
Abstract
A quantitative trait locus (QTL) for grain weight (GW) was detected near SSR marker RM210 on chromosome 8 in backcross populations derived from a cross between the Korean japonica cultivar Hwaseongbyeo and Oryza rufipogon (IRGC 105491). The O. rufipogon allele increased GW in the Hwaseongbyeo background despite the fact that O. rufipogon was the small-seeded parent. Using sister BC3F3 near-isogenic lines (NILs), gw8.1 was validated and mapped to a 6.1 cM region in the interval between RM42 and RM210 (P≤0.0001). Substitution mapping with eight BC3F4 sub-NILs further narrowed the interval containing gw8.1 to about 306.4 kb between markers RM23201.CNR151 and RM30000.CNR99. A yield trial using homozygous BC3F4 sister sub-NILs and the Hwaseongbyeo recurrent parent indicated that the NIL carrying an O. rufipogon chromosome segment across the entire gw8.1 target region out-yielded its sister NIL (containing Hwaseongbyeo chromosome in the RM42–RM210 interval) by 9% (P=0.029). The higher-yielding NIL produced 19.3% more grain than the Hwaseongbyeo recurrent parent (P=0.018). Analysis of a BC3F4 NIL indicated that the variation for GW is associated with variation in grain shape, specifically grain length. The locus, gw8.1 is of particular interest because of its independence from undesirable height and grain quality traits. SSR markers tightly linked to the GW QTL will facilitate cloning of the gene underlying this QTL as well as marker-assisted selection for variation in GW in an applied breeding program.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Grain or seed weight is an important trait in rice. It is directly associated with yield, and indirectly correlated with the cooking quality and the physical appearance of rice, which is important both aesthetically and commercially (Takeda and Saito 1980; Webb 1991). The influence of grain weight (GW) on yield lies in the fact that GW and grain number are the main determinants of sink size for the products of photosynthesis in the plant. The importance of GW or seed size to the human eye is revealed by the fact that larger seeds are consistently selected in most cultivated species during the domestication of crops (Simmonds 1979; Doganlar et al. 2000).
The morphological development of rice seed has been extensively studied (reviewed in Takeda 1991). Seed or grain length, as a quantitative trait, is regarded as the strongest determinant of grain size. Prior to the development of molecular marker technology, most studies focused on the characterization of mutants and the expression of major genes associated with grain size, e.g., the Lk-f gene, which confers long kernel size (Takeda and Saito 1980), or Mi conferring short kernel size (Takeda and Saito 1977).
With the progress of molecular genetic maps, many quantitative trait loci (QTLs) for GW/size have been identified in populations from crosses between divergent cultivars or accessions of rice (Lin et al. 1996; Zhuang et al. 1997; Li et al. 1998; Cui et al. 2003; Ishimaru 2003). A total of 26 QTLs for GW have been detected in populations derived from crosses between the wild rice Oryza rufipogon (IRGC 105491) and diverse cultivars. While the seed of O. rufipogon is small in size, it is interesting to note that 58% of the QTLs reported in these interspecific crosses identify O. rufipogon alleles as contributing to an increase in seed or GW (Xiao et al. 1998; Moncada et al. 2001; Thomson et al. 2003; Septiningsih et al. 2003). Similar phenomena were also reported in wheat (Huang et al. 2003) and barley (Pillen et al. 2003), where nearly half of the wild relative alleles for kernel weight acted to increase seed weight compared to elite cultivar alleles. On the other hand, a recent study by Li et al. (2004b) reported the fine mapping of gw3.1 in the pericentromeric region of chromosome 3. In this case, the O. rufipogon allele was dominant for small grain size (Li et al. 2004b).
In a previous study from our lab (Cho et al. 2003), a total of 12 QTLs for GW were detected in BC2F2 progenies derived from a cross between the temperate japonica cultivar, Hwaseongbyeo, and O. rufipogon (IRGC 105491). O. rufipogon alleles at four of the loci increased 1,000-grain weight (TGW) in the Hwaseongbyeo background. The percent of phenotypic variance explained by the QTL gw8.1 (11%) was the largest of the four QTLs (Cho et al. 2003). The fact that the O. rufipogon allele at gw8.1 contributed to increased seed size, while the dominant O. rufipogon allele at gw3.1, as described by Li et al. (2004b) decreased seed size made it an attractive target for study. Understanding the mode of action of different genes from the same donor and how they interact to generate a phenotype will help better to manipulate them in a plant breeding program aimed at optimizing yield while balancing concerns about GW and grain quality in rice where large, bold grains do not have the cooking and eating quality desired for commercial production (Juliano and Villareal 1993; Takeda 1991; Webb 1991). A few studies have reported QTL associated with grain or seed weight in the grasses. Interestingly, comparative mapping seems to suggest that this GW QTL is homologous to a major QTL for test weight on wheat 7AS (Sorrells et al. 2003; Elouafi and Nachit 2004). As increasing numbers of genes associated with GW are identified and cloned with the positional correspondence across the grasses, structural and functional conservation of orthologs underlying these QTLs in the grasses will be revealed.
In this study we developed near-isogenic lines (NILs) containing 1–2 introgressions from O. rufipogon using advanced backcross QTL analysis with BC3F3 and BC3F4 progenies. The objectives of the study were to (1) construct a high-resolution map for gw8.1 and (2) understand the characteristics associated with the gw8.1 locus in terms of its affect on GW and yield potential in rice.
Materials and methods
Population development and field trials
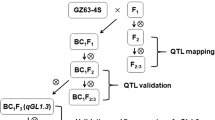
The wild rice O. rufipogon (IRGC 105491) was used as a pollen parent in crosses with Oryza sativa spp. japonica cv. Hwaseongbyeo, an elite Korean cultivar. Two successive backcrosses were made with Hwaseongbyeo as the recurrent parent. Among the 820 BC2F1 plants, 172 individuals selected based on desirable plant type, maturity and fertility were advanced to BC2F2 families. QTLs for yield, yield components and other agronomic traits were analyzed in the 172 BC2F2 as reported by Cho et al. (2003). Two plants were chosen from a BC2F2 line, F101, which contained an O. rufipogon introgression carrying the gw8.1 QTL on chromosome 8 and seven additional introgressions representing approximately 18.5% of the genome located on 5 of the 12 chromosomes (non-target regions). In 2002, NILs were developed by backcrossing two plants from F101 to Hwaseongbyeo, followed by selfing to eliminate O. rufipogon introgressions in non-target genomic regions. In 2003, two BC3F3 NIL families, CR1448 and CR1449, with 223 and 229 individuals, respectively, were used for further analysis and a series of BC3F4 sub-NILs (i.e., substitution lines containing recombination breakpoints across the target region) were developed from CR1448 and CR1449 (Fig. 1). In 2004, a yield trial was performed using sub-NILs with the Hwaseongbyeo parent as a control. All the materials were grown in the experimental plots at Chungnam National University in Deajeon, Korea. The field was equipped with a bird-net to prevent bird damage.

A scheme showing how plant materials were developed
Trait evaluation
At the BC3F2 and BC3F3 generations, only GW was evaluated. Each plot of 30-day-old seedlings was laid out with 15 cm between plants and 30 cm between rows. Grains harvested from individual plant were dried naturally after harvesting, and then water was used to remove those that had not fully filled. The fully filled grains were re-dried in an oven at 30°C for 24 h. After drying, GW was measured based on three replications of 100 randomly selected grains. The estimate of GW was converted to TGW by multiplying the average weight of 100 randomly selected grains by 10. TGW is the measure most commonly used by rice geneticists and breeders and the trait will be referred to as TGW throughout this manuscript. Simultaneously, moisture content of the grain was also measured on three replications using a Grain Moisture Meter (GMK-303). At BC3F4, sub-NILs were evaluated for the selected traits. Each line was represented by two rows of thirty 30-day-old seedlings planted with 15 cm between plants and 30 cm between rows in a randomized complete block design with three replications. For 48 individual plants from one selected NIL, CR4363, mean grain length, grain width and grain thickness were measured on 100 randomly selected fully filled grains using the digimatic caliper (Mitutoyo Corp., Tokyo, Japan). The ratio of grain length to grain width and grain volume (length × width × thickness) was calculated. Days to heading was evaluated as the number of days from seeding until 50% of the panicles (on all 60 plants) flowered. For culm length, panicles per plant and spikelets per panicle, ten individual plants per line in the middle were measured and the average of the measurements was used as the phenotype of each line. Culm length was measured in centimeters from the soil surface to the tip of the tallest panicle. Panicle number was calculated as the number of panicles per plant. Spikelets per panicle were measured by averaging two major panicles per plant. Grain yield (measured in grams of seed per plant) was measured on 15 plants harvested from the middle of one plot per block. Amylose content was measured as described by Williams et al. (1958) and Juliano (1971) using a sample of 100 g 100-mesh sieved-rice flour. TGW and yield per plant were corrected for the 10% grain moisture content.
Marker analysis
Micro-quantities of DNA were extracted by the method described in McCouch et al. (1988) with minor modifications. SSR markers (Panaud et al. 1996; Chen et al. 1997; Temnykh et al. 2000, 2001; McCouch et al. 2002) were used to survey for polymorphism between the two parents, Hwaseongbyeo and O. rufipogon, and among genotypes within segregating populations. Polymerase chain reaction (PCR) conditions, PCR product electrophoresis and silver staining procedures were as described in Panaud et al. (1996). The silver staining kits were purchased from Bioneer Co., Korea. Published SSR maps (Temnykh et al. 2000, 2001) were adopted for surveying the presence and map positions of introgressions from O. rufipogon. A total of 223 polymorphic SSR markers distributed on all 12 chromosomes were used to detect introgressed segments. The linkage map used for QTL analysis was constructed using MAPMAKER (Macintosh v2.0) and Kosambi centiMorgans (cM) were used to calculate genetic distances.
Development of new SSR markers
SSR marker development followed the protocols described by Li et al. (2004b). Newly developed SSR markers were designated using the “RM” locus identifier (as described by McCouch et al. 2002), with the “marker reagent” specified by the suffix “CNR,” indicating the particular primer pairs used in this study. Marker order in the target region on chromosome 8 was based on the publicly available rice genome sequence information (www.gramene.org) and confirmed by MAPMAKER (Macintosh v2.0). Primers were synthesized by Bioneer Co., Korea (www.bioneer.co.kr).
Data analysis
QGENE (Nelson 1997) was used for single point and interval mapping in the F3 and F4 generations derived from BC3 NILs. A QTL was declared if the phenotype was associated with a marker locus at LOD>4.0. Phenotypic means were compared using t-test, Duncan’s multiple range test and one-way ANOVA. The proportion of observed phenotypic variation attributable to a particular QTL was estimated by the coefficient of determination (R 2). Pearson Product-Moment correlation coefficients were calculated using the SAS program.
Results
Mapping of gw8.1 QTL
A total of 308 SSR markers distributed throughout the genome (Temnykh et al. 2001; McCouch et al. 2002) were used to survey for polymorphism between the parents, Hwaseongbyeo and O. rufipogon, and 223 (75.6%) were polymorphic. These polymorphic markers were used to confirm the genotype of F101, the BC2F2 line that was used as the source material for NIL development in this study. F101 had large seeds and contained an homozygous O. rufipogon introgression across the entire 25.8 cM interval defining gw8.1 in the study (Cho et al. 2003). To eliminate the non-target introgressions, F101 was backcrossed to Hwaseongbyeo for one generation to generate BC3 lines. The lines were genotyped using markers flanking gw8.1 to ensure retention of the target O. rufipogon introgression and also genotyped to select for individuals in which the background introgressions had been eliminated.
In the BC3F2 families derived from F101, the difference in TGW between plants having O. rufipogon introgressions in the target region and those having Hwaseongbyeo alleles was as large as 4.4 g under field conditions. These results confirmed primary QTL observations based on field evaluations (Cho et al. 2003) and demonstrated that transgressive segregants could be identified in subsequent generations. To identify recombinants for fine mapping, a BC3F2 population (F11) was selected. The F11 population had an O. rufipogon/Hwaseongbyeo heterozygous segment in the target region and one additional heterozygous introgression in the genetic background flanked by SSR markers RM234 and RM478 on chromosome 7 (Fig. 2). Segregating populations of 223 (CR1448) and 229 (CR1449) individual BC3F3 plants (Fig. 1) were evaluated both phenotypically and genotypically using ten markers in the target region on chromosome 8 flanked by RM42 (RM515) and RM210 (RM531) (Fig. 2). These populations were also genotyped with two SSR markers on chromosome 7 showing segregation. Phenotypes were compared among the three genotypic classes defined by the allele constitution of markers in the interval RM42 (RM515)–RM210 (RM531).

Linkage maps of chromosomes 7 and 8 showing introgressions from O. rufipogon and location of gw8.1 on chromosome 8 in CR1449. Gray regions indicate introgressed segments, open rectangles indicate approximate positions of centromeres. Arrows show the position of peak LOD score by interval analysis
The distribution of TGW based on the genotypes in the CR1449 BC3F3 population is shown in Fig. 3. In CR1449, the mean TGW of the homozygous O. rufipogon class (26.23 g) was significantly different from that of the heterozygous (25.51 g) and the Hwaseongbyeo homozygous class (24.85 g), and the mean TGW of the heterozygous class was again significantly different from that of the Hwaseongbyeo homozygous class based on the t-test. Similarly, in CR1448, there was a significant difference in mean TGW between each of the genotypic classes; the homozygous O. rufipogon class had a mean TGW of 27.29 g, the heterozygous class of 26.34 g and the Hwaseongbyeo homozygous class had a mean of 25.36 g based on the t-test. These results demonstrated that the O. rufipogon allele increased GW in the Hwaseongbyeo background in an additive fashion. The additive effect of the O. rufipogon allele was 0.69–0.97 g in the two BC3F3 populations and gw8.1 explained 30.2–39.0% of the total phenotypic variation (Table 1). Mapping also confirmed a QTL peak between markers RM7556 and RM7356 with a LOD score of 24.08 and 18.3 in CR1448 and CR1449, respectively (data not shown). There was no association between TGW and any of the two markers in the introgression on chromosome 7 in either population.

Frequency distribution of TGW for the three genotypic groups, Hwaseongbyeo/Hwaseongbyeo (HH), Hwaseongbyeo/O. rufipogon (HR) and O. rufipogon/O. rufipogon (RR) for BC3F3 progeny derived from CR1449. Arrows show the mean TGW of each group
To further refine the position of gw8.1, 36 additional SSR markers were developed within the target region on chromosome 8 based on Nipponbare sequence information. Among them, 21 SSR markers (58.3%) produced polymorphic bands between the parents and 9 amplified strongly and were used to fine map gw8.1 (Table 2). Their order and distances were based on the pseudomolecules of japonica cv. Nipponbare (TIGR v. 3 pseudomolecules available at www.gramene.org and at sliver.plbr.cornell.edu/SSR).
Substitution mapping of gw8.1
A series of BC3F4 sub-NILs were developed from CR1448 and CR1449 based on the identification of recombinants using SSRs. Eight sub-NILs representing different recombination breakpoints across the gw8.1 region were identified and their graphical genotypes are shown in Fig. 4. There were no remaining O. rufipogon introgressions on chromosome 7 in any of these sub-NILs. CR4362 (n=15 progeny) and CR4365 (n=15) were used as controls because they had Hwaseongbyeo and O. rufipogon chromosome segments, respectively, across the entire target region. The rest of the lines all contained O. rufipogon introgressions in the heterozygous state across a specific sub-region of interest. Progeny from each of the sub-NILs were evaluated for segregation of TGW. No segregation for TGW was observed in populations derived from CR4382 (n=25), CR4383 (n=30) or CR4360 (n=30), but significant phenotypic differences were observed among the three genotypic classes within CR4357 (n=50), CR4363 (n=53) and CR4377 (n=79) (ANOVA, P≤0.0001) (Fig. 4). Comparisons among the three genotypic classes of the three NIL-derived populations suggested that gw8.1 was located in the interval RM23201.CNR151–RM7556 and explained between 35.2 and 58.3% of the phenotypic variation (Fig. 4). The TGW of sub-NILs, CR4383 and CR4360 were similar to that of CR4362 while that of sub-NIL CR4382 was similar to CR4365. Based on the recombination breakpoint in CR4360, the position of gw8.1 was narrowed to the interval defined by RM23201.CNR151–RM30000.CNR99 (Fig. 4). This data also demonstrated that there was no TGW QTL outside of the interval defined by the markers RM284 and RM7556, and therefore, the candidate position for gw8.1 was in the 306.4-kb interval RM23201.CNR151–RM30000.CNR99. This interval was comprised of four bacterial artificial chromosome clones, P0528B09, P0708B04, P0479C12 and P0429B05, based on the physical map of the region (http://www.rgp.dna.affrc.go.jp/cgi-bin/statusdb/stattable.pl?chr=8&lab=RGP) (Table 2).

Graphical genotypes of lines used in substitution mapping of gw8.1. White portions of the graph indicate homozygous Hwaseongbyeo chromosome segments, black regions indicate homozygous O. rufipogon chromosomes, diagonal slashes indicate heterozygous regions and gray areas are regions where crossing-over occurred. The table to the right of the graphical genotypes indicates mean TGW for each of the three genotypic classes of progeny derived from CR4357, CR4363 and CR4383. The broken vertical lines define the interval containing the gw8.1 locus. #Markers within the heterozygous regions were tested and the ones with the highest R 2 scores are shown. % nd not determined. Numbers followed by the different letter in each row are significantly different at P=0.0001 based on the t-test. HH Hwaseongbyeo homozygous, HR Hwaseongbyeo and O. rufipogon heterozygous, RR O. rufipogon homozygous, n number of evaluated individuals in each line
Analysis of QTLs for grain shape traits at BC3F4
To understand which aspect of grain shape trait was responsible for the increase in GW at gw8.1, progeny from CR4363 was used for QTL analysis. The data indicated that three grain shape QTLs, grain length (gl8.1), grain width (gwd8.1) and grain thickness (gt8.1) were all located near marker RM30002.CNR104 in the target region (P≤0.0001, P=0.0061 and P=0.0028, respectively) (Table 3). Three markers (RM30002.CNR104, RM23202.CNR153 and RM515) within the heterozygous regions were tested and RM30002.CNR104 and RM23202.CNR153 showed the highest P-value. Correlation analysis also indicated that there were significant correlations between TGW and grain length, grain width, grain thickness, length/width ratio and grain volume (Table 4).
Effect of the Oryza rufipogon introgression harboring gw8.1 on TGW and grain yield
To determine whether the large introgressed segment in CR4365 was associated with an effect on grain yield in addition to TGW, a yield trial was conducted using seed from CR4365 (which was homozygous for O. rufipogon across the entire region), CR4362 (a line that was homozygous for Hwaseongbyeo alleles in the same region), and Hwaseongbyeo, the recurrent parent. Plots for all three genotypes were laid out in a completely randomized block design with three replications. The results showed that the grain yield of CR4365 was 9.0% (P=0.029) and 19.3% (P=0.009) higher than those of CR4362 and the recurrent parent, respectively (Table 5). Also, the number of spikelets per panicle of CR4365 and CR4362 were higher than that of the recurrent parent, respectively (Table 5). In addition, a population derived from CR4363 segregating for the O. rufipogon introgression in the RM42–RM210 interval showed significant differences in average grain yield between the three genotypic classes. The O. rufipogon homozygous class (31.29 g, n=26) was significantly higher-yielding than the Hwaseongbyeo homozygous class (26.56 g, n=30). These independent experiments support the assertion that the 2.2-Mb introgressed segment between RM42 (RM515) and RM210 (RM531) is associated with a QTL for enhanced grain yield, spikelets per panicle as well as TGW. No QTL for heading date or plant height was detected in this region (Table 5).
Discussion
A few studies have reported QTL associated with GW located on the long arm of chromosome 8 using both inter- (Brondani et al. 2002) and intraspecific populations (Cui et al. 2003; Lu et al. 1997). While it is difficult to interpret whether any of the QTLs in these studies correspond to the same locus as gw8.1, due to a lack of common markers used for mapping, it appears that the map locations differ, making it unlikely that they are allelic. Also, studies using the same O. rufipogon accession (IRGC 105491) in combination with other recurrent parents failed to detect gw8.1 (Moncada et al. 2001; Thomson et al. 2003; Septiningsih et al. 2003; Xiao et al. 1998). This may be due to the use of different mapping populations and genotypes [none of the previous studies that utilized the same O. rufipogon donor used a temperate japonica variety (i.e., with short, round grains) as the recurrent parent]. The heritability of GW and grain length in rice is estimated at 71.9 and 80.0%, respectively (Takeda 1990; Takeda and Saito 1983), and though variation is observed among seeds on the same plant, i.e., among seeds on the main or secondary tillers, or on primary or secondary branches of a panicle (Takeda 1991), the trait is tractable to genetic analysis via the development of high-resolution NILs. As documented in this study, R 2 values steadily increased with advanced generations of backcrossing, from 11% in the BC2F2 generation to between 30 and 39% in the BC3F3 generation and up to 58% in the BC3F4 generation of NILs. As the number of spurious donor (i.e., O. rufipogon) introgressions in the genetic background decreased and the linkage between markers and the target gene(s) increased, the proportion of the phenotypic variation that could be explained by the markers was greatly enhanced. This situation was similar to that discussed by Li et al. (2004b) for gw3.1 in rice, or by Paterson et al. (1990) for soluble solids concentration, fruit mass and yield in tomato. The use of high-resolution NILs in a substitution mapping strategy means that only a small number of genetic differences distinguish the NILs, making phenotypic comparisons much more straightforward and avoiding the need for large-scale replication over years and environments.
Substitution mapping has been applied in diverse plant species to facilitate the fine mapping of QTLs (Paterson et al. 1990; Eshed and Zamir 1995; Wissuwa et al. 2002; Korff et al. 2004; Li et al. 2004b). The advantage of the method lies in its ability to clearly visualize the borders of an interval containing the QTL(s) (Wissuwa et al. 2002). Based on substitution mapping, the TGW QTL, gw8.1 was successfully mapped to a 306.4-kb interval between markers RM23201.CNR151 and RM30000.CNR99 on chromosome 8. To facilitate the fine mapping of gw8.1 eight new SSR markers were developed in the region using publicly available rice (cv. Nipponbare) genome sequence. As discussed by Li et al. (2004a) and other studies (IRGSP 2005; Yu et al. 2002; Li et al. 2004b), a large number of highly polymorphic SSR markers have been developed for rice and provide a useful tool for high-density mapping.
Having identified a QTL for increased GW coming from an accession of O. rufipogon not widely utilized in rice breeding programs, the aim of our marker-assisted introgression program was to fix the favorable allele (from O. rufipogon) of this QTL in a Hwaseongbyeo genetic background with as little additional O. rufipogon (donor) chromosome as possible. The locus, gw8.1 is of particular interest because of its independence from undesirable height and maturity effects. The additive gene effect of gw8.1 is relatively high, with the O. rufipogon allele being associated with a 0.65–0.95 g increase in TGW. Ishimaru (2003) also identified a GW QTL, tgw6 on chromosome 6 which was responsible for increased yield potential without any negative effect on plant type or grain quality in the Nipponbare genetic background. The colocalization of QTLs for GW and grain shape implies that the increase in grain length, width and thickness might be responsible for increased GW in the nearly isogenic background of Hwaseongbyeo. Takeda (1991) and Li et al. (2004b) also reported that grain length was the main cause of the variation of TGW based on the highly significant correlation between TGW and grain length at gw3.1. Although the gw8.1 QTL was not responsible for an increase in amylose content in this study, additional experiments are needed to determine the effect of gw8.1 on other grain quality traits because increased kernel length/weight may have negative effect on japonica rice grain quality.
A yield trial in the BC3F4 generation demonstrated that the introgressed segment from O. rufipogon defined by SSR markers, RM42 and RM210, has a significant and positive effect on yield potential. This finding proves the possibility that the introgression might harbor other QTLs for yield components (i.e., spikelets per panicle) in addition to gw8.1. This assertion is consistent with results from two independent studies which employed the same O. rufipogon accession (IRGC 105491) as the donor parent in AB-QTL analysis. Xiao et al. (1998) detected QTLs for grain yield, panicle length and grains per plant associated with the marker, RM210. Thomson et al. (2003) also identified a QTL for grain number per panicle near the marker RM210 which is located near to gw8.1. In all cases, the O. rufipogon alleles were beneficial in the diverse cultivated backgrounds. To better understand the genes contributing to these beneficial effects, we are pursuing a fine mapping strategy aimed at cloning the genes underlying these yield QTLs. It is of interest to better define their linkage relationships and to understand the molecular mechanisms responsible for yield in diverse genetic backgrounds. As increasing numbers of genes associated with agriculturally relevant phenotypes are cloned, plant breeders and geneticists confront an expanding array of choices and opportunities for utilizing the information for applied plant improvement.
Although it is not clear why this gw8.1 region, which harboris several useful alleles, appears not to have been selected during rice domestication, several articles by Thomson et al. (2003), Lee et al. (2005) and Li et al. (2006) provide clues that suggest a relationship between the gw8.1 region and domestication. A QTL for awns was detected in this region by both Thomson et al. (2003) and Lee et al. (2005) in crosses between O. sativa and O. rufipogon. More recently, a shattering gene was detected close to gw8.1 Li et al. (2006) using an F2 population derived from a cross between O. sativa and the annual wild species, O. nivara. These studies suggest that the gw8.1 region on chromosome 8 may not have been selected during rice domestication because of linkage with these deleterious alleles.
References
Brondani C, Rangel PHN, Brondani RPV, Ferreira ME (2002) QTL mapping and introgression of yield-related traits from Oryza glumaepatula to cultivated rice (Oryza sativa) using microsatellite markers. Theor Appl Genet 104:1192–1203
Chen X, Temnykh S, Xu Y, Cho YG, McCouch SR (1997) Development of a microsatellite framework map providing genome-wide coverage in rice (Oryza sativa L.). Theor Appl Genet 95:553–567
Cho YC, Suh JP, Choi IS, Hong HC, Baek MK, Kang KH, Kim YG, Ahn SN, Choi HC, Hwang HG, Moon HP (2003) QTLs analysis of yield and its related traits in wild rice relative Oryza rufipogon. Treat Crop Res Korea 4:19–29
Cui KH, Peng SB, Xing YZ, Yu SB, Xu CG, Zhang Q (2003) Molecular dissection of the genetic relationships of source, sink and transport tissue with yield traits in rice. Theor Appl Genet 106:649–658
Doganlar S, Frary A, Tanksley SD (2000) The genetic basis of seed-weight variation: tomato as a model system. Theor Appl Genet 100:1267–1273
Elouafi I, Nachit MM (2004) A genetic linkage map of the Durum × Triticum dicoccoides backcross population based on SSRs and AFLP markers, and QTL analysis for milling traits. Theor Appl Genet 108:401–413
Eshed Y, Zamir D (1995) An introgression-line population of Lycopersicon pennellii in the cultivated tomato enables the identification and fine mapping of yield-associated QTLs. Genetics 141:1147–1162
Huang XQ, Cöster H, Ganal MW, Röder MS (2003) Advanced backcross QTL analysis for the identification of quantitative trait loci alleles from wild relatives of wheat (Triticum aestivum L.). Theor Appl Genet 106:1379–1389
International Rice Genome Sequencing Project (2005) The map-based sequence of the rice genome. Nature 436:793–800
Ishimaru K (2003) Identification of a locus increasing rice yield and physiological analysis of its function. Plant Physiol 133:1083–1090
Juliano BO (1971) A simplified assay for milled-rice amylose. Cereal Sci Today 16:334–360
Juliano BO, Villareal CP (1993) Grain quality evaluation of world rices. International Rice Research Institute, Manila, pp 205
Korff MV, Wang H, Léon J, Pillen K (2004) Development of candidate introgression lines using an exotic barley accession (Hordeum vulgare ssp. Spontaneum) as donor. Theor Appl Genet 109:1736–1745
Lee SJ, Oh CS, Suh JP, McCouch SR, Ahn SN (2005) Identification of QTLs for domestication-related and agronomic traits in an Oryza sativa × O. rufipogon BC1F7 population. Plant Breed 124:209–219
Li Z, Pinson SRM, Stansel JW, Paterson AH (1998) Genetic dissection of the source–sink relationship affecting fecundity and yield in (Oryza sativa L.). Mol Breed 4:419–426
Li C, Zhang Y, Ying K, Liang XL, Han B (2004a) Sequence variations of simple sequence repeats on chromosome-4 in two subspecies of Asian cultivated rice. Theor Appl Genet 108:392–400
Li JM, Thomson M, McCouch SR (2004b) Fine mapping of a grain-weight quantitative trait locus in the pericentromeric region of rice chromosome 3. Genetics 168:2187–2195
Li CB, Zhou AL, Sang T (2006) Genetic analysis of rice domestication syndrome with the wild annual species, Oryza nivara. New Phytol 170:185–194
Lin HX, Qian HR, Zhuang JY, Lu J, Min SK, Xiong ZM, Huang N, Zheng KL (1996) RFLP mapping of QTLs for yield and related characters in rice (Oryza sativa L.). Theor Appl Genet 92:920–927
Lu CF, Shen LH, Tan ZB, Xu YB, He P, Chen Y, Zhu LH (1997) Comparative mapping of QTLs for agronomic traits of rice across environments by using a doubled-haploid population. Theor Appl Genet 94:145–150
McCouch SR, Kochert G, Yu ZH, Wang ZY, Khush GS, Coffman WR, Tanksley SD (1988) Molecular mapping of rice chromosomes. Theor Appl Genet 76:815–829
McCouch SR, Teytelman L, Xu Y, Lobos KB, Clare K, Walton M, Fu B, Maghirang R, Li Z, Xing Y, Zhang Q, Kono I, Yano M, Fjellstrom R, DeClerck G, Schneider D, Cartinhour S, Ware D, Stein L (2002) Development and mapping of 2240 new SSR markers for rice (Oryza sativa L.). DNA Res 9:199–207
Moncada P, Martinez CP, Borrero J, Chatel M, Gauch H, Guimaraes E, Tohme J, McCouch SR (2001) Quantitative trait loci for yield and yield components in an Oryza sativa × Oryza rufipogon BC2F2 population evaluated in an upland environment. Theor Appl Genet 102:41–52
Nelson JC (1997) QGENE: software for marker-based genomic analysis and breeding. Mol Breed 3:239–245
Panaud O, Chen X, McCouch SR (1996) Development of microsatellite markers and characterization of simple sequence length polymorphism (SSLP) in rice (Oryza sativa L.). Mol Gen Genet 252:597–607
Paterson AH, DeVerna JW, Lanini B, Tanksley SD (1990) Fine mapping of quantitative trait loci using selected overlapping recombinant chromosomes in an interspecies cross of tomato. Genetics 124:735–742
Pillen K, Zacharias A, Léon J (2003) Advanced backcross QTL analysis in barley (Hordeum vulgare L.). Theor Appl Genet 107:340–352
Septiningsih EM, Prasetiyono J, Lubis E, Tai TH, Tjubaryat T, Moeljopawiro S, McCouch SR (2003) Identification of quantitative trait loci for yield and yield components in an advanced backcross population derived from the Oryza sativa variety IR64 and the wild relative O. rufipogon. Theor Appl Genet 107:1419–1432
Simmonds NW (1979) Principles of crop improvement. Longman Inc., New York, pp 16–19
Sorrells ME, Rota ML, Bermudez-Kandianis CE, Greene RA, Kantety R, Munkvold JD, Miftahudin, Mahmoud A, Ma X, Gustafson PJ, Qi LL, Echalier B, Gill BS, Matthews DE, Lazo GR, Chao S, Anderson OD, Edwards H, Linkiewicz AM, Dubcovsky J, Akhunov ED, Dvorak J, Zhang D, Nguyen HT, Peng J, Lapitan NL, Gonzalez-Hernandez JL, Anderson JA, Hossain K, Kalavacharla V, Kianian SF, Choi DW, Close TJ, Dilbirligi M, Gill KS, Steber C, Walker-Simmons MK, McGuire PE, Qualset CO (2003) Comparative DNA sequence analysis of wheat and rice genomes. Genome Res 13:1818–1827
Takeda K (1990) Heritability for grain size of rice estimated from parent–offspring correlation and selection response. Jpn J Breed 40:313–320
Takeda K (1991) Inheritance of grain size and its implications for rice breeding. In: Rice genetics II. IRRI, Manila, pp. 181–189
Takeda K, Saito K (1977) The inheritance and character expression of the minute gene derived from a rice genetic tester “Minute”. Bull Fac Agric Hirosaki Univ 271:1–29
Takeda K, Saito K (1980) Major genes controlling grain size of rice. Jpn J Breed 30:280–282
Takeda K, Saito K (1983) Heritability and genetic correlation of kernel weight and white belly frequency in rice. Jpn J Breed 33:468–480
Temnykh S, Park W, Ayres N, Cartinhour S, Hauck N, Lipovich L, Cho YG, Ishii T, McCouch SR (2000) Mapping and genome organization of microsatellite sequences in rice (Oryza sativa L.). Theor Appl Genet 100:697–712
Temnykh S, DeClerk G, Lukashova A, Lipovich L, Cartinhour S, McCouch SR (2001) Computational and experimental analysis of microsatellites in rice (Oryza sativa L.): frequency, length, transposon associations, and genetic marker potential. Genome Res 11:1441–1452
Thomson MJ, Tai TH, McClung AM, Lai XH, Hinga ME, Lobos KB, Xu Y, Martinez CP, McCouch SR (2003) Mapping quantitative trait loci for yield, yield components and morphological traits in an advanced backcross population between Oryza rufipogon and the Oryza sativa cultivar Jefferson. Theor Appl Genet 107:479–493
Webb BB (1991) Rice quality and grades. In: Luh BS (ed) Rice, vol 2, Utilization, 2nd edn. An AVI Book, New York, pp. 89–119
Williams VR, Wu WT, Tsai HY, Bates HG (1958) Varietal differences in amylose content of rice starch. J Agric Food Chem 8:47–48
Wissuwa M, Wegner J, Ae N, Yano M (2002) Substitution mapping of Pup1: a major QTL increasing phosphorus uptake of rice from a phosphorus-deficient soil. Theor Appl Genet 105:890–897
Xiao J, Li J, Grandillo S, Ahn SN, Yuan L, Tanksley SD, McCouch SR (1998) Identification of trait-improving quantitative trait loci alleles from a wild rice relative, Oryza rufipogon. Genetics 150:899–909
Yu J, Hu S, Wang J, Wong GK, Li S, Liu B, Deng Y, Dai L, Zhou Y, Zhang X, Cao M, Liu J, Sun J, Tang J, Chen Y, Huang X, Lin W, Ye C, Tong W, Cong L, Geng J, Han Y, Li L, Li W, Hu G, Huang X, Li W, Li J, Liu Z, Li L, Liu J, Qi Q, Liu J, Li L, Li T, Wang X, Lu H, Wu T, Zhu M, Ni P, Han H, Dong W, Ren X, Feng X, Cui P, Li X, Wang H, Xu X, Zhai W, Xu Z, Zhang J, He S, Zhang J, Xu J, Zhang K, Zheng X, Dong J, Zeng W, Tao L, Ye J, Tan J, Ren X, Chen X, He J, Liu D, Tian W, Tian C, Xia H, Bao Q, Li G, Gao H, Cao T, Wang J, Zhao W, Li P, Chen W, Wang X, Zhang Y, Hu J, Wang J, Liu S, Yang J, Zhang G, Xiong Y, Li Z, Mao L, Zhou C, Zhu Z, Chen R, Hao B, Zheng W, Chen S, Guo W, Li G, Liu S, Tao M, Wang J, Zhu L, Yuan L, Yang H (2002) A draft sequence of the rice genome (Oryza sativa L. ssp. indica). Science 296:79–92
Zhuang JY, Lin HX, Lu J, Qian HR, Hittalmani S, Huang N, Zheng KL (1997) Analysis of QTL × environment interaction for yield components and plant height in rice. Theor Appl Genet 95:799–808
Acknowledgments
This study was supported by grants from the Bio Green 21 project of the Rural Development Administration and from the Crop Functional Genomics Center of the 21st Century Frontier Research Program (Project code no. CG3112) funded by the Ministry of Science and Technology, Republic of Korea.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by Y. Xue
Rights and permissions
About this article
Cite this article
Xie, X., Song, MH., Jin, F. et al. Fine mapping of a grain weight quantitative trait locus on rice chromosome 8 using near-isogenic lines derived from a cross between Oryza sativa and Oryza rufipogon . Theor Appl Genet 113, 885–894 (2006). https://doi.org/10.1007/s00122-006-0348-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00122-006-0348-5