
Abstract
Soil salinity is a major constraint to rice production. Understanding the genetic basis of salt tolerance is crucial for the improvement of salt tolerance through breeding. Previous quantitative trait locus (QTL) studies for salt tolerance were mainly derived from bi-parental segregating populations and relatively little is known about the results from natural populations. Understanding the genetic diversity, population structure and linkage disequilibrium (LD) in an association panel can effectively avoid spurious associations in association mapping. In this study, 341 japonica rice (Oryza sativa L. subsp. japonica) accessions worldwide were genotyped with 160 simple sequence repeat (SSR) markers to identify marker-trait associations with salt tolerance at the seedling stage. Salt tolerance was evaluated by survival days of seedlings and shoot K+/Na+ ratio. A total of 872 alleles ranging from 2 to 9 per locus were identified from all collections. Population structure analysis identified three main subpopulations for the accessions. Of the SSR pairs in these accessions, 40.05 % marker pairs showed significant LD (P < 0.01). The LD level for linked markers is significantly higher than that for unlinked markers, and LD level was elevated when the panel was classified into subpopulations. The LD decayed to the background at approximately 20–50 cM within the total panel and each subpopulation. A total of ten marker loci associated with salt tolerance were identified using MLM (Q + K) models in TASSEL 3.0. Among which nine marker loci confirmed or narrowed the genomic region reported to harbor QTLs for salt tolerance by linkage mapping in previous reports, and four salt tolerance-related genes were located in the QTL regions in the present study. According to phenotypic effects for alleles of the detected QTLs, favorable alleles were mined. These favorable alleles could be used to design parental combinations and the expected results would be obtained by pyramiding or substituting the favorable alleles per QTL (apart from possible epistatic effects). Our results demonstrate that association mapping can complement and enhance previous QTL information for marker-assisted selection and breeding by design.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Salt stress is a major constraint to agricultural food production because it decreases crop yield and restricts the use of agricultural land. It is estimated that salt-affected soils occur within the sovereign borders of at least 75 countries and occupy more than 20 % of the global irrigated area. In some countries, salt-affected land occurs on more than half of the irrigated land. The problem is increasing annually due to climatic change and poor irrigation management (Qadir et al. 2014). Rice (Oryza sativa L.) is one of the most important food crops in the world, and salinity is the most widespread soil problem limiting rice production. Approximately 30 % of rice-growing area in the world is affected by salinity (Takehisa et al. 2004). Rice threshold for salt stress is 3 dS/m, with a 12 % reduction in yield, per dS/m, beyond this value (Wallender and Tanji 2011), which makes rice a salt-sensitive crop. Therefore, understanding the genetic mechanisms controlling salt tolerance can enhance efforts to develop tolerant cultivars. Rice sensitivity to salt varies with the growth stage (Zeng and Shannon 2000), seedling and reproductive stages being the most sensitive. Varietal tolerance at these two stages is not directly related (Moradi et al. 2003). The high sensitivity during seedling stage set strongly compromises plant survival and good yields (Negrão et al. 2011). Therefore, understanding the genetic mechanisms controlling salt tolerance can enhance efforts to develop tolerant cultivars. To date, high genetic variability has been reported in salt tolerant rice varieties (Gregorio et al. 2002). The most famous tolerant varieties, Pokkali and Nona Bokra, belong to the indica subspecies, varieties of which have been used as the source of salt tolerance in almost all the studies published to date. However, little is known about the genetic variation for salt tolerance among japonica subspecies.
Salt tolerance in crop plants is a genetic and physiological complex trait (Flowers 2004). The seedling stage is one of the most sensitive stages in rice. Salinity stress affects photosynthesis and metabolic activities leading to the inhibition of cell division and expansion, accelerated cell senescence and, ultimately, reduced growth and even resulted in death, so survival days of seedlings under high salinity is the final index of salt tolerance. As NaCl is a major constituent of saline soil, shoot Na+ toxicity is associated with a reduction of stomatal conductance, and then reduce photosynthetic capacity, whereas potassium ions (K+) are essential to reduce the uptake of Na+ (Wu et al. 2009), plant ability to maintain high K+/Na+ ratio is a key feature of salt tolerance (Ahmadi et al. 2011; Wang et al. 2012a). Moreover, leaf K+/Na+ ratio predicts salinity-induced yield loss in rice (Asch et al. 2000). Therefore, shoot K+/Na+ ratio (SKN) is also an important indicator to evaluate salt tolerance in rice. In recent decades, using the rice seedling salt tolerance indicators mentioned above and other indicators, many studies have been reported with linkage mapping of QTL for salt tolerance at the seedling stage (Kim et al. 2009; Bonilla et al. 2002; Koyama et al. 2001; Lee et al. 2007; Lin et al. 2004; Prasad et al. 2000; Sabouri et al. 2009; Zang et al. 2008). For example, 11 QTLs for survival days of seedlings and the Na+ and K+ concentrations were detected on chromosomes 1, 4, 6, 7 and 9 using the Nona Bokra × Koshihikari mapping population (Lin et al. 2004). Ten QTLs for salt tolerance parameters, including Na+ and K+ uptake, Na+ and K+ concentrations and Na+/K+ ratio in shoots were identified by Koyama et al. (2001). Bonilla et al. (2002) mapped the SALTOL locus, which is linked to QTLs for Na+ and K+ uptake and Na+/K+ ratio, on chromosome 1, and Zang et al. (2008) reported 13 QTLs affecting survival days of seedlings, score of salt toxicity of leaves, shoot K+ concentration and shoot Na+ concentration on chromosomes 1, 3, 6, 8 and 11 at the seedling stage. Despite success of QTL analysis for salt tolerance in rice in previous study, traditional bi-parental segregating populations showed several disadvantages, including limited genetic variation and recombination (Wang et al. 2008; Xu and Crouch 2008; Dang et al. 2014).
Association mapping is an alternative for QTL mapping using diverse germplasm resources in plants, it utilizes the higher number of historical recombination events in natural population, thus a higher resolution of QTL mapping can be achieved than using the biparental segregating populations, and has recently become a new and powerful tool for the genetic dissection of complex quantitative traits (Yu and Buckler 2006; Yan et al. 2011; Le et al. 2012; Niu et al. 2013). In addition, association mapping takes shorter research time, and investigates a greater number of alleles when compared with linkage analysis (Flint-Garcia et al. 2003).
The principle of association mapping is to detect correlations between phenotypes and linked markers on the basis of LD (Ersoz et al. 2007). Therefore, it is important to characterize LD levels and patterns in a population analyzed and to infer evolutionary forces including genetic drift, population structure, population admixture, levels of inbreeding, and selection that contribute to the emergence and maintenance of LD. Patterns of LD have been characterized in several crop species. The decay or decrease of LD with increasing map distance between markers in outcrossing plants is usually faster than that in inbreeding plants (Flint-Garcia et al. 2003). For instance, LD decays rapidly within 1–5 kb in maize diverse inbred lines (Yan et al. 2009), 1.1 kb in cultivated sunflower (Liu and Burke 2006), 300 bp in wild grapevine (Lijavetzky et al. 2007), whereas LD decays slowly within 250 kb (1 cM) in Arabidopsis (Nordborg et al. 2002), 10–50 cM in soybean (Zhu et al. 2003; Jun et al. 2008) and 10–50 cM in barley (Kraakman et al. 2004; Malysheva-Otto et al. 2006). In rice, Mather et al. (2007) reported that the extent of the LD was greatest in temperate japonica (>500 kb), followed by tropical japonica (>150 kb), indica (~75 kb) and O. rufipogon (~40 kb). Moreover, it has been reported that the extent of LD in rice can vary from 20 to 50 cM depending on the assayed set of a germplasm (Agrama et al. 2007; Jin et al. 2010; Li et al. 2011). As a self-pollination species, the extent of LD is higher in rice, which is more suitable for genome-wide association mapping.
In recent years, association mapping for rice was used to identify favorable alleles for various traits such as yield (Agrama et al. 2007; Wen et al. 2009; Huang et al. 2010; Ordonez et al. 2010; Vanniarajan et al. 2012); outcrossing ratios (Yan et al. 2009; Huang et al. 2010); quality (Huang et al. 2010; Jin et al. 2010); resistance (Jia et al. 2012). However, there have been few studies to dissect the QTLs associated with tolerance under abiotic stresses especially salt stress by association mapping. Therefore, our study on association mapping of salt tolerance in japonica rice would be a beneficial supplementary and verification for current QTLs and genes of salt tolerance in rice. In this study, 341 japonica rice accessions were used to conduct association mapping for salt tolerance at the seedling stage combining information of 160 SSR markers. SDS and SKN were measured to indicate salt tolerance. The aims were (1) to evaluate the population structure and genetic diversity in the japonica rice panel; (2) to detect the extent of LD between pairs of SSR markers on a whole genome in rice; (3) to detect QTLs controlling salt tolerance and mine elite alleles; (4) to explore design of parental combinations for cultivar improvement.
Materials and methods
Plant materials
A total of 341 japonica rice accessions representing the genetic diversity among different geographic groups were collected from the Crop Science Research Institute, Chinese Academy of Agricultural Sciences, Liaoning Academy of Agricultural Sciences, Heilongjiang Academy of Agricultural Sciences and Northeast Agricultural University, and assembled to construct an association mapping panel. The population consisted of 209 accessions developed in China, 92 in Japan, 22 in Korea, 7 in the Democratic People’s Republic of Korea (DRPK), 7 in Russia and 4 in France. All accessions have been strictly self-pollinated during the past decades for germplasm renewing and the residual heterozygosity have been decreased remarkably. Detailed information about the accessions is summarized in Supplementary Table 1.
Evaluation of salt tolerance
Evaluation of rice for salt tolerance was done in a hydroponics solution at Northeast Agriculture University’s experimental station in 2013. The procedure of evaluation of salt-tolerance-related traits was carried out according to the modified system of Lin et al. (2004). Trials with the association panel were laid out in a randomized complete-block design with three replications. A sample of fifty healthy grains of each accession were placed in an oven at 50 °C for 3 days to break any possible dormancy, then germinated at 30 °C for 3 days after surface-sterilizing seeds with 1 % sodium hypochlorite solution for 10 min and rinsing three times with distilled water. The most uniform 20 germinated seeds were sown in holes of thin Styrofoam board with a nylon net bottom in a plastic tray, which floated on water up to the two leaf stage, then the seedlings were transferred to Yoshida’s cultural solution (Yoshida et al. 1976) containing 50 mmol/L of NaCl for 3 days, then NaCl concentration increased to 140 mmol/L. The seedlings were grown in the phytotron with 28 °C/21°Cday/night temperature and minimum relative humidity of 70 %. The solution was changed every 5 days and the pH was adjusted at 5.0 every other day by adding either 1 M NaOH or 1 M HCl. (Wang et al. 2012a, b). Survival days of seedlings (SDS) were recorded for each individual plant in days from seeding to death according to the standard evaluation system (Gregorio et al. 1997).
A separate second experiment was concurrently conducted to analyze the ratio of K+/Na+ in shoots (SKN). The procedure and management of the experiment was the same as the above-mentioned experiment. After 9 days of salinity stress with 140 mmol/L of NaCl, shoots were harvested and rinsed with distilled water several times. The shoots were then dried at 80 °C for 48 h, weighed, and extracted in acetic acid (100 mmol/L) at 90 °C for 2 h, and the concentrations of Na+ and K+ in shoots were analyzed by Flame Photometer (Sherwood410, Cambridge, UK).
SSR marker genotyping
Genomic DNA was extracted and purified from the young leaves using a modified CTAB method (Doyle and Doyle 1990). One thousand simple sequence repeat (SSR) primer pairs were designed from SSR-contained sequences on rice genome retrieved from http://www.gramene.org, and synthesized by the Sangon Biotech Co., Ltd. (Shanghai). To screen for polymorphisic loci, 12 genotypes (Laotoudao 1, Baidadu, Kendao 12, Xiaobaijingzihuadianbai, Jijing 61, Danjing 8, Shennong 91, Fushiguang, Kongyu 131, Yuntoudao, Zaojinfu, and Pingrang 10), three from Heilongjiang, two each from Jilin, Liaoning, Japan, and Korea, one from DPRK, were selected for primer amplification. Finally, 160 SSR markers were selected to genotype the panel of japonica rice, including 25 markers known to be linked to salt-tolerant QTLs from previous studies (Ammar et al. 2009; Mohammadi-Nejad et al. 2008; Sabouri et al. 2009; Thomson et al. 2010; Wang et al. 2012a, b).
PCR reaction was conducted in 10 μL volumes mixed with 1 μL of genomic DNA (25 ng/μL), 0.75 μL MgCl2 (25 mM), 0.15 μL dNTP mixtures (10 mM), 1 μL 10 × PCR buffer, 1 μL SSR primer pairs (2 μM), 0.1 μL Taq polymerase (10 U/μL), and 6 μL ddH20. The PCR amplification profile was 94 °C for 2 min, followed by 35 cycles of 94 °C for 30 s, 47 °C for 30 s, 72 °C for 30 s, then extended at 72 °C for 5 min. PCR products were mixed with loading buffer (2.5 mg/ml bromophenol blue, 2.5 mg/ml diphenylamine blue, 10 mM EDTA, 95 % formamide) and denatured at 94 °C for 5 min, and then incubated on ice for 5 min. The denatured PCR products were separated on 6 % denaturing polyacrylamide gel and visualized using silver staining. The stained bands were analyzed based on their migration distance relative to the pBR322 DNA Marker (Fermentas) using Quantity One software.
Statistical analysis
Phenotypic data analysis
All the basic statistical analyses were performed using the SPSS 13.0 for Windows (SPSS, Inc., Chicago, IL USA). The broad-sense heritability (H 2) was calculated and expressed as the ratio of the total genetic variance (V G ) over the phenotypic variance (V P ): H 2 = V G /V P .
Genetic diversity
Allelic information for SSR markers among accessions, including allele number, the Nei’s gene diversity (Nei and Takezaki 1983) and polymorphism information content (PIC) were estimated using PowerMarker V3.25 software (Liu and Muse 2005).
Population structure and differentiation analyses
The program Structure V2.3.2 (Pritchard et al. 2000) was used to test the hypotheses for 2–10 subpopulations (K) with an admixture model and correlated allelic frequencies, length of burn-in period equal to 10,000 iterations and a run of 100,000 replications of Markov Chain Monte Carlo after burn in. 10 runs of the Structure program were performed and an average likelihood value, LnP(D), across all runs was calculated for each K. The most likely number of clusters (K) was selected by comparing the logarithmized probabilities of data LnP(D) and ΔK according to Evanno et al. (2005). The CLUMPP software (Jakobsson and Rosenberg 2007) was used to integrate the results of replicate runs from STRUCTURE. Based on the correct K, each accession was assigned to a subpopulation for which the membership value (Q value) was >0.65 (Cui et al. 2013) and the population structure matrix (Q) was adopted for the association analysis. Using inferred subpopulations, an analysis of molecular variance (AMOVA) was performed under GenAlEx6.2 (Peakall and Smouse 2006), genetic distance and pair-wise FST among subpopulations were estimated with POPGENE version 1.31 developed by Yeh et al. (1999).
Relative kinship
The kinship matrix (K) was constructed on the basis of 160 SSRs using SPAGeDi (Hardy and Vekemans 2002), in order to estimate the genetic relatedness among individuals, with the negative value of kinship set to zero (Yu and Buckler. 2006).
Linkage disequilibrium
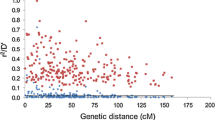
LD was estimated by calculating the square value of correlation coefficient (r 2) between all pairs of markers with the software package TASSEL 3.0 (http://www.maizegenetics.net). P values for each r 2 estimate were obtained with a two-sided Fisher’s exact test as implemented in TASSEL. Each pair of loci was categorized as linked (marker loci located on the same chromosome) or unlinked (marker loci located on different chromosomes). The LD was estimated for global, linked and unlined markers in the entire panel and each subpopulation, respectively. Heterozygous genotypes and rare alleles with minor allele frequency (MAF) < 0.05 were treated as missing data in further analysis (Bradbury et al. 2007). The pairs of loci were considered to have a significant LD if P < 0.01. To determine the average LD decay in the whole genome, significant intrachromosomal r 2 values were plotted against the genetic distance (cM) between markers in Microsoft Excel. The 99th percentile of r 2 distribution for unlinked markers was considered as the background level of LD, which determined whether LD is due to physical linkage (Mather et al. 2007). The r 2 values for pairs of SSR loci were plotted as a function of map distance (cM), and LD decay was estimated. The estimated genetic distance (cM) between loci was inferred from http://www.gramene.org.
Association mapping and favorable allele identification
Because the mixed linear model (MLM) accounts for the effects both of population structure information (Q-matrix) and pair-wise relatedness coefficients ‘kinship’ (K-matrix), and can significantly reduce spurious associations (Yu and Buckler. 2006), the marker-trait association mapping was carried out with the MLM model as implemented in the program TASSEL 3.0. The P value (P ≤ 0.01) determined whether a trait was associated with a marker and the r 2 indicating the fraction of the total variation explained by the marker was recorded. MapChart 2.2 was used to draw the map (Voorrips 2002).
Based on the results of association mapping, QTL alleles of loci significantly associated with the target traits were further analyzed. The computational procedure was carried out according to Zhang et al. (2013). The phenotypic allele effect was estimated through comparison between the average phenotypic value over accessions with the specific allele and that of all accessions:
where a i is the value of the phenotypic effects of the i-th allele, x ij is the phenotypic value over the j-th accession with the i-th allele, n i is the number of accessions with the i-th allele, N k is the phenotypic value over all accessions, and n k is the number of accessions. If a i > 0, the allele is considered to have a positive effect. In contrast, if a i < 0, the allele is he allele is considered to have a negative effect. The favorable alleles were then identified according to the breeding objective of each target trait.
Results
Phenotypic evaluations
Under salt stress of 140 mmol/L NaCl, mean value, coefficient of variation, kurtosis, and skewness for SDS and SKN were calculated in 341 japonica rice accessions (Table 1). Continuous distributions were observed in the two traits, and the phenotypic data followed a normal distribution based on the values of skewness and kurtosis statistics. A two-way analysis of variance (ANOVA) showed that differences among accessions for each trait were highly significant (P < 0.01), indicating a large amount of genetic variation existed in the population. The broad-sense heritability (H 2) for SDS and SKN was 82.82 and 90.51 %, respectively. Analysis of linear correlations showed that SDS was significantly positively correlated with SKN (r = 0.54, n = 341, P < 0.001).
Genetic diversity in the japonica rice panel
The allele number, gene diversity and polymorphism information content (PIC) were calculated to estimate the genetic diversity in the japonica rice panel. A total of 160 SSR markers were used to measure the genetic diversity of the population. All of them showed polymorphism with a total of 872 alleles which were detected across the 341 rice accessions. The allele number, gene diversity and PIC value of the 160 loci ranged from 2 to 9, 0.0233 to 0.8441, and 0.0236 to 0.8598, with an average of 5.4500, 0.5801 and 0.5366, respectively (Supplementary Table 2).
Population structure and kinship in the panel of 341 japonica rice accessions
STRUCTURE analysis with 55 unlinked SSR markers showed that the log-likelihood increased with the elevation of model parameter K, so the statistic ∆K was used to determine a suitable value for K. Here, the ∆K value was much higher for the model parameter K = 3 than for other values of K (Supplementary Fig. 1), suggesting that the total panel could be divided into three major subpopulations, designated as P1, P2 and P3, respectively. Figure 1 showed the population structure indicated by the Q matrix. P1 contained 91 accessions including 83 cultivars from Heilongjiang and Jilin province of China, and 8 cultivars from Japan. P2 consisted of 154 accessions including 64 cultivars from the rice cropping regions of Northeastern China, the remaining cultivars were from other countries, such as Japan, DPRK, Korea, and France. P3 included 27 accessions including 20 cultivars from Heilongjiang and Jilin province of China, and 7 cultivars from Russia. The remaining 69 accessions showed membership probabilities less than 0.65 in any given subpopulation and were thus classified as a mix group (Supplementary Table 1). Then, the corresponding Q matrix at K = 3 was used for the following association analysis.

Population structure of 341 japonica rice accessions based on 55 SSR markers (K = 3). The population is partitioned into three color-coded subpopulations. Each bar represents a single accession, and the colored subsections within each bar reflect the proportional contributions of each subpopulation to that accession. Colors of more than 65 % in red, green and blue represent P1, P2 and P3, respectively. Mixed colors with <65 % each color were grouped to MIX. The figures in the abscissa were the accession number in Supplementary Table 1
The pairwise kinship estimates based on 160 informative molecular markers showed that the majority of the pairs of japonica rice accessions (57.4 %) had zero estimated kinship values, while 21.8 % kinship estimates ranged from 0 to 0.05, 11.8 % from 0.05 to 0.1, and 26.4 % of the pairs had a value 0.10–0.20. The remaining pairs of accessions (2.3 %) had >0.25 kinship values, suggesting involvement of some common parental genotypes in the breeding history of these germplasm groups (Supplementary Fig. 2). These results indicated that most accessions in the panel have no or very weak kinship, which might be attributed to the broad range collection of genotypes and the exclusion of similar genotypes before analysis.
Population differentiation
AMOVA was performed to investigate population differentiation (Supplementary Table 3). It showed that a significant difference existed among subpopulations, within subpopulations and within accessions (P < 0.01). 10.30 % of the total molecular variation in the panel was attributed to the differentiation among subpopulations, 89.70 % of total genetic variance was attributed to the difference within subpopulations. It indicated that the main genetic variation occurred within subpopulations. Therefore, it is necessary to do further analysis of the genetic diversity in different subpopulations.
The pair-wise F ST value ranged from 0.0492 (P1 with P2) to 0.1067 (P2 with P3) among subpopulations, with an average of 0.0834 (P < 0.01). The genetic distance among subpopulations had the same trend. The genetic distance between P1 and P2 was 0.1209, while it was 0.3835 between P2 and P3 (Supplementary Table 4).
Genetic diversity within subpopulations
The genetic diversity is presented for each subpopulation in Supplementary Table 5. Among subpopulations, P2 had the highest gene diversity index (0.5951) with 5.01 alleles per locus and the PIC value of 0.5456. However, P1 had the lowest gene diversity index (0.4999) with 4.64 alleles per locus and the PIC value of 0.4545. A total of 264 (30.27 %) of the 872 alleles that were detected in the total populations were subpopulation-specific alleles. Moreover, P2 had more subpopulation-specific alleles (91 or 11.79 %) than others (Supplementary Table 5).
Linkage disequilibrium and LD decay
As the 341 japonica rice accessions could be divided into three subpopulations, pairwise LD estimates were performed in the total panel and in each subpopulation using a total of 160 SSR markers (Table 2). In the total panel, the average r 2 of locus pairs was 0.0107, and 40.05 % were significant (P ≤ 0.01), suggesting that the LD level is very high in the panel. Moreover, the average r 2 of linked marker pairs was 0.0148, and the percentage of linked marker pairs in significant LD was 49.39 %, both of which were higher than those for unlinked marker pairs (0.0104 and 40.44 %, respectively), demonstrating that physical linkage is predominant in determining LD compared with random forces. Further analysis of the LD in the subpopulation P1, P2 and P3 showed that the average r 2 in subgroups (ranging from 0.0131 to 0.1009) was larger than that in the total panel, suggesting that the LD level was elevated when the panel was classified into subpopulation. Comparing the sample size and the average r 2 of P1, P2 and P3, it revealed that the average r 2 of the global, linked, and unlinked markers decreased with the increase of sample size. Further analysis of the LD in subpopulations showed that both average r 2 and proportion of significant LD for linked loci were still higher than those for unlinked markers, which reinforced the view that physical linkage strongly influences LD in this panel of japonica rice accessions.
The distribution of data points in the plot of LD (r 2) decay against distance (cM) for the significant pairs showed that LD was not a simple monotonic function of the distance between markers. However, r 2 decreased as genetic distance between loci pairs increased, indicating that the probability of LD is low between distant locus pairs (Supplementary Fig. 3). In the total panel, the 99th percentile of r 2 distribution for unlinked markers, which determined the background level of LD, was 0.0918, and LD decayed to the background level within about 20 cM. Among the subpopulation P1, P2 and P3, the 99th percentile of r 2 distribution for unlinked markers was 0.1025, 0.0889, and 0.4186, and LD decayed to the background level within about 30, 20 and 50, respectively. A much slower decay of LD was observed within subpopulations, which might be attributed to the limited population size and narrow genetic background that inhibit LD decay.
Association between SSR markers and salt tolerance-related traits
Marker–trait associations (MTAs) for salt tolerance-related traits, SDS and SKN, were performed with the MLM model, considering both population structure (Q) and kinship (K), implemented in TASSEL 3.0 (Table 3). A total of 10 marker loci with the R 2 ranged of 4.76–11.82 % were identified to be significantly (P ≤ 0.01) associated with salt tolerance at the seedling stage, three on Chr.1, two on Chr.2, and one each on Chr.3, 4, 6, 7, 8. Of them, 5 and 7 markers were significantly associated with SDS and SKN, respectively. Two markers, including RM1324 on Chr.3, and RM418 on Chr.7, were significant for both traits. 9 out of the 10 detected marker loci confirmed or narrowed the genomic region reported to harbor QTLs for salt tolerance by linkage mapping in previous reports. RM283 is new unreported markers revealing associations with salt tolerance in this set of japonica rice germplasm. In addition, four salt tolerance-related genes were located in the QTL regions in the present study (Table 3; Supplementary Fig. 4).
Favorable allele mining
The higher SDS and SKN indicate stronger salt tolerance. Thus, favorable allele mining was focused on the loci that had positive phenotypic effects. Phenotypic effects of each QTL allele for the 10 associated loci were measured according to the method mentioned above. For each allele, not all but only the maximum of three carriers are listed. A summary of phenotypic effects and typical carrier accessions for each favorable allele are shown in Supplementary Table 6. A total of 11 and 19 favorable alleles were identified for SDS and SKN, respectively. Among the alleles associated with SDS, RM1324-160 bp had the greatest positive phenotypic effect and increased SDS by 6.53, which can be found in cultivars Kelista, Tiantai and Mudanjiang 2. Similarly, RM283-161 bp had the greatest positive phenotypic effect for SKN and was able to increase SKN by 0.35, which can be found in cultivars Qiuguang, Hejiang 18 and Tengxi 180. Pearson correlation analysis between the number of favorable alleles and each salt tolerance trait was carried out, and highly positive significance was found in both SDS (r = 0.644, P < 0.001) and SKN (r = 0.588, P < 0.001). It suggested that complementary recombination with the favorable alleles had the potential for salt tolerance in rice to further improve.
Prediction for novel parental combination
Favorable cultivars tolerant to salt could be potential donor parents in breeding. In this study, according to the phenotypic values and the number of favorable alleles, the five best cultivars of each trait were selected. Detailed information about each cultivar is summarized in Supplementary Table 7. In addition, based on the number of positive alleles that could be pyramided into an individual plant and the expected phenotypic effects, the five best cross combinations for improving SDS and SKN, respectively, were proposed (Table 4). It was found that some cultivars present repeatedly in the novel parental combinations: for example, Derta in the combinations for SDS, Huangbaiguang for SKN, Mudanjiang 2 and Tixi 180 for both SDS and SKN, indicating these cultivars possess unique elite alleles.
Discussion
Genetic diversity in the rice panel
The appropriate sample size, abundant diversity in phenotype and genotype of an association mapping panel are critical to the success of association study. In this study, the association mapping panel was a collection of 341 japonica rice accessions released over many years in China, Japan, Korea, the Democratic People’s Republic of Korea, Russia, and France representing elite lines bred for genes and QTL alleles for yield and abiotic stress resistance. An average of 5.45 alleles per locus which ranged from 2 to 9, the value was significantly lower than 11.9 alleles per locus ranging from 2 to 34, in the world collections of rice materials (Xu et al. 2005). However, this value exceeded most of the previous reports (Jin et al. 2010; Zhang et al. 2011) and close to Lapitan et al. (2007) (5.89 over 151 SSR loci in Philippine rice cultivars). The gene diversity in this study ranged from 0.0236 to 0.8598 with an average of 0.5801. The value is much higher than that determined by Agrama and Eizenga (2008) in US rice collection (0.43), and Thomson et al. (2007) in improved indica rice varieties (0.46). The PIC values of the rice accessions ranged from 0.0233 to 0.8441 with an average of 0.5366 in japonica rice varieties. It was higher than the collection of 416 rice accessions including landrace, cultivars and breeding lines (Jin et al. 2010, PIC = 0.4214) and 347 improved japonica rice varieties (Cui et al. 2013, PIC = 0.3137), but lower than the collection of 236 rice materials (Xu et al. 2005, PIC = 0.74). The differences of genetic diversity in different reports are related with the number of rice accessions analyzed and the choice of rice germplasm, the number of SSR loci and the SSR repeat type. A higher number of accessions in the sample lead to a more diverse range of germplasm simply by sampling, and a larger number of loci will lead to a higher number of alleles and thus a higher apparent level of genetic diversity. The higher genetic diversity in our study indicated that huge phenotypic variations increased detection power and allowed the quantification of more allelic effects. The results showed that these alleles are more suitable for association mapping.
Among subpopulations, the group P1, P2 and P3 showed significant differences in gene diversity and PIC. Significant differentiation assessed in AMOVA was observed between the three subpopulations. When compared with P1, P2 and P3 had a higher level of gene diversity and PIC. Moreover, P1 subpopulation has 33 population-specific alleles, while P2 and P3 had 91 and 78 population-specific alleles, respectively (Supplementary Table 5). The reason might be due to that P2 and P3 subpopulations in this study contained more accessions from abroad than P1.
Population structure and differentiation in the association panel
Understanding the population structure is important to avoid identifying spurious associations between phenotype and genotype in association mapping (Pritchard et al. 2000). Many studies have been conducted about genetic structure of rice (Garris et al. 2005; Wang et al. 2014). Garris et al. (2005) detected five major groups from a diverse sample of 234 rice accessions including indica, aus, tropical japonica, temperate japonica, and aromatic. Wang et al. (2014) discussed the population structure among 278 improved japonica rice varieties and identified nine main clusters that corresponded to the major geographic regions. In this study, three main subpopulations were detected among 341 japonica rice accessions (Fig. 1) including P1, P2 and P3, and it was found that most of the japonica rice accessions from the same or nearby geographic regions had a closer genetic relationship and were clustered in the same subpopulation. However, a few accessions were not consistent with the geographic regions. This could be due to their long breeding history, the intercrossing and introgressing of accessions from diverse backgrounds and the presence of different ancestries with more than one background in the same geographic region. It’s worth noting that the accessions from Heilongjiang and Jilin province of China were distributed in three subgroups, indicating the exchange and domestication of germplasm between the two provinces and other geographic regions.
F ST values were significant for all the subpopulations (P < 0.01), indicating the real existence of a genetic differentiation. The differentiation between P1 and P2 was the lowest (F ST = 0.0492), while the differentiation between P2 and P3 was the highest (F ST = 0.1067). It indicated that higher level of differentiation was present between P2 and P3, which indicated that the accessions in P2 and P3 had further genetic relationship. It also revealed that during the breeding procedure, the narrow genetic base might have been broadened for crosses between the accessions in P2 and P3.
Linkage disequilibrium in the rice panel
The extent of LD can provide information for the needed marker density and mapping resolution in association mapping study. If LD declines rapidly, the genome scan would require excessive marker density for a feasible identification of the candidate genes. If LD is too large, the resolution may be low, but a genome scan with a lower marker density would be possible (Garris et al. 2003). In this study, The average r 2 in our association panel is 0.0107, which is lower than 0.027 and 0.08 reported by Jin et al. (2010) and Li et al. (2011), respectively. This can be explained due to different significance thresholds and different plant materials that were used in these studies. The LD decayed to the background level varied from about 20 to 50 cM among the total panel and three subpopulations in our study, suggesting that the mapping resolutions could possibly be achieved between 20 and 50 cM with variation among different genetic groups and different chromosomes. In some studies, LD decay was observed at 1 cM or less in rice based on nucleotide polymorphism markers, such as SNP (Garris et al. 2003; Mather et al. 2007), whereas other studies indicated that LD decays at 20–50 cM using SSR markers (Agrama et al. 2007; Jin et al. 2010; Li et al. 2011), being these reports consistent with the result of this study. In a special case for a worldwide collection of O. sativa and its wild relatives, the LD varied in a large range of 50–225 cM (Agrama and Eizenga 2008). These studies suggest that the extent of LD varies among different genomic regions, different types of marker used and different materials with different reproductive modes and different accessions studied. For the approximately 389 Mb rice genome, the 160 SSRs that cover the rice genome at a density of approximately 10 cM (assuming an average of 250 kb/cM across the genome) provided a reasonable resolution for the association mapping in this study.
Physical linkage that determines LD between molecular marker and causative polymorphisms is the genetic basis for association mapping of genes or QTLs underlying traits of interest (Flint-Garcia et al. 2003). In this study, the extent of LD of linked markers in the entire panel and subpopulations is significantly higher than that of unlinked markers (Table 2), suggesting that physical linkage strongly influences LD in this japonica rice panel, and indicating that this japonica rice panel is suitable for association analysis.
Population structure is one of several important factors that have strong influences on LD (Flint-Garcia et al. 2003). In our LD estimations, we took into account the effect of population structure by subdividing the total panel into three subpopulations. Various levels of LD in subpopulations were observed, indicating that population structure has significant impact on LD (Table 2). Based on LD analyses in the whole genome level, the LD level was elevated when the panel was classified into subpopulations (Table 2), comparing the sample size and the average r 2 of P1, P2 and P3, it revealed that the average r 2 of the global, linked, and unlinked markers decreased with the increase of sample size, indicating that the impact of population structure on LD is at least partially attributed to the effect of sample size. Therefore, it implied that variable extents of LD are expected within the different genetic groups and highlight the fact that different marker densities will be required if association studies are planned in the different genetic groups.
Comparison of QTLs for salt tolerance with previous reports
The MTAs identified by association mapping in this study were compared with the QTLs identified by linkage mapping in previous studies, according to the same SSR markers and physical location of markers linked with the QTLs. 9 out of the 10 marker loci in this study were consistent with salt-tolerance-related QTLs in previous studies (Table 3). The locus RM292 on chromosome 1 was located within the region of qKLV-1.1 (Pandit et al. 2010); RM213 on chromosome 2 was located within the region of QSst2 (Zang et al. 2008); RM539 on chromosome 6 was located within the region of qDRW6 (Wang et al. 2012a, b), qDTF6.1 s (Mohammadi et al. 2013) and QSkc6 (Zang et al. 2008) simultaneously; RM1287 on chromosome 1 was the same marker for Saltol (Mohammadi-Nejad et al. 2008), and located within the region of qSKC-1 (Lin et al. 2004) and qSNC1 (Thomson et al. 2010); RM1379 on chromosome 2 was mapped together with three QTLs (i.e. qPH2, qRKC2 and qCHL2), flanked by SSR markers RM13197 and RM6318 (Thomson et al. 2010), and located within the region for salt tolerance reported by Sabouri et al. (2009) and Wang et al. (2011) simultaneously; RM551 on chromosome 4 and RM281 on chromosome 8 shared the same marker for salt tolerance reported by Mohammadi et al. (2013) and Ammar et al. (2009); Remarkably, RM1324 on chromosome 3 and RM418 on chromosome 7 associated with both salt-tolerance-related traits (SDS and SKN) at the seedling stage in this study, were also associated with salt tolerance at the germination and early seedling stage in our previous study (Zheng et al. 2014), and there were no reports to date for salt tolerance in other studies, suggesting that they were novel markers associated with salt tolerance. Therefore, the markers for salt tolerance mentioned above, which were detected in different mapping populations, different growth period and various environments, were significant markers for salt tolerance in rice. Furthermore, the markers that were localized within a QTL interval for salt tolerance not only validated the QTL but also provided a more closely linked marker. These markers may be useful for breeding programs in rice based on MAS and may accelerate the development of salt tolerant rice varieties. In addition, RM283 was reported for the first time, potentially indicating the novel markers associated with salt tolerance.
Co-localization of salt tolerance-related genes in the QTL regions
Four salt tolerance-related genes previously reported were found to coincide with the QTL regions in the present study (Table 3; Supplementary Fig. 4). OsEREBP1, a AP2/ERF transcription factor that significantly affected the transcript level by salt, ABA or severe cold (5 °C) (Serra et al. 2013) was located in the same region as the marker loci RM213. The locus RM539 was co-located with OsABF2 identified by Hossain et al. (2010), which was a bZIP transcription factor gene, and induced by different types of abiotic stress treatments such as salinity, drought, cold, oxidative stress and ABA. The locus RM1287 was close to the HKT1;5 gene, which encoded a member of HKT-type transporters, was involved in regulating K+/Na+ homeostasis under salt stress (Ren et al. 2005). OsAHP1, a rice authentic histidine phosphotransfer protein that mediated cytokinin signaling pathway and salt and drought stress responses in rice (Sun et al. 2014a, b), was located in the same region as the marker loci RM281. The markers for salt tolerance mentioned above, validated these genes further, and will accelerate the development of gene-based functional markers for molecular breeding applications.
Pyramiding favorable alleles to improve salt tolerance in rice
Among the association mapping population that contains a large number of genotypes, the molecular markers that are associated with the target trait can be analyzed at an allelic level by association analysis. In addition, the contributions of different alleles that are carried by different germplasms to the target trait can be determined to identify cultivars that carry multiple favorable alleles. Next, by hybridizing different cultivars and selecting the RIL that carry the maximum number of favorable alleles, the RIL that have improved target traits can be bred. The predicted results can be further confirmed by crop breeding. For example, using 27 detected QTL for rice seed vigor, Dang et al. (2014) designed 15 elite parental combinations for improving seed vigor in rice. Similarly, by designed QTL pyramiding based on combinations of qgw8 and qgs3 alleles with molecular marker–assisted selection, Mather et al. (2007) developed a new elite indica variety, Huabiao1, with substantially improved grain quality, indicating that novel alleles from different cultivars can be pyramided into a new cultivar. In this study, by comparing the average phenotypic value of each allele in the 12 detected MTAs, 11 and 19 favorable alleles for SDS and SKN were identified, respectively, suggesting that the favorable alleles and their carrier materials have great potential for developing salt tolerant rice varieties in future molecular breeding programs. In addition, we also predict some parental combinations based on the number of positive alleles that could be pyramided into an individual plant and the expected phenotypic effects. In these combinations, some cultivars are repeatedly present, of which, two favorable cultivars, Mudanjiang 2 and Texi 180, can be used to improve SDS and SKN simultaneously, are considered important salt tolerance donor parents in breeding. If we want to improve multiple traits, we might pyramid all the favorable alleles into one cultivar as far as possible, it suggested that a multi-parent population should be constructed using cultivars that possessed most of the favorable alleles, and a ranking system for MAS should be developed based on the results of association mapping.
References
Agrama HA, Eizenga GC (2008) Molecular diversity and genomewide linkage disequilibrium patterns in a worldwide collection of Oryza sativa and its wild relatives. Euphytica 160:339–355
Agrama HA, Eizenga GC, Yan W (2007) Association mapping of yield and its components in rice cultivars. Mol Breed 19:341–356
Ahmadi N, Negrão S, Katsantonis D, Frouin J, Ploux J, Letourmy P, Droc G, Babo P, Trindade H, Bruschi G, Greco R, Oliveira MM, Piffanelli P, Courtois B (2011) Targeted association analysis identified japonica rice varieties achieving Na+/K+ homeostasis without the allelic make-up of the salt tolerant indica variety Nona Bokra. Theor Appl Genet 123:881–895
Ammar MHM, Pandit A, Singh RK, Sameena S, Chauhan MS, Singh AK, Sharma PC, Gaikwad K, Sharma TR, Mohapatra T, Singh NK (2009) Mapping of QTLs controlling Na+, K+ and Cl− ion concentrations in salt tolerant indica rice variety CSR27. J Plant Biochem Biotechnol 18:139–150
Asch F, Dingkuhn M, DörZing K, Miezan K (2000) Leaf K/Na ratio predicts salinity induced yield loss in irrigated rice. Euphytica 113:109–118
Bonilla P, Mackill D, Deal K, Gregorio G (2002) RFLP and SSLP mapping of salinity tolerance genes in chromosome 1 of rice (Oryza sativa L.) using recombinant inbred lines. Philipp Agric Sci 85:68–76
Bradbury PJ, Zhang Z, Kroon DE, Casstevens TM, Ramdoss Y, Buckler ES (2007) TASSEL: software for association mapping of complex traits in diverse samples. Bioinformatics 23:2633–2635
Cui D, Xu CY, Tang CF, Yang CG, Yu TQ, Xin-xiang A, Cao GL, Xu FR, Zhang JG, Han LZ (2013) Genetic structure and association mapping of cold tolerance in improved japonica rice germplasm at the booting stage. Euphytica 193:369–382
Dang XJ, Thi TGT, Dong GS, Wang H, Edzesi WM, Hong DL (2014) Genetic diversity and association mapping of seed vigor in rice (Oryza sativa L.). Planta 239:1309–1319
Doyl JJ, Doyle JL (1990) Isolation of plant DNA from fresh tissue. Focus 12:13–15
Ersoz ES, Yu J, Buckler ES (2007) Applications of linkage disequilibrium and association mapping in crop plants. Genomics-assisted crop improvement Springer, Dordrecht, pp 97–120
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Flint-Garcia S, Thornsberry J, Buckler ES (2003) Structure of linkage disequilibrium in plants. Annu Rev Plant Biol 54:357–374
Flowers TJ (2004) Improving crop salt tolerance. J Exp Bot 55:307–319
Garris AJ, McCouch SR, Kresovich S (2003) Population structure and its effects on haplotype diversity and linkage disequilibrium surrounding the xa5 locus of rice (Oryza sativa L.). Genetics 165:759–769
Garris AJ, Tai TH, Coburn J, Kresovich S, McCouch SR (2005) Genetic structure and diversity in Oryza sativa L. Genetics 169:1631–1638
Gregorio GB, Senadhira D, Mendoza RD (1997) Screening rice for salinity tolerance. IRRI Discuss Paper Ser 22:1–30
Gregorio GB, Senadhira D, Mendoza RD, Manigbas NL, Roxas JP, Guerta CQ (2002) Progress in breeding for salinity tolerance and associated abiotic stresses in rice. Field Crop Res 76:91–101
Hardy OJ, Vekemans X (2002) SPAGEDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol Ecol Notes 2:618–620
Hossain MA, Cho JI, Han M, Ahn CH, Jeon JS, An G, Park PB (2010) The ABRE-binding bZIP transcription factor OsABF2 is a positive regulator of abiotic stress and ABA signaling in rice. J Plant Physiol 167:1512–1520
Huang XH, Wei XH, Sang T et al (2010) Genome-wide association studies of 14 agronomic traits in rice landraces. Nat Genet 42:961–967
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 21:1801–1806
Jia LM, Yan WG, Zhu CS, Agrama HA, Jackson A, Yeater K, Li XB, Huang BH, Hu BL, McClung A, Wu DX (2012) Allelic analysis of sheath blight resistance with association mapping in rice. PLoS One 7:e32703
Jin L, Lu Y, Xiao P, Sun M, Corke H, Bao JS (2010) Genetic diversity and population structure of a diverse set of rice germplasm for association mapping. Theor Appl Genet 121:475–487
Jun TH, Van K, Kim MY, Lee SH, Walker DR (2008) Association analysis using SSR markers to find QTL for seed protein content in soybean. Euphytica 162:179–191
Kim DM, Ju HG, Kwon TR, Oh CS, Ahn SN (2009) Mapping QTLs for salt tolerance in an introgression line population between japonica cultivars in rice. J Crop Sci Biotech 12:121–128
Koyama ML, Levesley A, Koebner RM, Flowers TJ, Yeo AR (2001) Quantitative trait loci for component physiological traits determining salt tolerance in rice. Plant Physiol 125:406–422
Kraakman ATW, Niks RE, Van den Berg PMMM, Stam P, Van Eeuwijk FA (2004) Linkage disequilibrium mapping of yield and yield stability in modern spring barley cultivars. Genetics 168:435–446
Lapitan VC, Brar DS, Abe T, Redona ED (2007) Assessment of genetic diversity of Philippine rice cultivars carrying good quality traits using SSR markers. Breed Sci 57:236–270
Le Gouis J, Bordes J, Ravel C, Heumez E, Faure S, Praud S, Galic N, Remoue C, Balfourier F, Allard V, Rousset M (2012) Genome-wide association analysis to identify chromosomal regions determining components of earliness in wheat. Theor Appl Genet 124:597–611
Lee SY, Ahn JH, Cha YS, Yun DW, Lee MC, Ko JC, Lee KS, Eun MY (2007) Mapping QTLs related to salinity tolerance of rice at the young seedling stage. Plant Breed 126:43–46
Li XB, Yan WG, Agrama H, Jia LM, Shen XH, Jackson A, Moldenhauer K, Yeater K, McClung A, Wu DX (2011) Mapping QTLs for improving grain yield using the USDA rice mini-core collection. Planta 234:347–361
Lijavetzky D, Cabezas JA, Ibanez A, Rodriguez V, Martinez-Zapater JM (2007) High throughput SNP discovery and genotyping in grapevine (Vitis vinifera L.) by combining a re-sequencing approach and SNPlex technology. BMC Genom 8:424
Lin HX, Zhu MZ, Yano M, Gao JP, Liang ZW, Su WA, Hu XH, Ren ZH, Chao DY (2004) QTLs for Na+ and K+ uptake of the shoots and roots controlling rice salt tolerance. Theor Appl Genet 108:253–260
Liu A, Burke JM (2006) Patterns of nucleotide diversity in wild and cultivated sunflower. Genetics 173:321–330
Liu K, Muse SV (2005) PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21:2128–2129
Malysheva-Otto LV, Ganal MW, Ro¨der MS (2006) Analysis of molecular diversity, population structure and linkage disequilibrium in a worldwide survey of cultivated barley germplasm (Hordeum vulgare L.). BMC Genet 7:6
Mather KA, Caicedo AL, Polato NR, Olsen KM, McCouch S, Purugganan MD (2007) The extent of linkage disequilibrium in rice (Oryza sativa L.). Genetics 177:2223–2232
Mohammadi R, Mendioro MS, Diaz GQ, Gregorio GB, Singh RK (2013) Mapping quantitative trait loci associated with yield and yield components under reproductive stage salinity stress in rice (Oryza sativa L.). J Genet 92:433–443
Mohammadi-Nejad G, Arzani A, Rezai AM, Singh RK, Gregorio GB (2008) Assessment of rice genotypes for salt tolerance using microsatellite markers associated with the saltol QTL. Afr J Biotech 7:730–736
Moradi F, Ismail AM, Gregorio GB, Egdane JA (2003) Salinity tolerance of rice during reproductive development and association with tolerance at the seedling stage. Ind J Plant Physiol 8:105–116
Negrão S, Courtois B, Ahmadi N, Abreu I, Saibo N, Oliveira MM (2011) Recent updates on salinity stress in rice: from physiological to molecular responses. Crit Rev Plant Sci 30:329–377
Nei M, Takezaki N (1983) Estimation of genetic distances and phylogenetic trees from DNA analysis. Proceedings of the 5th world congress. Genet Appl Livest Prod 21:405–412
Niu Y, Xu Y, Liu XF, Yang SX, Wei SP, Xie FT, Zhang YM (2013) Association mapping for seed size and shape traits in soybean cultivars. Mol Breed 31:785–794
Nordborg M, Borevitz JO, Bergelson J, Berry CC, Chory J, Hagenblad J, Kreitman M, Maloof JN, Noyes T, Oefner PJ, Stahl EA, Weigel D (2002) The extent of linkage disequilibrium in Arabidopsis thaliana. Nat Genet 30:190–193
Ordonez SA Jr, Silva J, Oard JH (2010) Association mapping of grain quality and flowering time in elite japonica rice germplasm. J Cereal Sci 51:337–343
Pandit A, Rai V, Bal S, Sinha S, Kumar V, Chauhan M, Gautam RK, Singh R, Sharma PC, Singh AK, Gaikwad K, Sharma TR, Mohapatra T, Singh NK (2010) Combining QTL mapping and transcriptome profiling of bulked RILs for identification of functional polymorphism for salt tolerance genes in rice (Oryza sativa L.). Mol Genet Genomics 284:121–136
Peakall R, Smouse PE (2006) GENALEX 6: genetic analysis in Excel. Population genetic software for teaching and research. Mol Ecol Notes 6:288–295
Prasad SR, Bagali PG, Hittalmani S, Shashidhar HE (2000) Molecular mapping of quantitative trait loci associated with seedling tolerance of salt stress in rice (Oryza sativa L.). Curr Sci 78:162–164
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Qadir M, Quillérou E, Nangia V, Murtaza G, Singh M, Thomas RJ, Noble AD (2014) Economics of salt-induced land degradation and restoration. Nat Resour Forum 38:282–295
Ren ZH, Gao JP, Li LG, Cai XL, Huang W, Chao DY, Zhu MZ, Wang ZY, Luan S, Lin HX (2005) A rice quantitative trait locus for salt tolerance encodes a sodium transporter. Nat Genet 37:1141–1146
Sabouri H, Rezai AM, Moumeni A, Kavousi A, Katouzi M, Sabouri A (2009) QTLs mapping of physiological traits related to salt tolerance in young rice seedlings. Biol Plant 53:657–662
Serra TS, Figueiredo DD, Cordeiro AM, Almeida DM, Lourenco T, Abreu IA, Sebastian A, Fernandes L, Contreras-Moreira B, Oliveira MM, Saibo NJM (2013) OsRMC, a negative regulator of salt stress response in rice, is regulated by two AP2/ERF transcription factors. Plant Mol Biol 82:439–455
Sun J, Zou DT, Luan FS, Zhao HW, Wang JG, Liu HL, Xie DW, Su DQ, Ma J, Liu ZL (2014a) Dynamic QTL analysis of the Na+ content, K+ content, and Na+/K+ ratio in rice roots during the field growth under salt stress. Biol Plant 58:689–696
Sun LJ, Zhang Q, Wu JX, Zhang LQ, Jiao XW, Zhang SW, Zhang ZG, Sun DY, Lu TG, Sun Y (2014b) Two rice authentic histidine phosphotransfer proteins, OsAHP1 and OsAHP2, mediate cytokinin signaling and stress responses in rice. Plant Physiol 165:335–345
Takehisa H, Shimodate T, Fukuta Y, Ueda T, Yano M, Yamaya T, Kameya T, Sato T (2004) Identification of quantitative trait loci for plant growth of rice in paddy field flooded with salt water. Field Crops Res 89:85–95
Thomson MJ, Septiningsih EM, Suwardjo F, Santoso TJ, Silitonga TS, McCouch SR (2007) Genetic diversity analysis of traditional and improved Indonesian rice (Oryza sativa L.) germplasm using microsatellite markers. Theor Appl Genet 114:559–568
Thomson MJ, Ocampo M, Egdane J, Rahman MA, Sajise AG, Adorada DL, Tumimbang-Raiz E, Blumwald E, Seraj ZI, Singh RK, Gregorio GB, Ismail AM (2010) Characterizing the Saltol quantitative trait locus for salinity tolerance in rice. Rice 3:148–160
Vanniarajan C, Vinod KK, Pereira A (2012) Molecular evaluation of genetic diversity and association studies in rice (Oryza sativa L.). J Genet 91:1–11
Voorrips RE (2002) MapChart: software for the graphical presentation of linkage maps and QTLs. J Hered 93:77–78
Wallender WW, Tanji KK (2011) Agricultural salinity assessment and management, 3rd edn. American Society of Civil Engineers (ASCE)
Wang J, McClean P, Lee R, Goos R, Helms T (2008) Association mapping of iron deficiency chlorosis loci soybean (Glycine max L. Merr.) advanced breeding lines. Theor Appl Genet 116:777–787
Wang Z, Wang J, Bao Y, Wu Y, Zhang H (2011) Quantitative trait loci controlling rice seed germination under salt stress. Euphytica 178:297–307
Wang Z, Chen Z, Cheng J, Lai Y, Wang J, Bao Y, Huang J, Zhang H (2012a) QTL analysis of Na+ and K+ concentrations in roots and shoots under different levels of NaCl stress in rice (Oryza sativa L.). PLoS One 7:e51202
Wang Z, Cheng J, Chen Z, Huang J, Bao Y, Wang J, Zhang H (2012b) Identification of QTLs with main, epistatic and QTL × environment interaction effects for salt tolerance in rice seedlings under different salinity conditions. Theor Appl Genet 125:807–815
Wang JG, Jiang TB, Zou DT, Zhao HW, Li Q, Liu HL, Zhou CJ (2014) Genetic diversity and genetic relationships of japonica rice varieties in Northeast Asia based on SSR markers. Biotechnol Biotec Equip 28:230–237
Wen WW, Mei HW, Feng FJ, Yu SB, Huang ZC, Wu JH, Chen L, Xu XY, Luo LJ (2009) Population structure and association mapping on chromosome 7 using a diverse panel of Chinese germplasm of rice (Oryza sativa L.). Theor Appl Genet 119:459–470
Wu Y, Hu Y, Xu G (2009) Interactive effects of potassium and sodium on root growth and expression of K/Na transporter genes in rice. Plant Growth Regul 57:271–280
Xu Y, Crouch J (2008) Marker-assisted selection in plant breeding: from publications to practice. Crop Sci 48:391–407
Xu YB, Beachell H, McCouch SR (2005) A marker-based approach to broadening the genetic base of rice in the USA. Crop Sci 44:1947–1959
Yan J, Shah T, Warburton ML, Buckler ES, McMullen MD, Crouch J (2009) Genetic characterization and linkage disequilibrium estimation of a global maize collection using SNP markers. PLoS One 4:e8451
Yan JB, Warburton M, Crouch J (2011) Association mapping for enhancing maize (Zea mays L) genetic improvement. Crop Sci 51:433–449
Yeh FC, Yang RC, Boyle T (1999) POPGENE: microsoft window-based freeware for population genetic analysis. Version 1.31. University of Alberta, Canada
Yoshida S, Forno DA, Cock JH, Gomez KA (1976) Laboratory manual for physiological studies of rice. International Rice Research Institute, Manila, p 38
Yu JM, Buckler ES (2006) Genetic association mapping and genome organization of maize. Curr Opin Biotechnol 17:1–6
Zang J, Sun Y, Wang Y, Yang J, Li F, Zhou YL, Zhu LH, Jessica R, Fotokian M, Xu JL, Li ZK (2008) Dissection of genetic overlap of salt tolerance QTLs at the seedling and tillering stages using backcross introgression lines in rice. Sci China C Life Sci 51:583–591
Zeng LH, Shannon MC (2000) Salinity effects on seedling growth and yield components of rice. Crop Sci 40:996–1003
Zhang P, Li J, Li X et al (2011) Population structure and genetic diversity in a rice core collection (Oryza sativa L.) investigated with SSR markers. PLoS One 6:e27565
Zhang T, Qian N, Zhu X, Chen H, Wang S, Mei H, Zhang Y (2013) Variations and transmission of QTL alleles for yield and fiber qualities in upland cotton cultivars developed in China. PLoS One 8:e57220
Zheng HL, Liu BW, Zhao HW, Wang JG, Liu HL, Sun J, Xing J, Zou DT (2014) Identification of QTLs for salt tolerance at the germination and early seedling stage using linkage and association analysis in japonica rice. Chin J Rice Sci 28:358–366 (in Chinese with an English Abstract)
Zhu YL, Song QJ, Hyten DL et al (2003) Single-nucleotide polymorphisms in soybean. Genetics 63(3):1123–1134
Acknowledgments
This work was supported by the National Science and Technology support programs of China (2011BAD35B02-01 and 2013BAD20B04), Science and Technology support programs of the Science and Technology Ministry of China (2011BAD16B11).
Author information
Authors and Affiliations
Corresponding author
Additional information
Hongliang Zheng and Jingguo Wang have contributed equally to this work.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zheng, H., Wang, J., Zhao, H. et al. Genetic structure, linkage disequilibrium and association mapping of salt tolerance in japonica rice germplasm at the seedling stage. Mol Breeding 35, 152 (2015). https://doi.org/10.1007/s11032-015-0342-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11032-015-0342-1