
Abstract
Soil salinity is a major constraint to rice production. Na+ and K+ concentrations and ion balance play important roles in the salt tolerance of rice. In the present study, linkage mapping and association mapping were used to identify the QTLs for the visual tolerance score (SES), and the concentrations of Na+ and K+ in shoots (SNC and SKC) and roots (RNC and RKC). A BC2F2:3 population with 137 SSR markers derived from Dongnong425 (a salt-sensitive and widely cultivated variety) as the recurrent parent and Changbai10 (a salt-tolerant variety) as the donor parent was used for linkage mapping. A total of 13 QTLs were identified by the inclusive composite interval mapping method, including 2 for SES, 4 for SNC, 3 for SKC, 3 for RNC, and 1 for RKC. This study was supplemented with association mapping, which was conducted using a panel of 341 japonica rice accessions from different geographical origins with 160 selected SSR markers. A total of 24 significant marker-trait associations (P ≤ 0.01) involving 20 markers were identified using the GLM (Q) and MLM (Q+K) models in TASSEL2.1. Among them, 10 of the SSR markers confirmed or narrowed the genomic regions for salt tolerance that were reported in linkage studies, including six QTLs identified during the present study. The QTLs identified through linkage and association mapping may be useful for marker-assisted selection in rice breeding programs and may accelerate the development of salt-tolerant rice varieties.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Rice (Oryza sativa L.) is a staple food for more than half of the world’s population, and soil salinity is a major abiotic stress that limits rice production in approximately 30 % of the rice growing area worldwide (Wu and Garg 2003). Salinity occurs in coastal areas, river delta soils and former flood plains around the world (Yeo et al. 1990), and the problem of salinity is expected to increase in the future due to global warming (Wassmann et al. 2004). In these particular areas, rice is one of the few crops that can thrive on salt-affected soils because of its ability to grow well in standing water, which can help leach salts from topsoil. Rice is also recommended as an entry crop for the desalinization of salt-affected lands (Ismail 2007; Singh et al. 2010). Therefore, understanding the genetic mechanisms controlling salt tolerance can enhance efforts to develop tolerant cultivars; the identification of QTLs related to tolerance can subsequently be integrated into rice breeding programs. To date, high genetic variability has been reported in salt-tolerant rice varieties (Gregorio et al. 2002). The most famous tolerant varieties, Pokkali and Nona Bokra, belong to the indica subspecies (Yeo et al. 1990), varieties of which have been used as the source of salt tolerance in almost all the studies published to date. On the other hand, little is known about the genetic variation for salt tolerance among japonica subspecies.
NaCl is a major constituent of saline soil. Sodium ions (Na+) are harmful to plants, whereas potassium ions (K+) are essential to reduce the uptake of Na+ (Wu et al. 2009). Thus, Na+ and K+ concentrations and ion balance play important roles in the salt tolerance of rice. With the development of modern molecular biology techniques, a large number of quantitative trait loci (QTLs) associated with the Na+ and K+ concentrations within roots and shoots have been identified (Prasad et al. 2000; Flowers et al. 2000; Koyama et al. 2001; Lin et al. 2004; Lee et al. 2007; Sabouri et al. 2009; Haq et al. 2010). For example, 11 QTLs for survival days of seedlings and the Na+ and K+ concentrations were detected on chromosomes 1, 4, 6, 7 and 9 using the Nona Bokra × Koshihikari mapping population. Two major QTLs for shoot Na+ content (qSNC-7) and shoot K+ content (qSKC-1) were found, which explained 48.5 and 40.1 % of the total phenotypic variance, respectively (Lin et al. 2004). Ammar et al. (2009) reported 25 QTLs for salt ion concentrations on rice chromosomes 1, 2, 3 and 8. Bonilla et al. (2002) mapped the SALTOL locus, which is linked to QTLs for Na+ and K+ uptake and Na+/K+ ratio, on chromosome 1. Pandit et al. (2010) reported eight QTLs for salt ion concentrations on rice chromosomes 1, 8 and 12, and Cheng et al. (2011) reported 12 QTLs for salt ion concentrations on rice chromosomes 1, 2, 3, 4, 7 and 11. In these studies, different populations and statistical approaches were used to discover the QTLs associated with salt tolerance. However, a few QTLs were uncovered in single populations, and they differed from those from the other populations because of their diverse parents. These populations represented a small fraction of tolerant rice genotypes, and the observed transgressive segregation was indicative of additive and epistatic relationships among the alleles of only two parents in a segregating population (Holland 2007).
Association mapping, an alternative to linkage analysis, has been widely used in maize, Arabidopsis, rice, wheat, and soybean (Weng et al. 2011; Li et al. 2010; Courtois et al. 2013; Le Gouis et al. 2012; Niu et al. 2013). Compared with QTL analysis, association analysis significantly increases the range of variation that can be investigated in a single experiment because of the high degree of natural variation caused by accumulated historic recombination events in natural populations. Additionally, association mapping has the ability to identify multiple alleles at genetic loci in germplasm and also provides higher resolution than QTL analysis (Famoso et al. 2011). However, the power of association analysis is complicated by the population structure present in germplasm sets (Flint-Garcia et al. 2003). Furthermore, association mapping fails to identify rare alleles due to limited statistical power (Lewis 2002).
The combination of linkage analysis and association mapping can provide both the power and resolution needed to detect QTLs of interest and may prove more successful than either strategy alone. For instance, Brachi et al. (2010) validated association peaks detected for flowering time in Arabidopsis thaliana when QTLs detected by linkage mapping co-localized with these peaks. Similarly, combined linkage analysis and association mapping made the fine mapping of QTLs in rice (Famoso et al. 2011) and wheat (Mir et al. 2012) possible. Therefore, this combined approach in a single experiment will provide sufficient power for the detection of QTLs and facilitate marker-assisted breeding programs. To our knowledge, although there have been extensive studies on QTL mapping for salinity tolerance in rice, few studies have used association mapping strategies to identify QTLs associated with salt tolerance in japonica rice. Furthermore, no studies have been reported that have examined linkage and association analyses at the same time in a single experiment. In the present study, the QTLs for Na+ and K+ concentrations in shoots and roots under NaCl stress were identified based on linkage and association analysis. The results of linkage mapping in the present and previous studies were compared with the results of whole-genome association mapping. The objective of our study was to identify the QTLs that were detected either by both approaches or by earlier linkage mapping, which will provide useful information to improve salt tolerance in rice by marker-assisted selection (MAS).
Materials and methods
Plant materials
Linkage analysis was conducted on 190 BC2F2 individuals developed from a cross between Changbai10 (a salt-tolerant japonica rice) as the donor parent and Dongnong425 (a salt-sensitive and widely cultivated japonica rice) as the recurrent parent. For the association analysis, 341 japonica rice accessions were used representing the genetic diversity among different heterotic groups. They were selected from the Heilongjiang, Jilin and Liaoning provinces in China and other countries including Japan, Korea, the Democratic People’s Republic of Korea, Russia and France. Both parents from the linkage analysis were included in the association panel. These accessions were collected from the Crop Science Research Institute, Chinese Academy of Agricultural Sciences, Liaoning Academy of Agricultural Sciences, Heilongjiang Academy of Agricultural Sciences and Northeast Agricultural University (Supplementary Table 1).
Evaluation of salt tolerance
The evaluation of the rice for salt tolerance was conducted in a hydroponic solution at Northeast Agriculture University’s experimental station in 2013. Trials with the BC2F2:3 population and the association panel were laid out in a randomized complete-block design with three replications. Samples of 50 healthy grains of each accession were placed at 50 °C for 3 days to break any possible dormancy, surface-sterilized with 1 % sodium hypochlorite solution for 10 min, and rinsed three times with sterile distilled water. The seeds were soaked in distilled water at 30 °C for 3 days to allow the seeds to germinate. The 20 most uniform germinated seeds were sown in holes in a thin Styrofoam board with a nylon net bottom, which floated on distilled water in a plastic tray until the two leaf stage. The water was then replaced by 15 L of nutrient solution (Yoshida et al. 1976) for 7 days. For salt treatment, the nutrient solution was replaced by a fresh solution containing 50 mmol/L NaCl for 3 days, after which the NaCl concentration was increased to 140 mmol/L. The seedlings were grown in a phytotron with 28/21 °C day/night temperatures, natural sunlight and 70 % RH. The solution was changed every 5 days, and the pH was adjusted daily to 5.0 by adding either 1 M NaOH or 1 M HCl (Wang et al. 2012a, b). After 12 days of salt stress, samples of 10 plants per line/accession were scored based on visual symptoms using IRRI’s SES for rice, with ratings from 1 (highly tolerant) to 9 (highly sensitive; IRRI 1996) (Thomson et al. 2010). The shoots and roots were then harvested, and the roots were rinsed with distilled water several times. The shoots and roots were dried at 80 °C for 48 h, weighed and extracted in acetic acid (100 mmol/L) at 90 °C for 2 h. The concentrations of Na+ and K+ in the shoots (SNC and SKC) and roots (RNC and RKC) were analyzed using a Flame Photometer (Sherwood410, Cambridge, UK).
Population genotyping with SSR markers
Total genomic DNA was extracted and purified from the young leaves using a modified CTAB method (Doyl and Doyle 1990). One thousand simple sequence repeat (SSR) primer pairs were designed from SSR-containing sequences in the rice genome, which was retrieved from www.gramene.org. The primers were synthesized by Sangon Biotech Co., Ltd. (Shanghai). For the BC2F2:3 population, 137 SSR markers that were polymorphic between the two parents were used to genotype the mapping population. For the association mapping, 160 SSR markers were used to genotype the panel of japonica rice, including 10 QTL-flanking markers from the present linkage map and 25 markers known to be linked to salt-tolerant QTLs from previous studies (Ammar et al. 2009; Mohammadi-Nejad et al. 2008; Sabouri et al. 2009; Thomson et al. 2010; Wang et al. 2012a, b).
The PCR reactions were conducted in 10 μL volumes containing 1 μL of genomic DNA (25 ng/μL), 0.75 μL of MgCl2 (25 mM), 0.15 μL of dNTP mixture (10 mM), 1 μL of 10 × PCR buffer, 1 μL of a SSR primer pair (2 μM), 0.1 μl of Taq polymerase (10 U/μL), and 6 μL of ddH2O. The PCR amplification profile was 94 °C for 2 min, followed by 35 cycles of 94 °C for 30 s, 47 °C for 30 s, and 72 °C for 30 s, with a final extension at 72 °C for 5 min. The PCR products were mixed with loading buffer (2.5 mg/ml bromophenol blue, 2.5 mg/ml diphenylamine blue, 10 mM EDTA, and 95 % formamide), denatured at 94 °C for 5 min, and then incubated on ice for 5 min. The denatured PCR products were separated on a 6 % denaturing polyacrylamide gel and directly detected by silver straining.
Statistical analysis
Phenotypic data analysis
The means, variances and correlation coefficients among the different salt tolerance parameters were determined using SPSS 13.0 for Windows (SPSS, Inc., Chicago, IL, USA). The broad-sense heritability (H 2) was calculated and expressed as the ratio of the total genetic variance (V G ) over the phenotypic variance (V P ): H 2 = V G /V P .
Construction of a genetic linkage map and QTL mapping
Physical maps obtained through BLAST searches or found in the Gramene Genome Browser (IRGSP 2005; www.gramene.org) were used as a reference to establish marker order. The recombination distances between the SSR markers were determined based on segregation analysis in the BC2F2 generation using JoinMap 3.0 (Ooijen and Voorrips 2002). The genetic distances were calculated in Kosambi centiMorgans (cM). A set of 137 SSR markers was constructed, covering 12 chromosomes and spanning 1742.4 cM of the rice genome with an average interval of 12.72 cM between the adjacent markers. The QTL analysis was conducted on the arithmetic mean values from three replicates for each trait and each family by the inclusive composite interval mapping (ICIM) method described by Li et al. (2007). The LOD score for declaring a QTL was 2.5 for each trait, and the walking speed was 1 cM. The method developed by McCouch et al. (1997) was used to name the QTLs, which were mapped on linkage groups using MapChart 2.2 (Voorrips 2002).
Association mapping
The marker loci with an allele frequency of less than 0.05 in the population were filtered and grouped together with the missing alleles in the population structure and LD analyses. The pairwise estimates of LD (r 2) based on 160 SSR loci were calculated using the TASSEL 2.1 software (Bradbury et al. 2007).The population structure was analyzed using the STRUCTURE program (Pritchard et al. 2000) with the admixture model. 55 unlinked SSR loci located over the 12 rice chromosomes were selected to analyze the population structure. 10 independent runs were performed at K levels, ranging from 1 to 10. The burn-in period was set at 10,000, and the number of iterations was 100,000. The likely number of sub-populations present was estimated following Evanno et al. (2005), where ΔK = m(|L(k + 1) − 2 L(k) + L(k − 1)|)/s[L(k)], where L(k) represents the kth LnP(D), m is the mean of 10 runs and s is their standard deviation. The Q values corresponding to the highest ΔK were adopted for the association analysis. The program SPAGeDi (Hardy and Vekemans 2002) was used to calculate the relative kinship matrix (K).The association mapping analyses were performed using both the general linear model (GLM, Q) and the mixed linear model (MLM, Q+K) functions in the TASSEL 2.1 software (Bradbury et al. 2007). In this analysis, we considered the Q matrix as a fixed effect and the K matrix as a random effect. The markers were considered to be significantly associated with traits if the P value was less than 0.01.
Results
Salt tolerance phenotypes
The statistical analyses of the phenotypic traits related to salt tolerance at the seeding stage for the parents, BC2F3 lines and 341 accessions are summarized in Table 1. There were significant differences in the SES, SNC, and SKC, but no significant difference in the RNC and RKC, between the two parents. The salt-tolerant Changbai10 had lower SES and SNC but higher SKC values than the sensitive Dongnong425. In both the BC2F3 lines and the rice accessions, the Na+ and K+ concentrations in the shoots were higher than in the roots under NaCl stress. All the parameters exhibited a normal distribution and significant transgressive segregations. The broad-sense heritability for all five traits was very high. The heritabilities of SES, SNC, SKC, RNC and RKC were variable in both populations, ranging from 75.3 to 92.2 % in the BC2F3 lines and from 76.3 to 94.2 % in the 341 japonica rice accessions.
Correlation of Na+ and K+ in the shoots and roots
The correlation coefficients between the SES and the four physiological traits in the shoots and roots under salt stress in the BC2F3 lines and 341 japonica rice accessions are shown in Table 2. The correlation analyses showed that the significance of correlation among the five traits was nearly identical in the two populations. The SES was only significantly positively correlated with SNC, while SNC was significantly negatively correlated with SKC. RNC was significantly positively correlated with RKC. The correlation among other traits showed no significance.
Linkage analysis
In the BC2F2:3 population, a total of 13 QTLs were detected for the five salt-tolerance-related traits using the ICIM method (Table 3; Fig. 1). These QTLs were distributed over chromosomes 1, 2, 5, 6, 7, 9, 11 and 12. The explanation for the phenotypic variation by a single QTL varied from 6.45 to 17.95 %. Of the 13 QTLs, two were associated with SES and explained 11.25 and 7.95 % of the phenotypic variance, with LOD scores of 4.05 and 3.12, respectively. The positive alleles of these QTLs were contributed by Dongnong425. Four QTLs were associated with SNC and explained 9.26 to 17.95 % of the phenotypic variance, with LOD scores ranging from 2.87 to 3.83. The positive alleles of qSNC-1, qSNC2-1 and qSNC-11 were contributed by Dongnong425, and the positive allele of qSNC-12 was contributed by Changbai10. Three QTLs were associated with SKC and explained 8.42 to 13.52 % of the phenotypic variance, with LOD scores ranging from 2.82 to 3.22. The positive alleles of these QTLs were contributed by Changbai10. Three QTLs were associated with RNC, with LOD scores ranging from 2.58 to 4.13; these QTLs were distributed over chromosomes 6, 7 and 9 and explained 6.45 to 16.58 % of the phenotypic variance. The positive alleles of qRNC-6 and qRNC-9 were contributed by Changbai10, and the positive allele of qRNC-7 was contributed by Dongnong425. Only one QTL was associated with RKC; this QTL was located on chromosome 6 and had a LOD score of 2.83 that explained 10.10 % of the phenotypic variance. Furthermore, qSES-2, qSNC-2 and qSKC-2 were co-localized in the RM1285 ~ RM423 marker interval, while qRNC-6 and qRKC-6 were located in an adjacent genomic region.

Locations of the QTLs for salt tolerance indices on linkage groups by ICIM. Each QTL interval is shown as a filled rectangle. The name of the QTL are shown on the left, and the marker loci are indicated on the right
Association mapping
Linkage disequilibrium
A total of 5094 SSR marker pairs had a significant LD (P < 0.01), representing 40.05 % of the pairwise comparisons between 160 polymorphic SSR marker loci. The distribution of data points in the plot of LD decay (threshold r 2 = 0.1) against distance (cM) for the significant pairs showed that the LD was not a simple monotonic function of the distance between markers; rather, it occurred between proximal marker loci and occasionally distant marker loci (Fig. 2). The LD rapidly decayed to a basal value at distances around 20 cM, suggesting it should be possible to achieve resolution down to the 20 cM level.

The pattern of LD for 160 SSR loci indicating the correlation values of the allele frequencies (r 2) against the genetic distance (cM) between all the loci pairs. LD decay was determined based on a threshold of r 2 < 0.1. A genetic distance of 250 cM was chosen to represent unlinked loci on different chromosomes. The LD decayed at approximately 20 cM
Population structure
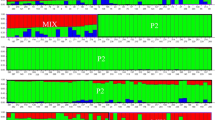
The population structure of the 341 accessions was calculated based on 55 unlinked SSR markers. The results showed a steadily increasing curve for LnP(D), indicating that the posterior probabilities for the number of subpopulations increased steadily. The method of Evanno et al. (2005) indicated that the value of ΔK was the highest at K = 3 (Fig. 3). Therefore, these accessions could be grouped into three subpopulations.

Estimated LnP(D) and ΔK over 10 repeats of STRUCTURE analysis a LnP(D) for K from 1 to 10 for 341 accessions. LnP(D) value of each hypothetical K kept increasing with K value and did not show any peak. b ΔK for K from 2 to 10 for 341 accessions. The ΔK value showed a highest likelihood at K = 3, suggesting that the total panel should be divided into three major subpopulations
Estimation of relative kinship
Relative kinship analysis of the 341 accessions showed that a total of 57.4 % of the pair-wise kinship estimates were equal to zero, and the remaining estimates decreased continuously with the increase in the number of categories, indicating that most accessions had a weak or no relationship with each other in this study. The kinship estimates indicated that a few accessions showed strong similarities, which suggested that the simple relatedness among accessions will decrease the frequency of spurious associations because of the effect of the population structure. Thus, the japonica rice panel was suitable for association mapping (Fig. 4).

Distribution of the pair-wise relative kinship estimates between 341 japonica rice accessions
Association between salt tolerance-related traits and SSR markers
During the present study, both whole-genome association and targeted regional association mapping were conducted. The main focus was on some important genomic regions that were identified through linkage mapping during the present study (see Table 3) and in other earlier studies conducted on SES, SNC, SKC, RNC and RKC. A total of 24 marker-trait associations (MTAs) were identified (P ≤ 0.01) with 20 different SSR markers (Table 4), and the R2 ranged from 4.31 to 12.35 %. Among these MTAs, 4 markers were associated with SES, 6 with SNC, 5 with SKC, 5 with RNC, and 4 with RKC. Four markers (RM1287, RM1350, RM226, and RM215) were co-associated with two or more traits. 10 SSR markers confirmed or narrowed the genomic region reported to harbor QTLs for salt tolerance by linkage studies, including 6 identified during the present study (Table 4).
Discussion
The rationality for the tested population in linkage and association mapping
Advanced backcross quantitative trait locus (AB-QTL) analysis was first introduced to simultaneously identify favorable QTL alleles and transfer those alleles from donor lines to the genomes of elite cultivars (Tanksley and Nelson 1996). In this study, we developed a population including 190 BC2F2 families using Changbai10 (a salt-tolerant variety) as the donor parent and Dongnong425 (a high-yield and high-quality variety) as the recurrent parent. Each family carried a small chromosomal segment from Changbai10, and the genetic background was basically consistent with Dongnong425. This approach can effectively eliminate genetic background variation, improve the accuracy of QTL detection, and allow rapid development of near isogenic lines (NILs) containing the QTL of interest for further genetic research (Xie et al. 2006). In addition, the comprehensive characteristics of the advanced backcross families were very superior, and this approach allowed the breeding of new lines while conducting the QTL mapping. Therefore, QTL mapping and practical breeding can be performed simultaneously, eventually improving the efficiency of breeding.
The association mapping panel in this study was a collection of 341 japonica rice accessions released over many years in China, Japan, Korea, the Democratic People’s Republic of Korea, Russia, and France representing elite lines bred for genes and QTL alleles for yield and abiotic stress resistance. The accessions varied widely in Na+ and K+ concentrations and exhibited high heritability, indicating good control of random errors during phenotyping. Furthermore, the relatively large sample size used for association mapping was necessary to improve population differentiation (Rosenberg et al. 2001), increase detection, and allow the determination of more alleles. Therefore, the japonica rice varieties in this study could provide the sufficient phenotypic and genotypic diversity suitable for association mapping.
Estimates of LD are important to determine the appropriate association mapping approach. If LD declines rapidly, the genome scan would require excessive marker density for a feasible identification of the candidate genes. If LD is too large, the resolution may be low, but a genome scan with a lower marker density would be possible (Garris et al. 2003). In this study, extensive LD was detected among both syntenic and nonsyntenic markers; the percentage of significant marker pairs in LD (P < 0.01) accounted for 40.05 % of the total loci pairs. The LD rapidly decayed to a basal value of r 2 = 0.1 at distances around 20 cM, suggesting that a mapping resolution of 20 cM could possibly be achieved. This observed pattern of LD is similar to that reported in a germplasm collection of worldwide accessions (Agrama et al. 2007) and another collection consisting mostly of Chinese accessions (Jin et al. 2010). The extent of LD determines the number of markers required to cover the genome and consequently the resolution level of the mapping. For the approximately 389 Mb rice genome, the 154 SSRs that cover the rice genome at a density of approximately 10 cM (assuming an average of 250 kb/cM across the genome) provided a reasonable resolution for the association mapping in this study.
Distribution of the QTLs and the relationship between Na+ and K+
The QTLs for correlated traits often cluster in the same chromosomal regions. This trend was observed in this study. For example, qSES2, qSNC2 and qSKC2 co-localized to RM1285 ~ RM423 based on the linkage mapping (Table 3). RM1287 on chromosome 1 was associated with SNC and SKC based on the association mapping, and RM586 on chromosome 6 was associated with RNC and RKC (Table 4). These traits were highly correlated. These results suggest that the trait correlation may be due to the effect of pleiotropy or to the very close linkage of these genes.
The processes of Na+ and K+ uptake in rice are considered to occur in parallel and not in direct competition during salt stress. This view is supported by the results of this study, which showed a significant positive correlation between RNC and RKC (r 2 = 0.338** and 0.531** in the two populations). However, there was a significant negative correlation between SNC and SKC (r 2 = −0.355** and −0.341** in the two populations), suggesting that competition occurred when Na+ and K+ in the roots were transported to the shoots. In this study, the QTLs detected in seedling shoots and roots were quite different. The QTLs for SNC and RNC did not map to the same locations as the QTLs for SKC and RKC. These results also suggest that the loci controlling the transport of the two ions, Na+ and K+, between the shoots and the roots of seedlings may be different or induced unequally by salt stress.
Combination of linkage and association analysis
The QTL mapping strategy of linkage mapping combined with association mapping has the potential of linkage mapping to locate the QTL positions in respective linkage groups with the added advantage of association mapping to find more loci and alleles. Thus, there is the potential to use each method simultaneously to verify the results of the other method. Due to the predicted high rate of false discovery in association mapping, it is important to test the markers identified through association mapping using linkage mapping for consistency. However, one advantage of the association mapping study is that it allowed a large number of recombination events to be sampled among the natural populations As a result, more novel genetic factors were discovered, and the resolution of those QTLs identified in association mapping was likely better than that of the QTLs identified in the linkage mapping studies.
In the present study, we report the results of whole-genome association mapping, which also includes targeted association mapping of those genomic regions that were identified by linkage mapping during the present and previous studies. Targeted association mapping for different traits has also been conducted previously (Breseghello and Sorrells 2006; Zheng et al. 2012). The targeting of selected markers linked with QTLs in previous studies was found to improve the power of association mapping studies to confirm or identify more closely linked markers of important QTLs (Breseghello and Sorrells 2006). In this study, the combination of linkage and association mapping identified 10 markers, RM1287, RM423, RM229, RM1340, RM560, RM292, RM1371, RM215, RM1350, and RM226, that were linked to salt tolerance in rice.
Comparison with previous reports
The MTAs identified by association mapping in this study were compared with the QTLs identified by linkage mapping in the present and previous studies, according to the same SSR markers and physical location of markers linked with the QTLs. Several salt-tolerance-related QTLs or the candidate genes whose functions have been known to affect salinity tolerance in previous studies were found to coincide with the QTLs identified in this study (Table 4). Seven QTLs (including 6 markers) identified by linkage mapping in our study were confirmed by the association mapping, and 5 SSR markers were also consistent with previous QTL reports. For instance, RM1287 was located within the Saltol segment on chromosome 1 (Bonilla et al. 2002) and was close to the gene SKC1 (Ren et al. 2005), which encoded a member of HKT-type transporters. RM560 on chromosome 7 was the same marker for three QTLs (qRRW7, qRSW7, and qRTW7) reported by Tian et al. (2011). Similarly, RM423, RM229, and RM1340 also mapped to the genomic region identified by Ammar et al. (2009), Rana et al. (2009), and Thomson et al. (2010), and RM1340 was co-located at the gene OsiSAP8 (Kanneganti and Gupta 2008), which encoded a zinc finger A20 and AN1 domain-containing stress-associated protein. RM215, which was identified by both mapping approaches, might be a novel marker associated with salt tolerance because there have been no reports to date of this marker in studies of salt tolerance. In addition, four markers (RM292, RM1371, RM1350, and RM226) identified by association mapping in this study were also mapped within the QTL interval for salt tolerance reported by earlier studies (Pandit et al. 2010; Sabouri et al. 2009; Lin et al. 2004). Among them, RM1350 was close to the gene OsBIHD1 (Luo et al. 2005), which encoded a homeodomain protein, and RM226 was co-located at the gene ZFP179 (Sun et al. 2010), which encoded a Cys2/His2-type zinc finger protein. Therefore, the markers for the salt-tolerance-related traits mentioned above, which were detected in different mapping populations and various environments, were significant markers for salt tolerance in rice. Furthermore, the markers that were localized within a QTL interval for salt tolerance not only validated the QTL but also provided a more closely linked marker. These markers may be useful for breeding programs in rice based on MAS and may accelerate the development of salt tolerant rice varieties.
References
Agrama HA, Eizenga GC, Yan W (2007) Association mapping of yield and its components in rice cultivars. Mol Breed 19:341–356
Ammar MHM, Pandit A, Singh RK, Sameena S, Chauhan MS, Singh AK, Sharma PC, Gaikwad K, Sharma TR, Mohapatra T, Singh NK (2009) Mapping of QTLs controlling Na+, K+ and Cl− ion concentrations in salt tolerant Indica rice variety CSR27. J Plant Biochem Biotechnol 18:139–150
Bonilla P, Mackell D, Deal K, Gregorio G (2002) RFLP and SSLP mapping of salinity tolerance genes in chromosome 1 of rice (Oryza sativa L.) using recombinant inbred lines. Philipp Agric Sci 85:68–76
Brachi B, Faure N, Horton M, Flahauw E, Vazquez A, Nordborg M, Bergelson J, Cuguen J, Roux F (2010) Linkage and association mapping of Arabidopsis thaliana flowering time in nature. PLoS Genet 6:e1000940
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635
Breseghello F, Sorrells ME (2006) Association mapping of kernel size and milling quality in wheat (Triticum aestivum L.) cultivars. Genetics 172:1165–1177
Cheng LR, Wang Y, Meng LJ, Hu X, Cui YR, Sun Y, Zhu LH, Ali J, Xu JL, Li ZK (2011) Identification of salt-tolerant QTLs with strong genetic background effect using two sets of reciprocal introgression lines in rice. Genome 55:45–55
Cheng YW, Qi YC, Zhu Q, Chen X, Wang N, Zhao X, Chen HY, Cui XJ, Xu L, Zhang W (2009) New changes in the plasma-membrane-associated proteome of rice roots under salt stress. Proteomics 9:3100–3114
Courtois B, Audebert A, Dardou A, Roques S, Ghneim-Herrera T, Droc G, Frouin J, Rouan L, Goze E, Kilian A, Ahmadi N, Dingkuhn M (2013) Genome-wide association mapping of root traits in a japonica rice panel. PLoS One 11:e78037
Doyl JJ, Doyle JL (1990) Isolation of plant DNA from fresh tissue. Focus 12:13–15
Dubouzet JG, Sakuma Y, Ito Y, Kasuga M, Dubouzet EG, Miura S, Seki M, Shinozaki K, Yamaguchi-Shinozaki K (2003) OsDREB genes in rice, Oryza sativa L., encode transcription activators that function in drought-, high-salt- and cold-responsive gene expression. Plant J 33:751–763
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Famoso AN, Zhao K, Clark RT, Tung CW, Wright MH, Bustamante C, Kochian LV, McCouch SR (2011) Genetic architecture of aluminum tolerance in rice (Oryza sativa) determined through genome-wide association analysis and QTL mapping. PLoS Genet 7:e1002221
Flint-Garcia SA, Thornsberry JM, Bucker ES (2003) Structure of linkage disequilibrium in plants. Annu Rev Plant Biol 54:357–374
Flowers TJ, Koyama ML, Flowers SA, Sudhakar C, Singh KP, Yeo AR (2000) QTL: their place in engineering tolerance of rice to salinity. J Exp Bot 51:99–106
Garris AJ, McCouch SR, Kresovich S (2003) Population structure and its effect on haplotype diversity and linkage disequilibrium surrounding the xa5 locus of rice (Oryza sativa L.). Genetics 165:759–769
Gregorio GB, Senadhira D, Mendoza RD, Manigbas NL, Roxas JP, Guerta CQ (2002) Progress in breeding for salinity tolerance and associated abiotic stresses in rice. Field Crop Res 76:91–101
Haq TU, Gorham J, Akhtar J, Akhtar N, Steele KA (2010) Dynamic quantitative trait loci for salt stress components on chromosome 1 of rice. Funct Plant Biol 37:634–645
Hardy OJ, Vekemans X (2002) SPAGEDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes 2:618–620
Holland JB (2007) Genetic architecture of complex traits in plants. Curr Opin Plant Biol 10:156–161
International Rice Genome Sequencing Project (IRGSP) (2005) The map-based sequence of the rice genome. Nature 436:793–800
IRRI (1996) Standard evaluation system for rice, 4th edn. International Rice Research Institute, Manila, p 52
Ismail AM, Heuer S, Thomson MJ, Wissuwa M (2007) Genetic and genomic approaches to develop rice germplasm for problem soils. Plant Mol Biol 65:547–570
Jin L, Lu Y, Xiao P, Sun M, Corke H, Bao J (2010) Genetic diversity and population structure of a diverse set of rice germplasm for association mapping. Theor Appl Genet 121:475–487
Kanneganti V, Gupta AK (2008) Overexpression of OsiSAP8, a member of stress associated protein (SAP) gene family of rice confers tolerance to salt, drought and cold stress in transgenic tobacco and rice. Plant Mol Biol 66:445–462
Koyama ML, Levesley A, Koebner RM, Flowers TJ, Yeo AR (2001) Quantitative trait loci for component physiological traits determining salt tolerance in rice. Plant Physiol 125:406–422
Le Gouis J, Bordes J, Ravel C, Heumez E, Faure S, Praud S, Galic N, Remoue C, Balfourier F, Allard V, Rousset M (2012) Genome-wide association analysis to identify chromosomal regions determining components of earliness in wheat. Theor Appl Genet 124:597–611
Lee SY, Ahn JH, Cha YS, Yun DW, Lee MC, Ko JC, Lee KS, Eun MY (2007) Mapping QTLs related to salinity tolerance of rice at the young seedling stage. Plant Breed 126:43–46
Lewis CM (2002) Genetic association studies: design, analysis and interpretation. Brief Bioinform 3:146–153
Li HH, Ye GY, Wang JK (2007) A modified algorithm for the improvement of composite interval mapping. Genetics 175:361–374
Li Y, Huang Y, Bergelson J, Nordborg M, Borevitz JO (2010) Association mapping of local climate-sensitive quantitative trait loci in Arabidopsis thaliana. Proc Natl Acad Sci 107:21199–21204
Lin HX, Zhu MZ, Yano M, Gao JP, Liang ZW, Su WA, Hu XH, Ren ZH, Chao DY (2004) QTLs for Na+ and K+ uptake of the shoots and roots controlling rice salt tolerance. Theor Appl Genet 108:253–260
Luo HL, Song FM, Zheng Z (2005) Overexpression in transgenic tobacco reveals different roles for the rice homeodomain gene OsBIHD1 in biotic and abiotic stress responses. J Exp Bot 56:2673–2682
McCouch SR, Cho YG, Yano M, Paul E, Blinstrub M, Morishima H, Kinoshita T (1997) Report on QTL nomenclature. Rice Genet Newsl 14(11):11–131
Mir RR, Kumar N, Jaiswal V, Girdharwal N, Prasad M, Balyan HS, Gupta PK (2012) Genetic dissection of grain weight in bread wheat through quantitative trait locus interval and association mapping. Mol Breed 29:963–972
Mohammadi-Nejad G, Arzani A, Rezai AM, Singh RK, Gregorio GB (2008) Assessment of rice genotypes for salt tolerance using microsatellite markers associated with the saltol QTL. Afr J Biotechnol 7:31
Niu Y, Xu Y, Liu XF, Yang SX, Wei SP, Xie FT, Zhang YM (2013) Association mapping for seed size and shape traits in soybean cultivars. Mol Breed 31:785–794
Ooijen JW, Voorrips RE (2002) JoinMap: version 3.0: software for the calculation of genetic linkage maps. University and Research Center
Pandit A, Rai V, Bal S, Sinha S, Kumar V, Chauhan M, Gautam RK, Singh R, Sharma PC, Singh AK (2010) Combining QTL mapping and transcriptome profiling of bulked RILs for identification of functional polymorphism for salt tolerance genes in rice (Oryza sativa L.). Mol Genet Genomics 284:121–136
Prasad SR, Bagali PG, Hittalmani S, Shashidhar HE (2000) Molecular mapping of quantitative trait loci associated with seedling tolerance of salt stress in rice (Oryza sativa L.). Curr Sci 78:162–164
Pritchard JK, Stephens M, Rosenberg NA, Donnelly P (2000) Association mapping in structured populations. Am J Hum Genet 67:170–181
Rana P, Jain S, Yadav S, Saini N, Jain RK (2009) Identification of SSR Markers for salt-tolerance in rice variety CSR10 by selective genotyping. J Plant Biochem Biotechnol 18:87–91
Ren ZH, Gao JP, Li LG, Cai XL, Huang W, Chao DY, Zhu MZ, Wang ZY, Luan S, Lin HX (2005) A rice quantitative trait locus for salt tolerance encodes a sodium transporter. Nat Genet 37:1141–1146
Rosenberg NA, Burke T, Elo K, Feldman MW, Freidlin PJ, Groenen MA, Hillel J, Mki-Tanila A, Tixier-Boichard M, Vignal A, Wimmers K, Weigend S (2001) Empirical evaluation of genetic clustering methods using multilocus genotypes from 20 chicken breeds. Genetics 159:699–713
Sabouri H, Rezai AM, Moumeni A, Kavousi A, Katouzi M, Sabouri A (2009) QTLs mapping of physiological traits related to salt tolerance in young rice seedlings. Biol Plant 53:657–662
Singh RK, Redoña E, Refuerzo L (2010) Varietal improvement for abiotic stress tolerance in crop plants: special reference to salinity in rice. In: Abiotic stress adaptation in plants. Springer, Dordrecht, pp 387–415
Sun SJ, Guo SQ, Yang X, Bao YM, Tang HJ, Sun H, Huang J, Zhang HS (2010) Functional analysis of a novel Cys2/His2-type zinc finger protein involved in salt tolerance in rice. J Exp Bot 61:2807–2818
Tanksley SD, Nelson JC (1996) Advanced backcross QTL analysis: a method for the simultaneous discovery and transfer of valuable QTLs from unadapted germplasm into elite breeding lines. Thero Appl Genet 92:191–203
Thomson MJ, de Ocampo M, Egdane J, Rahman MA, Sajise AG, Adorada DL, Tumimbang-Raiz E, Blumwald E, Seraj ZI, Singh RK, Gregorio GB, Ismail AM (2010) Characterizing the Saltol quantitative trait locus for salinity tolerance in rice. Rice 3:148–160
Tian L, Tan LB, Liu FX, Cai HW, Sun CQ (2011) Identification of quantitative trait loci associated with salt tolerance at seedling stage from Oryza rufipogon. J Genet Genomics 38:593–601
Voorrips RE (2002) MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered 93:77–78
Wang ZF, Chen ZW, Cheng JP, Lai YY, Wang JF, Bao YM, Huang J, Zhang HS (2012a) QTL analysis of Na+ and K+ concentrations in roots and shoots under different levels of nacl stress in rice (Oryza sativa L.). PLoS One 7:e51202
Wang ZF, Cheng JP, Chen ZW, Huang J, Bao YM, Wang JF, Zhang HS (2012b) Identification of QTLs with main, epistatic and QTL × environment interaction effects for salt tolerance in rice seedlings under different salinity conditions. Theor Appl Genet 125:807–815
Wassmann R, Hien NX, Hoanh CT, Tuong TP (2004) Sea level rise affecting the Vietnamese Mekong Delta: water elevation in the flood season and implications for rice production. Clim Chang 66:89–107
Weng JF, Xie CX, Hao ZF, Wang JJ, Liu CL, Li MS, Zhang DG, Bai L, Zhang SH, Li XH (2011) Genome-wide association study identifies candidate genes that affect plant height in Chinese elite maize (Zea mays L.) inbred lines. PLoS One 6:e29229
Wu R, Garg A (2003) Engineering rice plants with trehalose-producing genes improves tolerance to drought, salt, and low temperature. ISB news report
Wu Y, Hu Y, Xu G (2009) Interactive effects of potassium and sodium on root growth and expression of K/Na transporter genes in rice. Plant Growth Regul 57:271–280
Xie XB, Song MH, Jin FX, Ahn SN, Suh JP, Hwang HG, McCouch SR (2006) Fine mapping of a grain weight quantitative trait locus on rice chromosome 8 using near-isogenic lines derived from a cross between Oryza sativa and Oryza rufipogon. Thero Appl Genet 113:885–894
Yeo AR, Yeo ME, Flowers SA, Flowers TJ (1990) Screening of rice (Oryza sativa L.) genotypes for physiological characters contributing to salinity resistance, and their relationship to overall performance. Theor Appl Genet 79:377–384
Yoshida S, Forno DA, Cock JH, Gomez KA (1976) Laboratory manual for physiological studies of rice. International Rice Research Institute, Manila, p 38
Zheng TQ, Yang J, Zhong WG, Zhai HQ, Zhu LH, Fan FJ, Ali AJ, Yang JH, Wang J, Zhu JY, Uzokwe VNE, Xu JL, Li ZK (2012) Novel loci for field resistance to black-streaked dwarf and stripe viruses identified in a set of reciprocal introgression lines of rice (Oryza sativa L.). Mol Breed 29:925–938
Acknowledgments
This study was supported by the Tackle Key Program in Science and Technology of the Science and Technology Ministry of China (2011BAD35B02-01) and by the Program in Science and Technology of the Science and Technology Ministry of China (2011BAD16B11).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zheng, H., Zhao, H., Liu, H. et al. QTL analysis of Na+ and K+ concentrations in shoots and roots under NaCl stress based on linkage and association analysis in japonica rice. Euphytica 201, 109–121 (2015). https://doi.org/10.1007/s10681-014-1192-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10681-014-1192-3