white circle indicates median vectors (i.e., unsampled or extinct haplotypes). The black line on the branches indicates the number of steps separating adjacent haplotypes. Three hypothetical haplotype groups are indicated as I, II, and III. (c) The distribution of E. agallocha (source Duke, 2013)
Distinct and non-homogeneous distribution of haplotypes in the three regions was observed. In EIO, haplotype C2 was widely shared by all populations except for the TLU population that was fixed for haplotype C1. Haplotype C1 was also present in two populations from the Bay of Bengal with high frequency and one population from the west Malay Peninsula with low frequency. In WPO, most populations from Southeast China, Japan, and the Philippines were fixed for haplotype C6, while populations from east Malay Peninsula and Borneo were dominated by haplotype C3. Differing by a single site with each other, haplotypes C3 and C6 coexisted in 10 populations located in east Malay Peninsula, Southeast China, the Philippines, and Indonesia. Haplotypes C4 and C5 were private to populations in Borneo and Taiwan, respectively, with low frequency. In NA, population ADR was fixed for haplotype C7, while haplotypes C9 and C10 were specific to population VT.
Genetic diversity and population structure
Haplotype diversity (Hd) was estimated to be 0.723 for the total samples, and was in the range of 0.00–0.69 within the 47 populations (Tables 1, 2). The overall genetic diversity (haplotype diversity and nucleotide diversity) was higher in the WPO populations (Hd = 0.525; π = 0.00010) than in the EIO populations (Hd = 0.379; π = 0.00007) (Table 2). Total genetic diversity H T (0.740) across all sampled populations was much higher than the average intra-population diversity H S (0.150), suggesting that the majority of cpDNA diversity is distributed among populations. Compared with that of EIO (H S = 0.075; H T = 0.344), H S and H T were each remarkably higher for WPO (H S = 0.155; H T = 0.597).
Population differentiation was high (G ST = 0.798; N ST = 0.850) for the entire IWP. Compared with that of EIO (G ST = 0.783; N ST = 0.783), population differentiation and number of substitution types were each slightly lower for WPO (G ST = 0.740; N ST = 0.756). The permutation test showed that N ST is not significantly greater than G ST either for the entire IWP or for EIO and WPO separately (P > 0.05).
Hierarchical AMOVA analysis revealed that 56.88% of the variation can be attributed to the differentiation among EIO, WPO, and NA, while 26.02 and 17.11% of the variation are among populations within the regions and within populations, respectively, supporting the geographical divergence of E. agallocha among these regions. When performing the analysis on each region independently, more than half of the total genetic variation (74.48, 59.10, and 56.95%) was found among populations with high F ST values (0.755, 0.591, and 0.570 for EIO, WPO, and NA, respectively). When all populations from the three regions were combined, there was more genetic variation among populations (75.03%) than within populations (24.97%), with a significant genetic differentiation coefficient of F ST = 0.750 (Table 3). Mantel test revealed weak significant correlation between population differentiation (F ST) and geographical distance for all sampled populations (r 2 = 0.0582, P = 0.001), while no significant correlation was observed when applied to EIO or WPO separately (r 2 = 0.215, P = 0.070 for EIO; r 2 = 0.398, P = 0.001 for WPO).
Phylogeographic subdivision and population demography
A genealogical network reflecting the relationship among populations and frequency of all cpDNA haplotypes was constructed (Fig. 1). The network showed that the centrally located nodes (i.e., haplotypes C2, C3, and C6) most likely represent hypothetical ancestral haplotypes, with C6 more closely related to C3 than to C2. The remaining haplotypes were linked to these central haplotypes by one to three steps in a star-like network, within which only haplotypes C9 and C10 were separated from C6 by unidentified haplotype(s). As shown in Fig. 1, haplotypes C2, C3, C6 and their derived haplotype(s) were named group I, group II, and group III hereafter, respectively.
Principal coordinate analysis (PCoA) resulted in a population grouping pattern generally consistent with the above haplotypes aggregation analysis (Fig. 2): Populations from EIO (fixed for group I haplotypes) were located close to each other; most populations from east Malay Peninsula, Borneo, and Bali (dominated by haplotype C3) formed a second group; and most populations from Southeast China, Japan, the Philippines, and Indonesia (dominated by haplotype C6) were clustered into a third group. In addition, population VT was placed relatively distant to population ADR, indicating barrier(s) of gene flow between these populations. Similar observation was also found in populations from the Bay of Bengal, which were placed relatively distant to other populations of west Malay Peninsula.

Principal coordinate analysis (PCoA) for all sampled populations of E. agallocha from the Indo-West Pacific (IWP) region. The 47 populations were clustered into three groups (i.e., I, II, and III), generally consistent with the three haplotype groups indicated in Fig. 1
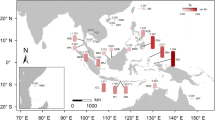
Further investigation based on the Monmonier’s algorithm identified potential geographical barriers associated with the genetic abruption between EIO and WPO, and between WPO and NA (Fig. 3), reflecting significant genetic isolation between them. Besides, barriers between the Bay of Bengal and west Malay Peninsula in EIO, between east Malay Peninsula and Southeast China in WPO, and between populations ADR and VT in NA were also identified, with the latter one having the highest bootstrap support of 96%.

Spatial separation and gene flow barriers of E. agallocha populations. Barriers (thick red lines) were detected and numbered A–E in the map. The thickness of the red line indicates the support of the barriers based on pairwise F ST matrix with 100 permutations. Black dots with ID numbers indicate sampled populations
Mismatch distribution analyses for both WPO and NA displayed multi-modal graphs (Fig. 4), which were significantly different from the expected sudden expansion model, as supported by the analyses of raggedness index (H Rag = 0.650, P = 0.019 for WPO; H Rag = 0.719, P = 0.015 for NA) although not by that of the sum-of-squared deviations (SSD = 0.170, P = 0.089 for WPO; SSD = 0.197, P = 0.062 for NA). Neutrality tests showed no significantly negative Fs and D values for samples from WPO and NA, thus no support for hypotheses of sudden population expansion in these two regions (Table 4). For EIO, although statistical tests for both SSD (SSD = 0.006, P = 0.119) and H Rag (H Rag = 0.202, P = 0.155) were not significant, the observed curve in the mismatch distribution analysis was not of a typical unimodal distribution. Neutrality tests of Tajima’s D and Fu’s Fs statistics also did not support the population expansion hypothesis in EIO. As for total samples from all locations, although the mismatch distribution showed a unimodal distribution, both SSD (SSD = 0.117, P = 0.031) and H Rag (H Rag = 0.385, P = 0.000) suggested that the observed distributions differed significantly from those expected under a sudden expansion model. Tajima’s D and Fu’s Fs tests were negative but not statistically significant, which also did not support the population expansion hypothesis. Collectively, no compelling evidence of recent expansion for the three regions or the total sampling locations was obtained in this current study.

Mismatch distribution analysis for (a) E. agallocha as a whole, (b) populations from EIO, (c) populations from WPO, and (d) populations from NA, respectively. The solid lines show observed distributions of pairwise differences among cpDNA haplotypes and the dashed lines represent the distributions expected for an expanding population
Discussion
Genetic diversity
This is the first report of chloroplast DNA sequence variation within and among natural populations of E. agallocha across most of its distribution range. At the species level, analysis of cpDNA sequences revealed a high degree of genetic variation in E. agallocha with a total genetic diversity (H T) of 0.740, higher than the average value (H T = 0.68) of 169 plant species which were also obtained from cpDNA-based studies (Petit et al., 2005). Compared with other mangrove species, the genetic variation found in E. agallocha cpDNA is higher than that of Ceriops decandra (H T = 0.270 using ISSR markers, Huang et al., 2008), Aegiceras corniculatum (H T = 0.039 using ISSR markers, Ge & Sun, 1999), and Kandelia candel (H T = 0.036 using allozyme markers, Sun et al., 1998) but lower than that of Avicennia germinans (H T = 0.87 using cpDNA markers, Nettel & Dodd, 2007). Undeniably, comparing diversity between marker types should be treated with caution given that they fit different mutational models. The current comparisons, however, could still provide some references for diversity levels between E. agallocha and other mangrove species since related studies that used cpDNA markers and simultaneously provided H T values are limited. These findings are also consistent with the earlier report on the same species using ISSR analysis, which revealed a higher species-level expected heterozygosity (H E = 0.277, Zhang et al., 2008) than that of the mangrove C. decandra (H E = 0.253, Tan et al., 2005) and mangrove associate Heritiera littoralis (H E = 0.236, Jian et al., 2004) using ISSR analysis. The high level of genetic diversity of E. agallocha populations could be attributed to its wide distribution and cross-pollinating nature given that predominately out-crossing woody plants with wide-range distribution usually show high genetic diversity (Hamrick et al., 1992).
At the population level, genetic variation was relatively low (H S = 0.150) compared with that at the species level, implying that the high level of genetic diversity of E. agallocha populations may be due to genetic divergence of the populations. In most populations, the levels of genetic variation within population were low. Ten out of 13 populations from EIO and 20 out of 32 populations from WPO, for example, showed no nucleotide variation for all sequences studied. Since E. agallocha is a cross-pollinated species, this reduces inbreeding as a cause of low genetic variation. Hence, a plausible explanation is that these local populations may have suffered from bottleneck(s) caused by the climatic oscillations during the Pleistocene. Glaciations, for example, especially the last glaciation and the resulting sea-level fluctuations during this time, are considered to have strongly impacted the coastal mangrove species (Chen et al., 2015). During the last glacial maximum, sea levels fell from + 6 m 120 Kya to approximately—120 m, resulting in the emergence of shelves and the disappearance of relevant coastal areas, e.g., the coastal areas of Hainan disappeared with the emergence of the Sunda Shelf in Southeast Asia, and thus the mangrove forests were mainly restricted to a narrow area on the outer margins of the shelf (Cannon et al., 2009; Woodruff, 2010; Chen et al., 2015). A general pattern of relatively high species-level diversity and relatively low and heterogeneous within-population variation in the cpDNA of E. agallocha seem to reflect the geological changes that the species has gone through in the past.
Population genetic structure
Permutation test (with 1,000 permutations) on the total samples showed that N ST was not significantly greater than G ST, indicating no significant phylogeographic structuring based on cpDNA variation in E. agallocha of IWP. However, genetic variation among populations was strong (G ST = 0.798), indicating that the total genetic diversity was primarily distributed among populations. Obvious genetic structure was also detected in E. agallocha by both AMOVA and haplotype network analysis. Like G ST, AMOVA performed on all sampling locations revealed that more genetic variation existed among population (75.03%) than within populations (24.97%) when regional clustering is being disregarded. This pattern is consistent with those observed in other mangrove species like C. decandra (Tan et al., 2005) and L. racemosa (Su et al., 2006), but in contrast to that observed by Zhang et al. (2008) for the same species using ISSR marker data. Given that cpDNA is haploid, maternally inherited, and evolve relatively slowly (Ye et al., 2014), while nuclear DNA is diploid and biparentally inherited, such inconsistent conclusions, which has been reported in other species (e.g., Feng et al., 2016), can be explained by the different inheritance patterns and evolutionary rates of the markers used.
AMOVA also revealed that a large proportion of variation (56.88%) was due to differences among regions (EIO, WPO, and NA) with significant genetic differentiation coefficient, F CT = 0.569, indicating long-term impediments to gene flow (i.e., seed dispersal) among regional populations. This agreed with previous observation that there was little seed dispersal of E. agallocha between regions (Zhang et al., 2008). Like other mangrove species with water-dispersed propagules, the gene flow (through propagule dispersal) of E. agallocha is largely determined by ocean currents and the species’ life history strategies (Duke, 1992; Dodd et al., 2002; Wee et al., 2014). E. agallocha has small seeds (about 3 mM in diameter) with an air space in the seed coat; these characteristics help the seeds to float and disperse via sea surface currents. Although the longevity of E. agallocha seeds in transit remains unclear, the absence of suitable currents seems more likely to be the main reason for the limited dispersal ability of seeds observed for this species, and the impacts of geographical distance and the presence of major landscape barriers should also be partially responsible.
An indication of isolation-by-distance (IBD) was observed in E. agallocha when taking into account all populations studied, but no such indication was observed when each region was tested separately. Previous findings suggested that genetic structure in mangroves is often more complex than predicted by a simple stepping-stone model (Maguire et al., 2000; Dodd et al., 2002; Ceron-Souza et al., 2015). In combination with the view that tests of IBD may be strongly biased by hierarchical population structure (Meirmans, 2012; Ngeve et al., 2016), the IBD signals observed for E. agallocha here is likely caused by mixed factors from both distance and resistance posed by an effective barrier to gene flow.
Phylogeographic subdivision and demographic history
The network analysis provided a well-resolved phylogenetic relationship of the haplotypes, which suggested three star-like evolutionary units separately dominating three lineages in E. agallocha of the IWP region. While EIO was found to be fixed for a single lineage (group I haplotypes), WPO comprised two lineages: group II haplotypes distributed in east Malay Peninsula, Borneo, and Bali, and group III distributed in Southeast China, Japan, the Philippines, and Indonesia. Populations in NA also had group III haplotypes. This clustering pattern was further supported by the PCoA analysis.
The split between group I and group II haplotypes, or simply the genealogical break across the Malay Peninsula, has been demonstrated in numerous other mangrove species, such as Rhizophora apiculata (Inomata et al., 2009), Bruguiera gymnorrhiza (Urashi et al., 2013), Ceriops decandra (Huang et al., 2008), and Lumnitzera racemosa (Su et al., 2006). During glacial periods in the Pleistocene, the Sundaland was largely exposed due to the lowering of sea levels, forming massive lowland connections between present-day islands in this region (i.e., the Malay Peninsula, Sumatra, Java, and Borneo) and the adjacent continents, impeding genetic exchange between the EIO and WPO regions (Voris, 2000). Our investigation based on Monmonier’s algorithm identified a significant barrier between EIO and WPO, indicating that the Malay Peninsula still acts as one of the most important present-day land barriers separating the Pacific and Indian oceans. Similarly, another barrier identified between WPO and NA could be attributed to the connection of New Guinea and Australia at the Torres Straits during the glacial periods in the Pleistocene (Jennings, 1972).
The closer relationship of group III haplotypes to group II instead of group I haplotypes echoed previous interpretation that mangrove populations in the EIO and WPO were derived from populations in two major refugia—one in the Andaman Sea and another in the South China Sea, during the Last Glacial Maximum (Flenley, 1998; Cannon et al., 2009). Since very different results may be observed when using different markers (Feng et al., 2016), the historical range expansion of E. agallocha in this region cannot be ruled out although no evidence of demographic expansion was observed using cpDNA data in this study. In fact, our additional work (data not shown) on the past range of E. agallocha by ecological niche modeling showed that E. agallocha populations in the region experienced frequent demographic fluctuations until the Last Glacial Maximum period and its range has been slowly expanding after that. Similar scenarios of population contraction and range expansion from a refugium after glaciations have also been frequently proposed or detected in other mangrove and non-mangrove species (Saenger & Bellan, 1995; Maguire et al., 2000; Hewitt, 2000; Nettel & Dodd, 2007; Kennedy et al., 2016).
Aside from the two land barriers mentioned above, three other barriers where no geographical obstacles exist were identified (i.e., the barrier between the Bay of Bengal and west Malay Peninsula in EIO, the barrier separating Southeast China from other populations of the South China Sea in WPO, and the barrier between populations ADR and VT in NA). One of the key forces involved in the dispersal of mangrove propagules is water surface currents. Thus, the lack of suitable ocean currents could be responsible for the genetic discontinuity observed in these regions. In WPO for example, when the seeds of E. agallocha ripen in summer, the surface current of the tropical Indian Ocean flows northward into the South China Sea and then through the Bashi Strait into the Pacific Ocean. The seeds of E. agallocha growing in Malaysia subsequently would have limited chances to disperse into Southeast China but can possibly reach the Philippines and Taiwan. On the other hand, the existence of two opposing currents, a northeastward current that originates from the southeast of the Hainan Island, flows along Guangdong and extends to the Taiwan Strait (Su & Wang, 1987; Hu & Liu, 1992), as well as a westward current which originates from the Kuroshio and intrudes the northern South China Sea through the Luzon Strait (Chan, 1970; Williamson, 1970; Nitani, 1972), provides opportunity for gene flow from Southeast China to Taiwan and from Iriomote Island to the Philippines. The pattern of ocean currents described above provides reasonable explanation for the coexistence of haplotypes C3 and C6 in the Philippines and Taiwan.
This study represents the most complete sampling of E. agallocha throughout their distribution range. In summary, our data based on four distinct cpDNA fragments suggested that E. agallocha displayed high genetic diversity at species level and conspicuous differentiation among the regions EIO, WPO, and NA. Genetic data and AMOVA analysis revealed significant genetic structure among E. agallocha populations. In contrast to findings by Zhang et al. (2008) using nuclear markers, the high cpDNA diversity in E. agallocha was likely due to genetic divergence among populations rather than variation within populations. Studies of genealogy, haplotype distribution, and PCoA suggested the existence of three haplotype groups distributed in distinct geographical locations. Although no compelling evidence for demographic expansion was obtained in the present study, the historical range expansion of E. agallocha cannot be fully ruled out. The genetic discontinuities found between distinct distributional areas of E. agallocha suggested that both land barrier and oceanic currents may have shaped the geographical distribution, genetic structure, and population demography of this species in the IWP region. These findings shall help to better understand the genetic diversity and evolution of mangroves, which would further contribute to the conservation and management of this important group of plants.
References
Ayre, D. J. & S. Dufty, 1994. Evidence for restricted gene flow in the viviparous coral Seriatopora hystrix on Australia’s Great Barrier Reef. Evolution 48: 1183–1201.
Bai, W. N., W. J. Liao & D. Y. Zhang, 2010. Nuclear and chloroplast DNA phylogeography reveal two refuge areas with asymmetrical gene flow in a temperate walnut tree from East Asia. New Phytologist 188: 892–901.
Bandelt, H. J., P. Forster & A. Rohl, 1999. Median-joining networks for inferring intraspecific phylogenies. Molecular Biology and Evolution 16: 37–48.
Cannon, C. H., R. J. Morley & A. B. Bush, 2009. The current refugial rainforests of Sundaland are unrepresentative of their biogeographic past and highly vulnerable to disturbance. Proceedings of National Academy of Sciences of the United States of America 106: 11188–11193.
Ceron-Souza, I., E. G. Gonzalez, A. E. Schwarzbach, D. E. Salas-Leiva, E. Rivera-Ocasio, N. Toro-Perea, E. Bermingham & W. O. McMillan, 2015. Contrasting demographic history and gene flow patterns of two mangrove species on either side of the Central American Isthmus. Ecology and Evolution 5: 3486–3499.
Ceron-Souza, I., E. Bermingham, W. O. McMillan & F. A. Jones, 2012. Comparative genetic structure of two mangrove species in Caribbean and Pacific estuaries of Panama. BMC Evolutionary Biology 12: 205.
Chan, K. M., 1970. The seasonal variation of hydrological properties in the northern South China Sea. In Marr, J. C. (ed.), The Kuroshio – A Symposium on the JAPAN Current. East-West Center Press, Honolulu: 143–162.
Chen, Y., Y. Hou, Z. Guo, W. Wang, C. Zhong, R. Zhou & S. Shi, 2015. Applications of multiple nuclear genes to the molecular phylogeny, population genetics and hybrid identification in the mangrove genus Rhizophora. PLoS ONE 10: e0145058.
Das, A. B., S. Jena, C. Pradhan & P. K. Chand, 2011. Genetic variability among male populations of a minor mangrove Excoecaria agallocha L. as evident by chromosome morphology and DNA markers. Nucleus 54: 39–47.
Dasgupta, N., P. Nandy, C. Sengupta & S. Das, 2015. RAPD and ISSR marker mediated genetic polymorphism of two mangroves Bruguiera gymnorrhiza and Heritiera fomes from Indian Sundarbans in relation to their sustainability. Physiology and Molecular Biology of Plants 21: 375–384.
Dodd, R. S., Z. Afzal-Rafii, N. Kashani & J. Budrick, 2002. Land barriers and open oceans: effects on gene diversity and population structure in Avicennia germinans L. (Avicenniaceae). Molecular Ecology 11: 1327–1338.
Doyle, J. J. & J. L. Doyle, 1990. A rapid total DNA preparation procedure for fresh plant tissue. Focus 12: 13–15.
Duke, N. C., 1992. Tropical mangrove ecosystems. In Robertson, A. I. & D. M. Alongi (eds), Mangrove Floristics and Biogeography. Wiley, Washington DC, USA: 63–100.
Duke, N. C., M. C. Ball & J. C. Ellison, 1998. Factors influencing biodiversity and distributional gradients in mangroves. Global Ecology and Biogeography Letters 7: 27–47.
Duke, N. C., 2013. World Mangrove ID: Expert Information at Your Fingertips. MangroveWatch Publications, Brisbane.
Excoffier, L. & H. E. Lischer, 2010. Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Molecular Ecology Resources 10: 564–567.
Feng, X., Y. Zheng & X. Gong, 2016. Middle-upper pleistocene climate changes shaped the divergence and demography of Cycas guizhouensis (Cycadaceae): evidence from DNA sequences and microsatellite markers. Scientific Reports 6: 27368.
Flenley, J. R., 1998. Tropical forests under the climates of the last 30,000 years. Climatic Change 39: 177–197.
Fu, Y. X., 1997. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 147: 915–925.
Ge, X. J. & M. Sun, 1999. Reproductive biology and genetic diversity of a cryptoviviparous mangrove Aegiceras corniculatum (Myrsinaceae) using allozyme and inter simple sequence repeat (ISSR) analysis. Molecular Ecology 8: 2061–2069.
Hamilton, M. B., 1999. Four primer pairs for the amplification of chloroplast intergenic regions with intraspecific variation. Molecular Ecology 8: 521–523.
Hamrick, J. L., M. J. W. Godt & S. L. Sherman-Broyles, 1992. Factors influencing levels of genetic diversity in woody plant species. New Forests 6: 95–124.
Harpending, H., 1994. Signature of ancient population growth in a low-resolution mitochondrial DNA mismatch distribution. Human Biology 66: 591–600.
Hewitt, G., 2000. The genetic legacy of the Quaternary ice ages. Nature 405: 907–913.
Hu, J. Y. & M. S. Liu, 1992. The current structure during summer in southern Taiwan Strait. Journal of Tropical Oceanography 11: 42–47.
Huang, Y. L., F. X. Tan, G. H. Su, S. L. Deng, H. H. He & S. H. Shi, 2008. Population genetic structure of three tree species in the mangrove genus Ceriops (Rhizophoraceae) from the Indo West Pacific. Genetica 133: 47–56.
Hudson, R. R., 1990. Gene genealogies and the coalescent process. In Futuyma, D. & J. Antonovics (eds), Oxford Surveys in Evolutionary Biology. Oxford University Press, Oxford: 1–44.
Inomata, N., X. R. Wang, S. Changtragoon & A. E. Szmidt, 2009. Levels and patterns of DNA variation in two sympatric mangrove species, Rhizophora apiculata and R. mucronata from Thailand. Genes Genetic Systems 84: 277–286.
Jennings, J. N., 1972. Some attributes of Torres Strait. In Walker, D. (ed.), Bridge and barrier: the natural and cultural history of the Torres Strait. Australian National University of Canberra, Canberra: 29–38.
Jian, S. G., T. Tang, Y. Zhong & S. H. Shi, 2004. Variation in inter-simple sequence repeat (ISSR) in mangrove and non-mangrove populations of Heritiera littoralis (Sterculiaceae) from China and Australia. Aquatic Botany 79: 75–86.
Kennedy, J. P., M. W. Pil, C. E. Proffitt, W. A. Boeger, A. M. Stanford & D. J. Devlin, 2016. Postglacial expansion pathways of red mangrove, Rhizophora mangle, in the Caribbean Basin and Florida. American Journal of Botany 103: 260–276.
Kimura, M. & G. H. Weiss, 1964. The stepping stone model of population structure and the decrease of genetic correlation with distance. Genetics 49: 561–576.
Lakshmi, M., M. Parani, N. Ram & A. Parida, 2000. Molecular phylogeny of mangroves VI. Intraspecific genetic variation in mangrove species Excoecaria agallocha L. (Euphorbiaceae). Genome 43: 110–115.
Liao, P. C., S. Havanond & S. Huang, 2007. Phylogeography of Ceriops tagal (Rhizophoraceae) in Southeast Asia: the land barrier of the Malay Peninsula has caused population differentiation between the Indian Ocean and South China Sea. Conservation Genetics 8: 89–98.
Librado, P. & J. Rozas, 2009. DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25: 1451–1452.
Maguire, T. L., P. Saenger, P. R. Baverstock & R. J. Henry, 2000. Microsatellite analysis of genetic structure in the mangrove species Avicennia marina (Forsk.) Vierh. (Avicenniaceae). Molecular Ecology 9: 1853–1862.
Manni, F., E. Guerard & E. Heyer, 2004. Geographic patterns of (genetic, morphologic, linguistic) variation: how barriers can be detected by using Monmonier’s algorithm. Human Biology 76: 173–190.
Meirmans, P. G., 2012. The trouble with isolation by distance. Molecular Ecology 21: 2839–2846.
Mori, G. M., M. I. Zucchi & A. P. Souza, 2015. Multiple-geographic-scale genetic structure of two mangrove tree species: the roles of mating system, hybridization, limited dispersal and extrinsic factors. PLoS ONE 10: e0118710.
Nettel, A. & R. S. Dodd, 2007. Drifting propagules and receding swamps: genetic footprints of mangrove recolonization and dispersal along tropical coasts. Evolution 61: 958–971.
Ngeve, M. N., V. S. Tom, D. Menemenlis, N. Koedam & L. Triest, 2016. Contrasting effects of historical sea level rise and contemporary ocean currents on regional gene flow of Rhizophora racemosa in Eastern Atlantic mangroves. PLoS ONE 11: e0150950.
Nitani, H., 1972. Beginning of the Kuroshi. In Stommel, H. & K. Yoshida (eds), Kuroshio: Its Physical Aspects. University of Tokyo Press, Tokyo, Japan: 129–163.
Parks, D. H., M. Porter, S. Churcher, S. Wang, C. Blouin, J. Whalley, S. Brooks & R. G. Beiko, 2009. GenGIS: a geospatial information system for genomic data. Genome Research 19: 1896–1904.
Peakall, R. & P. E. Smouse, 2012. GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 28: 2537–2539.
Petit, R. J., J. Duminil, S. Fineschi, A. Hampe, D. Salvini & G. G. Vendramin, 2005. Comparative organization of chloroplast, mitochondrial and nuclear diversity in plant populations. Molecular Ecology 14: 689–701.
Pons, O. & R. J. Petit, 1996. Measuring and testing genetic differentiation with ordered versus unordered alleles. Genetics 144: 1237–1245.
Rousset, F., 1997. Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145: 1219–1228.
Ruan, Y., B. Huang, S. Lai, Y. Wan, J. Li, S. Huang & P. Liao, 2013. Population genetic structure, local adaptation, and conservation genetics of Kandelia obovata. Tree Genetics & Genomes 9: 913–925.
Saenger, P., 1998. Mangrove vegetation: an evolutionary perspective. Marine and Freshwater Research 49: 277–286.
Saenger, P. & M. F. Bellan, 1995. The Mangrove Vegetation of the Atlantic Coast of Africa: A Review. Universite de Toulouse Press, Toulouse, France.
Saintilan, N., N. C. Wilson, K. Rogers, A. Rajkaran & K. W. Krauss, 2014. Mangrove expansion and salt marsh decline at mangrove poleward limits. Global Change Biology 20: 147–157.
Shaw, J., E. B. Lickey, J. T. Beck, S. B. Farmer, W. Liu, J. Miller, K. C. Siripun, C. T. Winder, E. E. Schilling & R. L. Small, 2005. The tortoise and the hare II: relative utility of 21 noncoding chloroplast DNA sequences for phylogenetic analysis. American Journal of Botany 92: 142–166.
Slatkin, M., 1987. Gene flow and the geographic structure of natural populations. Science 236: 787–792.
Small, R. L., J. A. Ryburn, R. C. Cronn, T. Seelanan & J. F. Wendel, 1998. The tortoise and the hare: choosing between noncoding plastome and nuclear Adh sequences for phylogeny reconstruction in a recently diverged plant group. American Journal of Botany 85: 1301–1315.
Su, G. H., Y. L. Huang, F. X. Tan, X. W. Ni, T. Tang & S. H. Shi, 2006. Genetic variation in Lumnitzera racemosa, a mangrove species from the Indo-West Pacific. Aquatic Botany 84: 341–346.
Su, G. H., Y. L. Huang, F. X. Tan, X. W. Ni, T. Tang & S. H. Shi, 2007. Conservation genetics of Lumnitzera littorea (Combretaceae), an endangered mangrove, from the Indo-West Pacific. Marine Biology 150: 321–328.
Su, J. L. & W. Wang, 1987. On the sources of the Taiwan warm current from the South China Sea. Chinese Journal of Oceanology Limnology 5: 299–308.
Sun, M., K. C. Wong & J. S. Y. Lee, 1998. Reproductive biology and population genetic structure of Kandelia candel (Rhizophoraceae), a viviparous mangrove species. American Journal of Botany 85: 1631–1637.
Taberlet, P., L. Fumagalli, A. G. Wust-Saucy & J. F. Cosson, 1998. Comparative phylogeography and postglacial colonization routes in Europe. Molecular Ecology 7: 453–464.
Tajima, F., 1989. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 123: 585–595.
Tan, F. X., Y. L. Huang, X. J. Ge, G. H. Su, X. W. Ni & S. H. Shi, 2005. Population genetic structure and conservation implications of Ceriops decandra in Malay Peninsula and North Australia. Aquatic Botany 81: 175–188.
Tomlinson, P. B., 1986. The Botany of Mangroves. Cambridge University Press, Cambridge, UK.
Triest, L., 2008. Molecular ecology and biogeography of mangrove trees towards conceptual insights on gene flow and barriers: a review. Aquatic Botany 89: 138–154.
Urashi, C., K. M. Teshima, S. Minobe, O. Koizumi & N. Inomata, 2013. Inferences of evolutionary history of a widely distributed mangrove species, Bruguiera gymnorrhiza, in the Indo-West Pacific region. Ecology and Evolution 3: 2251–2261.
Voris, H. K., 2000. Maps of Pleistocene sea levels in Southeast Asia: shorelines, river systems and time durations. Journal of Biogeography 27: 1153–1167.
Waters, J. M., 2008. Marine biogeographical disjunction in temperate Australia: historical landbridge, contemporary currents, or both? Diversity and Distributions 14: 692–700.
Wee, A. K. S., K. Takayama, T. Asakawa, B. Thompson, S. Onrizal, N. X. Sungkaew, M. Tung, K. K. Nazre, H. T. W. Soe, Y. Tan, S. Watano, T. Kajita Baba & E. Webb, 2014. Oceanic currents, not land masses, maintain the genetic structure of the mangrove Rhizophora mucronata Lam. (Rhizophoraceae) in Southeast Asia. Journal of Biogeography 41: 954–964.
Wiley, E. O., 1988. Vicariance biogeography. Annual Review of Ecology Evolution and Systematics 19: 513–542.
Williamson, G. R., 1970. The hydrography and weather of the Hong Kong fishing ground. Hong Kong Fisheries Bulletin 1: 43–49.
Woodruff, D. S., 2010. Biogeography and conservation in Southeast Asia: how 2.7 million years of repeated en environmental fluctuations affect today’s patterns and the future of the remaining refugial-phase biodiversity. Biodiversity and Conservation 19: 919–941.
Ye, C., Z. Lin, G. Li, Y. Wang, J. Qiu, F. Fu, H. Zhang, L. Chen, S. Ye, W. Song, G. Jin, J. Zhu, Y. Lu, L. Guo & L. Fan, 2014. Echinochloa chloroplast genomes: insights into the evolution and taxonomic identification of two weedy species. PLoS ONE 9: e113657.
Zhang, Z. H., R. C. Zhou, T. Tang, Y. L. Huang, Y. Zhong & S. H. Shi, 2008. Genetic variation in central and peripheral populations of Excoecaria agallocha from Indo-West Pacific. Aquatic Botany 89: 57–62.
Acknowledgements
This study is supported by grants from the National Natural Science Foundation of China (Grant No.41776166 and 31700178), the Natural Science Foundation of Guangdong Province (Grant No. 2015A030313136 and 2017A030313159), National Key Research and Development Program of China (Grant No. 2017FY100705), Science and Technology Program of Guangzhou (Grant No. 201707020035), and Chang Hungta Science Foundation of Sun Yat-sen University. The authors wish to thank Dr. Sirajul Haque, Dr. Kalimuddin Bhuiyan, and Dr. Mosharraf Hossen (Institute of Forestry and Environmental Sciences, Chittagong University, Chittagong, Bangladesh); Dr. Sonjai Havanond (Department of National Parks, Wildlife and Plant Conservation, Bangkok, Thailand); Mr. Chanop Jaengjai (Department of Marine and Coastal Resources, Phetchaburi, Thailand); Ms. Jayasri Lakshminarayanan and Mr. Koh Kwan Siong (National Biodiversity Centre, Singapore); Dr. Yubin Yan (the University of Hong Kong, Hong Kong, China); and Dr. Xinnian Li, Dr. Ying Liu, Dr. Suhua Shi, and Dr. Zihong Zhang (Sun Yat-sen University, China) for their assistance in collecting and providing plant material.
Author information
Authors and Affiliations
Corresponding author
Additional information
Handling editor: Ken Krauss
Rights and permissions
About this article

Cite this article
Guo, W., Ng, W.L., Wu, H. et al. Chloroplast phylogeography of a widely distributed mangrove species, Excoecaria agallocha, in the Indo-West Pacific region. Hydrobiologia 807, 333–347 (2018). https://doi.org/10.1007/s10750-017-3409-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10750-017-3409-7