
Abstract
The decline of the eastern oyster (Crassostrea virginica) has prompted various restoration and aquaculture efforts. Recent field surveys in Rhode Island suggest that wild populations are increasing, yet the factors contributing to expansion are unknown. We used a population genetic approach to characterize genetic differences between wild and cultured oyster populations and explore the extent of connectivity and admixture between groups. Individual oysters from four wild, three farmed, and two restored populations were collected within or just outside Ninigret Pond, a coastal lagoon highly influenced by human activity, and genotyped at 13 microsatellite loci. Results from the multi-locus genotype data showed that wild populations were more genetically diverse than the cultured populations. We also observed significant genetic differentiation between paired wild and cultured populations but not between pairs of wild populations. A cluster analysis detected substantial admixture between wild and cultured groups. As oyster aquaculture and restoration activities are forecasted to increase in the future, this study highlights the potential degree of genetic introgression between remnant wild populations and less diverse, hatchery-reared stocks. Those tasked with preserving our living natural resources should carefully consider how the juxtaposition of aquaculture, restored, and wild populations at small spatial scales will impact the genetic composition and evolutionary trajectories of species in decline for generations to come.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Environmental landscapes and the ecosystems they support have been and continue to be impacted by human activity. Many estuarine ecosystems are casualties of centuries of overfishing and subsequent habitat degradation (Jackson et al. 2001). Oysters, which are ecological and economic staples of coastal communities, have experienced an 85% reduction in biomass worldwide and 70% of natural oyster reefs are in poor condition (Beck et al. 2011). Along the Western Atlantic coast of the US, many eastern oyster, Crassostrea virginica, populations that once supported wild fisheries exceeding 5–35 million kg in total landings annually are now practically extinct (Kirby 2004; Zu Ermgassen et al. 2012). The dramatic decline of the eastern oyster and the inherent and economic benefits it provides, have prompted efforts to conserve and augment this valuable living resource.
Over the last three decades, the services gained from wild eastern oyster populations have been supplemented by both aquaculture and restoration activities. Aquaculture practices vary somewhat by region, but typically involve transplanting wild or hatchery-produced seed to designated grow-out areas. Seed sourced from commercial hatcheries are proprietary strains, often selected for fast growth and increased survival (Proestou et al. 2016). Oysters are grown either on-bottom or in suspended gear and filter food from the surrounding water until they reach market size (Naylor et al. 2000). Common oyster restoration strategies include protecting broodstock from harvest and planting hard substrate (e.g. cultch) to create artificial reefs on which oyster larvae can settle (Peters et al. 2017). At sites with low natural recruitment, cultch is also seeded with hatchery-derived oysters. Several abiotic and biotic factors, including reef composition, sedimentation dynamics, food supply, and the presence of natural enemies, are considered when establishing artificial reefs in order to maximize the potential for sustained success (Coen and Humphries 2017).
While much emphasis has been placed on optimal site selection for restored and aquaculture oyster populations, the importance of genetic attributes has received relatively little attention despite the key role genetic composition plays in the performance and longevity of all species (Schindler et al. 2010). Numerous studies have demonstrated that genetic diversity, measured as heterozygosity, is significantly, positively correlated with population fitness (Reed and Frankham 2003). For example, Hughes and Stachowicz (2004) showed that resistance to environmental disturbance is higher in genetically diverse sea grass populations. Variable populations are also better able to adapt to local conditions and respond to long term environmental change (Frankham et al. 2014).
Eastern oysters are broadcast spawners with a dispersive larval stage that lasts several weeks (Thorpe et al. 2000). These life history characteristics facilitate high levels of gene flow among populations at both small and large spatial scales (Palumbi 2003). Extensive gene flow via larval transport is expected to have a homogenizing effect; however, a recent survey of the literature found that genetic differences are common among marine invertebrate populations and that the differences reflect adaptation along fine-scale environmental gradients (Sanford and Kelly 2011). The potential for significant local adaptation, coupled with high rates of larval exchange, can complicate efforts that aim to restore oyster reefs to their original structure and function (Grabowski and Peterson 2007).
Although ‘domesticated’ aquatic species are only a few generations removed from the wild, hatchery-derived stocks used to supplement declining populations have lower genetic diversity than their wild counterparts (Lind et al. 2009). The degree to which diversity is lost depends on hatchery protocols, including the number, size, and source of broodstock as well as spawning methods (strip-spawning followed by controlled single pair crosses vs. mass broadcast spawning), which can vary significantly among commercial hatcheries. Release of non-native, genetically-limited hatchery stocks that can hybridize with natural populations risks reduced fitness, lower effective population sizes, and reversing the effects of local adaptation in the wild (Grant et al. 2017).
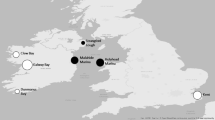
Supplementation of wild fish and shellfish populations through commercial aquaculture and restoration is commonplace (Kitada 2018), yet the extent and impact of exchange among wild, restored, and cultured groups are not measured routinely (Gaffney 2006; Levin 2006; Camara and Vadopalas 2009; Guo 2009). Rhode Island coastal lagoons or ‘salt ponds’, which support remnant native populations and have become a focal area for oyster aquaculture and conservation (Duball 2017), are an ideal setting to examine genetic structure and gene flow dynamics among wild and cultured stocks at small spatial scales. Ninigret Pond, located within the Ninigret National Wildlife Refuge, is the largest of the Rhode Island lagoons, covering approximately 1700 acres. In 2013, eight aquaculture leases totaling 22 acres were concentrated in the center of the pond (Buetel 2014) while a state-designated shellfish spawner sanctuary, which is closed to harvest, occupied 173 acres on the western side. Two separate oyster restoration projects flanking the spawner sanctuary commenced in 2008 and annual restoration plantings continued through 2010 and 2013, respectively (Griffin 2016). Additional small, wild oyster reefs were distributed throughout the pond at the time of this study (Fig. 1).

Location of sampled populations in southeastern Rhode Island. The wild control population is labeled C_NR, wild populations are W_FC, W_FN, and WW_SS. F_A, F_B, and F_C are farmed populations, and restored populations are R_A and R_D
Recent field surveys conducted in Ninigret Pond indicate that wild eastern oyster populations are increasing in size; however, it is uncertain what has contributed to this growth. Are the populations self-seeding or has recruitment been enhanced by the presence of restored and aquaculture populations? In this study, we genotyped individual oysters from wild, farmed, and restored populations at 13 microsatellite loci to characterize levels of genetic diversity within and genetic differentiation between wild populations and the aquaculture and restored populations derived from hatchery-reared seed. Patterns of diversity and genetic differentiation were then used to explore the extent of connectivity and admixture among groups. By providing a more complete understanding of the genetic composition of populations and the extent of genetic exchange within Ninigret Pond, we hope to better inform future eastern oyster management and conservation efforts that occur at small spatial scales.
Materials and methods
Sample collection
In early June of 2013, adult oysters (shell height ~ 75 mm) were collected by hand from eight populations within Ninigret Pond and one population from the Narrow River, also known as the Pettaquamscutt River (Fig. 1). To assess the effect of cultured stocks on the genetic composition of naturally-occurring eastern oyster populations, we focused on three wild populations within Ninigret Pond including Foster Cove (W_FC), Fort Neck Cove (W_FN), and a population within the state-designated Shellfish Spawner Sanctuary at Reeds Point (W_SS). The restored and aquaculture populations sampled in this study originated from one of four distinct commercial hatcheries (A, B, C, and D) located in four different states. Because the commercial seed sources used to stock aquaculture operations in the pond substantially overlapped across farms during the study period, adult, market-size oysters representing the most prevalent strains [derived from hatchery A (F_A), hatchery B (F_B), and hatchery C (F_C)] were sampled from just two of the eight farms. Samples were also collected from restored oyster reefs created by two separate restoration projects that differed greatly in scale. One of the reefs, located immediately adjacent to W_SS, was stocked with 30,000–60,000 seed from Hatchery D (R_D) each year between 2008 and 2013. The second restoration project seeded two reefs (one next to R_D and the other along the southern boundary of the Shellfish Spawner Sanctuary) with a total of nine million of Hatchery A’s restoration strain oysters (R_A) between 2008 and 2010 (Griffin 2016). As a wild control, we also included the Narrow River population (C_NR), where direct influences from oyster farming and restoration activities are absent. In late September 2013, oyster spat (shell height ≤ 25 mm) were collected from the four wild populations. We harvested tissues from no fewer than 30 individuals per population, preserved them individually in 70% ethanol, and stored them at 4 °C until DNA extraction.
DNA extraction and genotyping
We extracted genomic DNA from approximately 10 mg of mantle or gill tissue following a high-throughput chelex DNA extraction method adapted from Aranishi and Okimoto (2006). DNA quality and quantity were assessed using a spectrophotometer (NANODROP 8000) before DNA was diluted to a standard concentration of 10 ng/µl. Individual oysters were genotyped at 13 previously published microsatellite loci that were selected based on reported allele size range, low frequency of null alleles (those that are present but not amplified in a sample), and high level of polymorphism (Table 1). All loci were amplified in individual 10 µl PCR reactions containing 20 ng DNA, 1.5 mM MgCl2, 100 µM dNTPs, 0.5 U Taq polymerase (QiagenTopTaq), 0.3 µM of fluorescently-labeled forward primer, and 0.3 µM unlabeled reverse primer under optimized cycling parameters (Table 2). PCR products were subsequently diluted and pooled into one of four plexes (A, B, C, and D), purified according to the Agencourt AMPure XP protocol (Beckman Coulter) and submitted to the DNA Analysis Facility at Yale University for fragment analysis with an internal size standard (Liz500, Applied Biosystems). We scored alleles by size (in bp) using the default parameters in GeneMarker (SoftGenetics) and manually checked all allele calls against trace data for accuracy. We used the TANDEM tool to bin alleles of similar size within each locus for consistency across all samples (Matschiner and Salzburger 2009). A small, random subset of samples (N = 10) were extracted, amplified, and genotyped at each locus in duplicate to calculate genotype error.
Data analysis
We used MICRO-CHECKER v2.23 (Van Oosterhout et al. 2004) to further identify genotyping errors such as stuttering, large allele dropout, and null alleles. Within a population, each of the 13 loci were evaluated for Hardy–Weinberg equilibrium (HWE) by applying pseudo-exact tests [100,000 Markov Chain Monte Carlo (MCMC) iterations and 10,000 dememorization steps] using the software package Arlequin v3.5 (Excoffier and Lischer 2010). Significant deviations from HWE were detected after sequential Bonferroni correction for multiple tests (α = 0.05; P value = 0.000296). We estimated the inbreeding coefficient (FIS) both globally and at each locus for each population separately according to Weir and Cockerham (1984), and evaluated linkage disequilibrium between all pairs of loci within each population using the GENEPOP 4.6 software package (Raymond and Rousset 1995). Pairwise tests for linkage disequilibrium (LD) were deemed to be significant at a Bonferroni-corrected P value of 0.000049. We also evaluated concordance of paired loci with low p-values across populations.
To characterize the level of genetic diversity for each population, we calculated summary statistics including observed heterozygosity, expected heterozygosity, and total number of alleles at each locus in Arlequin v3.5. Private alleles, the alleles found in only one population, were identified using the popgen function in the R package ‘PopGenKit’ (Paquette 2011). We also applied the rank-based Kruskal–Wallis test to evaluate differences in genetic diversity between wild and cultured groups in R v3.4.2.
Effective population size (Ne), or the number of individuals that contribute genetically to future generations, provides insight to population structure, migration rates, and the genetic health of populations (Hare et al. 2011; He et al. 2012). We estimated Ne for all populations using the single-sample estimator based on linkage disequilibrium as implemented in NeEstimator v2.01 (Do et al. 2014). Ne for the four wild populations was also estimated with the temporal method (Waples 1989) by applying the Plan II default sampling strategy and calculating the standardized variance in allele frequency (FS) according to Jorde and Ryman (2007). Pair-wise comparisons were made between adults and spat from each wild population, representing generations 0 and 1 respectively. Alleles with frequencies < 0.02 were excluded from the analysis (Waples 2006).
Population structure in Ninigret Pond was inferred from three separate analyses: pairwise FST, model-based Bayesian clustering, and discriminant analysis of principle components (DAPC) methods. Null alleles are very common in oyster genomes due to exceptionally high levels of polymorphism (Reece et al. 2004; Wang and Guo 2007). High null allele frequencies may lead to pairwise FST values that overestimate the extent of genetic differentiation among populations (Chapuis and Estoup 2007). We therefore calculated pairwise FST values using both the original data and those adjusted for null alleles and performed a Mantel test (vegan package in R v3.4.2) with 1000 permutations to test whether the resulting FST matrices were correlated. We calculated Weir and Cockerham’s unbiased FST values for each population pair in Arlequin v3.5 and used 10,000 permutations of the data to test for statistically significant differences among populations at an α level of 0.05 with standard Bonferroni correction (P = 0.00064).
Our multi-locus genotype data were also analyzed with the model-based clustering program STRUCTURE v2.3.4 (Pritchard et al. 2000), assuming the admixture model with correlated allele frequencies among populations and no prior information about population locations. By choosing these options, we were better able to assess the extent of admixture among closely related populations. We performed separate analyses with all populations, wild populations only, and hatchery-derived populations only. For each population subset, the number of genetic clusters (K) represented in our data was inferred from five independent simulations of the data for K = 1–10 for all populations, 1–8 for wild populations, and 1–5 for hatchery populations. All simulations were performed using 100,000 MCMC iterations after a burn-in period of 50,000. We assessed the most probable value for K with both the mean likelihood method described in Pritchard et al. (2000) and the Delta K (ΔK) method (Evanno et al. 2005). We also considered the similarity of cluster matrices across runs (H′) for any given K. Individual admixture proportions, or the proportion of an individual’s genome that originated from each potential source population (Q) were also estimated. We used Structure Harvester v0.6.93 (Earl and vonHoldt 2011) to calculate ΔK and CLUMPP v. 1.1.2 (Jakobsson and Rosenberg 2007) to optimize the clustering results across runs. Graphical displays of the optimal cluster patterns were created with Distruct v1.1 (Rosenberg 2004).
To further confirm patterns of genetic structure among Ninigret Pond populations, we analyzed individual genotypes with the DAPC methods described by Jombart (2008) and implemented in the R package ADEGENET (Jombart et al. 2010). This multivariate analysis uses K-means clustering to identify genetic similarities among individuals, and unlike STRUCTURE, it does not assume explicit population genetics models to infer population structure. Again, we tested Ks 1 through 10 and used the find.clusters function and the lowest associated bayesian information criterion (BIC) to identify the optimal number of clusters. We also applied the XvalDAPC function to retain the appropriate number of principal components.
Results
Genetic variability
Within the subset of samples used to quantify genotype error, GeneMarker called conflicting genotypes/alleles between duplicates for just one sample at a single locus. This resulted in a combined error rate across all loci of 0.0046 per reaction and 0.0046 per allele (Hoffman and Amos 2005). Multi-locus microsatellite genotypes with ≤ 3 missing loci were obtained for 385 of the 390 oysters sampled in this study. Our MICRO-CHECKER analysis of the data revealed moderate null allele frequencies for most loci in all populations. For any given population, the number of loci with null alleles present ranged from 8 to 13. Consequently, heterozygote deficiencies associated with null alleles resulted in significant deviations from HWE for 66% of the tests conducted after Bonferroni correction (Online Resource 1). However, when genotypes were adjusted according to the Oosterhout correction algorithm (Van Oosterhout et al. 2004) the majority of loci within each population (75%) met HWE expectations (Online Resource 1). We conducted all subsequent analyses with both the uncorrected and corrected allele frequencies and emergent patterns of genetic diversity, Ne, and pairwise genetic differentiation were very similar between the two data sets. Therefore, we report results based on the corrected data in the main body of this manuscript, but have included the results based on the uncorrected data as an online resource (Online Resource 2).
We detected significant heterozygote deficiencies (as reflected in Global FIS estimates) for all populations and these deficiencies persisted even after the data were adjusted for null alleles (Table 3). Absence of significant pairwise comparisons for LD and lack of consistently low p-values for particular pairs of loci across several populations confirmed that the loci used in this study were independent of one another.
Levels of genetic diversity differed across populations. Allelic richness (NA), averaged across all loci for each population, ranged from 4.8 to 14.1 alleles per locus. NA was highest in the wild adult Fort Neck Cove population (W_FN) and lowest in the farmed stock produced by hatchery A (F_A) (Table 3). Average expected heterozygosity (HE) also varied by population, with population F_A again exhibiting the lowest HE (0.65) and the wild adult control population from Narrow River (C_NR) the highest (0.89). Overall, wild populations were significantly more genetically diverse than hatchery-derived populations (Kruskal–Wallis test; P = 0.0034 and P = 0.012 for NA and HE respectively).
The mean number of private alleles (NP) per locus for each population ranged from 0 to 0.8 (Table 3). No private alleles were detected in restored populations R_A and R_D, while the most private alleles were found in C_NR. We also observed high NP (0.5) in both W_FN and spat from Foster Cove (W_FC_S); however, few (0.1) were detected in the wild Shellfish Sanctuary population (W_SS). The relatively low number of private alleles detected in most of the sampled populations precluded us from inferring patterns of gene flow in Ninigret Pond using Slatkin’s (1985) private allele theory.
Our estimates of Ne based on the single-sample, bias-corrected, LD method suggest large effective population sizes (most estimates unbounded) for all wild populations except the spat samples from Fort Neck and Shellfish Sanctuary (W_FN_S and W_SS_S) (Table 4). Among the hatchery-derived stocks, Ne estimates ranged from 6.4 to 94.9 and were significantly larger for farmed stocks B and C than farmed and restored stocks derived from hatchery A and the restored stock from hatchery D. Because precision in estimating Ne decreases as the true Ne exceeds several thousand individuals (Waples 2016), we have shown that the Ne for most of the wild populations in this study is large, but not accurately quantifiable. In contrast, the temporal method severely underestimated Ne in the four wild populations because the time interval between samples was too short (Waples and Do 2010).
Population differentiation
The pairwise FST matrices obtained from the original data and those adjusted for null alleles were highly correlated (Mantel test; r = 0.9543, P = 0.001), indicating that the detection of population differentiation was not compromised by the presence of null alleles. Pairwise comparisons across the 13 populations generated FST values ranging from 0 (no differentiation between samples) to 0.215 (high differentiation between samples). Spat were not significantly differentiated from their adult counterparts in any of the wild populations. For the most part, the wild populations were not different from one another, with the exception of W_SS_S (Table 5). Oysters from this population were significantly differentiated from W_FN_S and those residing in the Narrow River, our control site with no culture efforts nearby.
The extent of genetic differentiation varied among hatchery-derived populations and between wild and hatchery groups. All farmed populations were significantly differentiated from one another, but no differentiation was apparent between the two restored populations (Table 5). Moderate to high (0.05 < FST < 0.215) genetic differentiation was detected between F_A and all other populations. F_B was moderately different from F_C, R_D, and all wild populations except W_FC and W_SS, while F_C differed only slightly from C_NR_S, W_FC_S, and W_FN, and moderately from W_FN_S, W_SS_S, and R_D. Moderate differences were also detected between R_A, W_FC, and F_A. Population R_D exhibited moderate to high differentiation from all populations except W_FC, W_SS, and R_A.
The STRUCTURE analysis identified three distinct genetic clusters (K = 3) as the best model when all 13 populations were included in the analysis. The mean likelihood and ΔK methods for estimating K were in agreement and H′ values were also highest for K = 3 (Fig. 2). For the most part, groupings were aligned with sample type (wild vs. hatchery-derived); however, the F_A population formed a cluster distinct from the other hatchery populations (Fig. 3). This result is not surprising given the high FST values detected between F_A and the rest of the populations in this study. No further genetic subdivision was detected when only hatchery-reared populations were included in the structure analysis (K = 2, Fig. 4). Although the overall analysis assigned all wild populations to a single cluster, when only these populations were included in a separate STRUCTURE analysis, the best-supported model suggests the W_SS_S population could be a genetically distinct cluster. While this result is consistent with the modest differences detected between W_SS_S and C_NR, C_NR_S, and W_FN_S using pairwise FST, the high degree of shared ancestry between the two potential clusters indicates very weak genetic structure among the wild populations (Fig. 5).

Plots depicting the most probable K value. a Mean likelihood L(K) and variance for each K value, b delta K for each value calculated with the Evanno method

Results of the STRUCTURE analysis when all 13 populations were included and corrected data were used. Bar plots for K values 2 through 4. The most probable value of K = 3 is based on both the mean likelihood and ΔK methods. Each vertical line represents a single individual and colors denote inferred ancestry. Black lines distinguish individuals collected from different sampling locations. (Color figure online)

Results of the STRUCTURE analysis when only hatchery-reared populations were included and corrected data were used. Bar plots for K values 2 and 3. The most probable value of K = 2 is based on both the mean likelihood and ΔK methods. Each vertical line represents a single individual and colors denote inferred ancestry. Black lines distinguish individuals collected from different sampling locations. (Color figure online)

Results of the STRUCTURE analysis when only wild populations were included and corrected data were used. Bar plots for K values 2 and 3. The most probable value of K = 2 is based on both the mean likelihood and ΔK methods. Each vertical line represents a single individual and colors denote inferred ancestry. Black lines distinguish individuals collected from different sampling locations. (Color figure online)
Population structure inferred from the DAPC analysis is consistent with our STRUCTURE and pairwise FST results. Individual oysters were distributed among three genetic clusters. Cluster 1 contains individuals derived from wild populations, cluster 2 contains F_A individuals, and cluster 3 represents the remaining hatchery-reared populations (Fig. 6). While small BIC values were observed with larger K, ordination plots do not really differ between K = 3 and K = 7. For example, when K = 5, the wild cluster is subdivided into three very closely related clusters while the other two clusters remain unchanged (Online Resource 3).

Ordination plots of discriminant analysis of principal components (DAPC) comparing all samples included in the study. Inferred genetic clusters are shown using numbers, colors, and inertia ellipses, and each dot represents an individual. Cluster 1 corresponds to the wild populations. Cluster 2 represents the F_A population and Cluster 3 consists of the remaining hatchery-reared populations. (Color figure online)
We found evidence of genetic exchange in all populations. Mixed ancestry was highest in population R_A where the membership coefficients (Q), averaged across all individuals in the population, were 0.63, 0.34, and 0.03 from hatchery-reared, wild, and F_A clusters respectively. Nearly one-third of R_A individuals were migrants from the wild (Qwild > 0.9). Genetic exchange was also high for population W_SS, where, on average, the level of ancestry associated with the hatchery-reared group was 0.29 and six individuals were migrants from the hatchery-reared group. Between 9 and 13% hatchery ancestry was observed in the remaining wild populations. Very low levels of F_A ancestry (1–5%) were detected in wild and other hatchery-reared clusters.
Discussion
As more and more restoration and aquaculture activities are initiated to supplement the economic and ecosystem services provided by depleted native populations, the risk of undermining the genetic composition of the very resources we aim to maintain increases (Conover 1998). According to Falk et al. (2006), “to overlook genetic variation is to ignore a fundamental force that shapes the ecology of living organisms.” For species such as the eastern oyster, that have experienced overexploitation as well as loss and degradation of habitat beyond the limits of natural recovery, knowledge of existing levels of genetic variation within, and genetic exchange between wild and hatchery-derived populations are needed to inform and improve current conservation and management programs (Weersing and Toonen 2009). Our analyses of genetic diversity, genetic differentiation, connectivity, and admixture among native, restored, and aquaculture eastern oyster populations inhabiting a coastal lagoon in southern Rhode Island provide insight into genetic exchange at small spatial scales and the extent to which hatchery populations could affect the genetic composition of their wild counterparts.
Due to the high polymorphism rate inherent in the eastern oyster genome, we observed a high frequency of null alleles across loci and populations. Null alleles can have profound effects on some genetic parameter estimates. The application of a null allele correction can reduce many analytical artifacts, but it did not fully address the extensive heterozygote deficits detected in our data set. Within each population, 25% of loci did not meet Hardy–Weinberg expectations and inbreeding coefficients (FIS) were large and statistically significant even after the null allele correction. Comparable levels of inbreeding have not been reported previously in the eastern oyster (e.g. Anderson et al. 2014) and are unusual for large, wild populations, suggesting that some error in genotype calls persisted. Waples (2018) recently showed FIS estimates to be particularly sensitive to null alleles; however, FST and LD estimates did not change significantly when null alleles were present. While we acknowledge the imperfections in our data set and the impact they may have on our analyses, the congruence of results from corrected and uncorrected data sets in this study supports the contention that the presence of null alleles does not preclude the analysis of genetic diversity and differentiation within and among populations (Waples 2018; Carlsson 2008).
First, we confirmed that, in southern Rhode Island, genetic diversity is significantly higher in the wild populations than the hatchery-reared populations. The four wild populations exhibited similar levels of diversity (measured as either allelic richness or heterozygosity); however, the average number of alleles per locus varied considerably among cultured populations, ranging from 4.8 to 9.5 (Table 3). We expected to see reduced genetic diversity in hatchery-reared stocks as the phenomenon has been observed in several commercially important cultured shellfish including the eastern oyster, the Pacific oyster (Crassostrea gigas), the pearl oyster (Pinctada maxima), abalone, and the hard clam (Mercenaria mercenaria) (reviewed in Araki and Schmid 2010; Carlsson et al. 2006; Lind et al. 2009; Kochmann et al. 2012).
Differences in allelic diversity among hatchery populations likely reflect inconsistencies in hatchery protocol. Seed oysters supplied to oyster growers and restoration practitioners operating in Ninigret Pond originated from four hatcheries located in four different states. In general, operating procedures are not standardized across commercial hatcheries, nor are they well reported. Furthermore, it is not known how hatchery practices were modified to accommodate aquaculture vs. restoration goals. While some commercial hatcheries in northeastern USA obtain broodstock from state-sponsored selective breeding programs, whether they are crossed with wild, local animals or hatchery-specific lines whose genetic make-up has not been studied extensively depends on the hatchery and the downstream purpose for the seed.
In addition, both the number of broodstock and the spawning technique used vary among hatcheries. Ideally, seed intended for restoration should derive from many single-pair matings of local, wild broodstock, whose offspring are reared separately until settlement and numbers equalized prior to planting, in order to maximize the capture of existing genetic variation and Ne (Camara and Vadopalas 2009; Cooper et al. 2009; Gruenthal et al. 2013). Unfortunately, many hatcheries do not have the infrastructure to support this approach and defer to mass spawns, where a modest number of broodstock are held in a single tank, gametes are released, and cross-fertilization among individuals is completely random. The problem with mass spawns, particularly in species with high fecundity but variable reproductive success, is that future generations can consist of offspring from only a small number of parents (sweepstakes reproductive success (SRS) per Hedgecock 1994). Lind et al. (2009) examined the genetic consequences of the mass spawning technique in pearl oysters and found that this method resulted in hatchery stocks with lower genetic variation and effective population sizes than stocks produced through controlled spawns. We know that at least two of the hatchery populations included in this study, F_A and R_D, were produced via mass spawn and they exhibit the lowest allelic richness and effective population size (Tables 3, 4).
While it has been suggested that the loss of genetic diversity in hatchery-reared populations can have substantial repercussions on their performance once released into the wild (e.g. Grant et al. 2017), few studies have successfully demonstrated a negative impact of reduced genetic diversity on fitness. Yet, there are two examples from the eastern oyster. Smee et al. (2013) combined adult oysters from one to three distinct populations on experimental trays and deployed them in the field to measure the effect of genetic diversity on recruitment. Larval recruitment to the trays containing a mixture of oysters from all three source populations was significantly higher than to the trays with just one population. To further characterize the relationship between diversity and key demographic traits and assess whether the relationship varies by environment, Hanley et al. (2016) produced oyster cohorts in a hatchery using broodstock from geographically distant natural populations, arranged them on tiles such that one to four cohorts were represented per tile, and placed the tiles at two sites that differed with respect to environmental stressors. Strong site and weaker diversity effects on short-term survival and growth were detected. Once again, a significant positive relationship between genetic diversity and recruitment was observed. Although we did not measure the relative fitness of hatchery-reared populations in our study, the works of Smee et al. (2013) and Hanley et al. (2016) underscore the potential risks associated with planting low-diversity stocks.
Marine species with planktonic larval stages (like oysters) often exhibit minimal population structure due to high levels of unrestricted gene flow (Palumbi 2003). There are several published examples where bivalve mollusc populations surveyed across varied spatial scales have shown very little genetic differentiation. Sea scallop populations within the Gulf of Maine are panmictic (Kenchington et al. 2006). Very weak population structure was detected among wild eastern oysters sampled throughout the Chesapeake Bay, although a significant pattern of isolation by distance suggests that local gene flow predominates there (Rose et al. 2006). Similarly, wild Sydney Rock oysters collected from separate bays within the Georges River estuary, New South Wales were not substantially different from one another (Thompson et al. 2017). Therefore, our results indicating genetic homogeneity among wild populations located within 10 km of one another and genetic similarity between wild adults and spat are not surprising. Natural populations in southern Rhode Island appear to be well-mixed.
However, we did detect considerable genetic differentiation between the wild and hatchery-derived populations residing within and just outside Ninigret Pond. Farmed stocks A, B, and restored stock D were moderately or highly differentiated from the wild populations. The differentiation observed between the two groups is likely due to differences in geographic origin between the hatchery-reared and wild strains and/or the extent to which the hatchery strains were subject to directional artificial selection. Genetic drift also can profoundly affect allele frequencies when acting within small hatchery populations founded with relatively few individuals.
Genetic differences between wild and hatchery strains associated with reduced fitness of hatchery strains in the wild may have limited the success of three separate eastern oyster restoration attempts in the Chesapeake Bay between 1999 and 2006. In each effort, several 100,000 to millions of disease-resistant oyster seed derived from either a wild Gulf of Mexico strain or a local selective breeding program were planted at multiple locations throughout the bay in close proximity to natural reefs. Oyster spat were collected from various reefs and screened at genetic loci that distinguished transplants from native stocks. Within 1 year of planting, Hare et al. (2006) deduced that the planting of 750,000 selected seed resulted in less than 10% enhancement of wild oyster reefs. By 3 years post-planting, < 2% of the oysters genotyped in the other two studies belonged to the strain used for supplementation, suggesting that the transplant survival and contribution to local recruitment was negligible (Milbury et al. 2004; Carlsson et al. 2008).
Alternatively, survival and subsequent interbreeding of hatchery-reared and wild stocks with genetically divergent backgrounds can have deleterious effects on the genetic composition of natural populations. These include the erosion of adaptive genetic structure, outbreeding depression (where hybrid animals are less fit than their native counterparts), and inbreeding depression (Ward 2006; Grant et al. 2017). For example, translocation of black-lipped pearl oyster (Pinctada margaritifera cumingii) seed across distinct locations within French Polynesia over the course of 10 years resulted in the homogenization of populations that previously had been adapted to specific environments (Arnaud-Haond et al. 2004). Several studies have documented the negative impacts of outbreeding depression in salmonid species and these have been reviewed in Ward (2006). The release of hatchery populations with a low effective number of breeders (Ne) can result in ‘genetic swamping,’ where large numbers of a few closely related genotypes swamp wild populations, and can lead to inbreeding depression (Ward 2006; Grant et al. 2017). The proximity of genetically-limited cultured populations to wild ones, the high connectivity among eastern oyster populations within both large and small embayments (Spires 2015), and the documented spillover of oyster larvae from protected areas (Peters et al. 2017) highlight the potential for genetic swamping of wild populations in Ninigret Pond.
The detection of low diversity, small Ne populations (R_A, R_D, and F_A) in our study suggest there is potential for genetic swamping of more genetically diverse wild populations in Ninigret Pond. At least one migrant was detected in most populations and the number of introgressed individuals in each population ranged from 2 to 11 (Fig. 3). The fewest number of migrants and the least represented ancestry among populations was from the F_A cluster. F_A exhibited the lowest genetic diversity and one of the smallest Ne among the populations sampled. Minimal genetic exchange between this farmed population and the wild suggests the risk of genetic swamping by F_A was low.
Curious results were observed in adult and spat collected from W_SS. Genetic diversity did not differ between the two generations and FST indicated no genetic differentiation, but estimated Ne was dramatically lower in the spat. W_SS also exhibited the greatest genetic exchange with hatchery-reared populations (Fig. 3). Furthermore, although the STRUCTURE analysis detected no population structure among wild populations when all 13 populations were included, W_SS_S formed a unique cluster when the subset of wild populations was analyzed separately, but admixture between W_SS_S and the remaining wild populations was relatively high (Fig. 5). This perplexing pattern may have been caused by either (1) erroneous collection and labeling of restored spat as W_SS (given the close proximity of the W_SS, R_A, and R_D populations) or (2) W_SS_S are a mixture of two genetically differentiated source stocks (wild and restored). We tested and rejected the first hypothesis by running STRUCTURE with W_SS_S and all hatchery-reared populations and detecting three distinct clusters: W_SS_S, F_A, and all other populations (data not shown). Support for the second explanation was similarly obtained from a STRUCTURE run that included a 14th ‘population’, created by sampling multi-locus genotypes from all original populations (data not shown).
Ninigret Pond is characterized by low flushing rates and high particle retention times (Nixon et al. 2001). These factors, in addition to the multiple uses the pond supports, facilitated ample genetic exchange among wild, restored, and cultured populations. We were primarily interested in the movement and introgression of hatchery-derived stocks into wild populations; however, we also observed substantial genetic exchange in the opposite direction. Contributions from wild were highest in R_A and the bulk of the exchange was due to migration. Extensive admixture was detected in F_C. The mechanism underlying the observed patterns of exchange must be inferred differently for restored and farmed populations. Three medium- to large-scale restored oyster reefs were established with hatchery-derived stocks during the 5 years prior to our study. Not only were these reefs closed to harvest, but a relatively rare, strong natural recruitment event was recorded in 2010 (Griffin 2016). Thus, the STRUCTURE results for R_A can be interpreted as migration from wild followed by introgression of the wild and hatchery stocks. Because the average residence time of farmed oysters in Ninigret Pond is 2 years, it is difficult to imagine a scenario where the admixture observed in F_C could be caused by the same mechanism. It is more likely that the genetic composition of the F_C stock prior to deployment was more similar to the wild populations than the other hatchery-derived stocks.
Implications
To offset the decline in wild eastern oyster harvests, the aquaculture industry has been steadily increasing since the 1970s and well-intentioned restoration efforts using hatchery-reared strains have been and continue to be initiated in Rhode Island (Rice et al. 2000). We applied population genetic methods to quantify genetic diversity and the degree of genetic differentiation among remnant wild and cultured populations in order to better understand the role anthropogenic manipulations play in the health and sustainability of wild eastern oyster populations living in Ninigret Pond, the largest multi-use coastal lagoon in southern Rhode Island. Our results indicate higher levels of genetic diversity within the wild populations compared to the cultured stocks and significant levels of differentiation between the wild and hatchery-reared populations. We also showed that genetic exchange does occur between wild and hatchery-derived oyster strains at small spatial scales. The recruitment of differentiated, less genetically diverse hatchery stocks to sympatric wild populations highlights the need for a policy change, particularly with respect to publicly-funded restoration projects that are closed to harvest. As a starting point, the origin and selection history of restoration seed should be well-documented prior to planting. Because the genetic composition of natural populations drives their evolutionary trajectory, management practices that minimize extensive introgression from and genetic swamping by low-diversity, domesticated strains, including the use of wild broodstock with known and well-matched geographic source(s), should be required. Furthermore, mandatory documentation of broodstock provenance will facilitate the use of genetics to better gauge the success of restoration efforts. Measures of successful restoration should extend beyond enhanced recruitment and ecosystem services in the short-term. Changes in genetic integrity (diversity, effective population size, and local adaptation) should also be used to evaluate the effects of restoration in the long term.
References
Anderson JD, Karel WJ, Mace CE, Bartram BL, Hare MP (2014) Spatial genetic features of eastern oyster (Cassostrea virginica Gmelin) in the Gulf of Mexico: northward movement of a secondary contact zone. Ecol Evol 4:1671–1683
Araki H, Schmid C (2010) Is hatchery stocking a help or harm? Aquaculture 308:S2–S11
Aranishi F, Okimoto T (2006) A simple and reliable method for DNA extraction from bivalve mantle. J Appl Genet 47:251–254
Arnaud-Haond S, Vonau V, Bonhomme F et al (2004) Spatio-temporal variation in the genetic composition of wild populations of pearl oyster (Pinctada margaritifera cumingii) in French Polynesia following 10 years of juvenile translocation. Mol Ecol 13:2001–2007
Beck MW, Brumbaugh RD, Airoldi L et al (2011) Oyster reefs at risk and recommendations for conservation, restoration, and management. Bioscience 61:107–116
Buetel D (2014) Aquaculture in Rhode Island 2014 annual status report. http://www.crmc.ri.gov/aquaculture/aquareport14.pdf. Accessed 19 Dec 2017
Camara MD, Vadopalas B (2009) Genetic aspects of restoring olympia oysters and other native bivalves: balancing the need for action, good intentions, and the risks of making things worse. J Shellfish Res 28:121–145
Carlsson J (2008) Effects of microsatellite null alleles on assignment testing. J Hered 99:616–623
Carlsson J, Morrison CL, Reece KS (2006) Wild and aquaculture populations of the eastern oyster compared using microsatellites. J Hered 97:595–598
Carlsson J, Carnegie RB, Cordes JF et al (2008) Evaluating recruitment contribution of a selectively bred aquaculture line of the oyster, Crassostrea virginica used in restoration efforts. J Shellfish Res 27:1117–1124
Chapuis M-P, Estoup A (2007) Microsatellite null alleles and estimation of population differentiation. Mol Biol Evol 24:621–631
Coen LD, Humphries AT (2017) Oyster-generated marine habitats: their services, enhancement, restoration and monitoring. In: Allison SK, Murphy SD (eds) The Routledge handbook of ecological and environmental restoration. Routledge, New York, pp 274–294
Conover DO (1998) Local adaptation in marine fishes: evidence and implications for stock enhancement. Bull Mar Sci 62:477–493
Cooper AM, Miller LM, Kapuscinski AR (2009) Conservation of population structure and genetic diversity under captive breeding of remnant coaster brook trout (Salvelinus fontinalis) populations. Conserv Genet 11:1087–1093
Do C, Waples RS, Peel D et al (2014) NeEstimator v2: re-implementation of software for the estimation of contemporary effective population size (Ne) from genetic data. Mol Ecol Resour 14:209–214
Duball CE (2017) Environmental impacts of oyster aquaculture on the coastal lagoons of southern Rhode Island. Master Thesis, University of Rhode Island
Earl DA, vonHoldt BM (2011) STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv Genet Resour 4:359–361
Evanno G, Regnaut S, Goudet J (2005) Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol Ecol 14:2611–2620
Excoffier L, Lischer HEL (2010) Arlequin suite ver 3.5: a new series of programs to perform population genetics analyses under Linux and Windows. Mol Ecol Resour 10:564–567
Falk DA, Richards CM, Zedler JB (2006) Integrating restoration ecology and ecological theory: a synthesis. In: Falk DA, Palmer MA, Zedler JB (eds) Foundations of restoration ecology. Island Press, Washington, DC, pp 341–346
Frankham R, Bradshaw CJA, Brook BW (2014) Genetics in conservation management: revised recommendations for the 50/500 rules, red List criteria and population viability analyses. Biol Conserv 170:56–63
Gaffney PM (2006) The role of genetics in shellfish restoration. Aquat Living Resour 19:277–282
Grabowski JH, Peterson CH (2007) Restoring oyster reefs to recover ecosystem services. In: Cuddington K, Byers JE, Wilson WG, Hastings A (eds) Theoretical ecology series. Academic Press, Burlington, pp 281–298
Grant WS, Stewart Grant W, Jasper J et al (2017) Responsible genetic approach to stock restoration, sea ranching and stock enhancement of marine fishes and invertebrates. Rev Fish Biol Fish 27:615–649
Griffin M (2016) Fifteen years of Rhode Island oyster restoration: a performance evaluation and cost-benefit analysis. Master Thesis, University of Rhode Island
Gruenthal KM, Witting DA, Ford T et al (2013) Development and application of genomic tools to the restoration of green abalone in southern California. Conserv Genet 15:109–121
Guo X (2009) Use and exchange of genetic resources in molluscan aquaculture. Rev Aquac 1:251–259
Hanley TC, Hughes AR, Williams B et al (2016) Effects of intraspecific diversity on survivorship, growth, and recruitment of the eastern oyster across sites. Ecology 97:1518–1529
Hare MP, Allen SK Jr, Bloomer P, Camara MD, Carnegie RB, Murfree J, Luckenbach M, Meritt D, Morrison C, Paynter K, Reece KS, Rose CG (2006) A genetic test for recruitment enhancement in Chesapeake Bay oysters, Crassostrea virginica, after population supplementation with a disease tolerant strain. Conserv Genet 7:717–734
Hare MP, Nunney L, Schwartz MK et al (2011) Understanding and estimating effective population size for practical application in marine species management. Conserv Biol 25:438–449
He Y, Ford SE, Bushek D et al (2012) Effective population sizes of eastern oyster Crassostrea virginica (Gmelin) populations in Delaware Bay, USA. J Mar Res 70:357–379
Hedgecock D (1994) Does variance in reproductive success limit effective population sizes of marine organisms? In: Beaumont AR (ed) Genetics and evolution of aquatic organisms. Chapman and Hall, London, pp 122–134
Hoffman JI, Amos W (2005) Microsatellite genotyping errors: detection approaches, common sources and consequences for paternal exclusion. Mol Ecol 14:599–612
Hughes AR, Stachowicz JJ (2004) Genetic diversity enhances the resistance of a seagrass ecosystem to disturbance. Proc Natl Acad Sci USA 101:8998–9002
Jackson JB, Kirby MX, Berger WH et al (2001) Historical overfishing and the recent collapse of coastal ecosystems. Science 293:629–637
Jakobsson M, Rosenberg NA (2007) CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23:1801–1806
Jombart T (2008) adegenet: a R package for the multivariate analysis of genetic markers. Bioinformatics 24:1403–1405
Jombart T, Devillard S, Balloux F (2010) Discriminant analysis of principal components: a new method for the analysis of genetically structured populations. BMC Genet 11:94
Jorde PE, Ryman N (2007) Unbiased estimator for genetic drift and effective population size. Genetics 177:927–935
Kenchington EL, Patwary MU, Zouros E, Bird CJ (2006) Genetic differentiation in relation to marine landscape in a broadcast-spawning bivalve mollusc (Placopecten magellanicus). Mol Ecol 15:1781–1796
Kirby MX (2004) Fishing down the coast: historical expansion and collapse of oyster fisheries along continental margins. Proc Natl Acad Sci USA 101:13096–13099
Kitada S (2018) Economic, ecological, and genetic impacts of marine stock enhancement and sea ranching: a systematic review. Fish Fish 19:511–532
Kochmann J, Carlsson J, Crowe TP, Mariani S (2012) Genetic evidence for the uncoupling of local aquaculture activities and a population of an invasive species—a case study of pacific oysters (Crassostrea gigas). J Hered 103:661–671
Levin LA (2006) Recent progress in understanding larval dispersal: new directions and digressions. Integr Comp Biol 46:282–297
Lind CE, Evans BS, Knauer J et al (2009) Decreased genetic diversity and a reduced effective population size in cultured silver-lipped pearl oysters (Pinctada maxima). Aquaculture 286:12–19
Matschiner M, Salzburger W (2009) TANDEM: integrating automated allele binning into genetics and genomics workflows. Bioinformatics 25:1982–1983
Milbury CA, Meritt DW, Newell RIE, Gaffney PM (2004) Mitochondrial DNA markers allow monitoring of oyster stock enhancement in the Chesapeake Bay. Mar Biol. https://doi.org/10.1007/s00227-004-1312-z
Naylor RL, Goldburg RJ, Primavera JH et al (2000) Effect of aquaculture on world fish supplies. Nature 405:1017–1024
Nixon SW, Buckley B, Granger S, Bintz J (2001) Responses of very shallow marine ecosystems to nutrient enrichment. Hum Ecol Assess 7:1457–1481
Palumbi SR (2003) Population genetics, demographic connectivity, and the design of marine reserves. Ecol Appl 13:146–158
Paquette SR (2011) PopGenKit: useful functions for (batch) file conversion and data resampling in microsatellite data sets. R package version 1.0. http://CRAN.R-project.org/package=PopGenKit. Accessed 22 Oct 2017
Peters JW, Eggleston DB, Puckett BJ, Theuerkauf SJ (2017) Oyster demographics in harvested reefs vs. no-take reserves: Implications for larval spillover and restoration success. Front Mar Sci. https://doi.org/10.3389/fmars.2017.00326
Pritchard JK, Stephens M, Donnelly P (2000) Inference of population structure using multilocus genotype data. Genetics 155:945–959
Proestou DA, Vinyard BT, Corbett RJ et al (2016) Performance of selectively-bred lines of eastern oyster, Crassostrea virginica, across eastern US estuaries. Aquaculture 464:17–27
Raymond M, Rousset F (1995) GENEPOP (version 1.2): population genetics software for exact tests and ecumenicism. J Hered 86:248–249
Reece KS, Ribeiro WL, Gaffney PM et al (2004) Microsatellite marker development and analysis in the eastern oyster (Crassostrea virginica): confirmation of null alleles and non-Mendelian segregation ratios. J Hered 95:346–352
Reed DH, Frankham R (2003) Correlation between fitness and genetic diversity. Conserv Biol 17:230–237
Rice MA, Valliere A, Caporelli A (2000) A review of shellfish restoration and management projects in Rhode Island. J Shellfish Res 19:401–408
Rose CG, Paynter KT, Hare MP (2006) Isolation by distance in the eastern oyster, Crassostrea virginica, in Chesapeake Bay. J Hered 97:158–170
Rosenberg NA (2004) Distruct: a program for the graphical display of population structure. Mol Ecol Notes 4:137–138
Sanford E, Kelly MW (2011) Local adaptation in marine invertebrates. Annu Rev Mar Sci 3:509–535
Schindler DE, Hilborn R, Chasco B et al (2010) Population diversity and the portfolio effect in an exploited species. Nature 465:609–612
Slatkin M (1985) Rare alleles as indicators of gene flow. Evolution 39:53–65
Smee DL, Overath RD, Johnson KD, Sanchez JA (2013) Intraspecific variation influences natural settlement of eastern oysters. Oecologia 173:947–953
Spires JE (2015) The exchange of eastern oyster (Crassostrea virginica) larvae between subpopulations in the Choptank and Little Choptank rivers: model simulations, the influence of salinity, and implications for restoration. Master Thesis, University of Maryland
Thompson JA, Stow AJ, Raftos DA (2017) Lack of genetic introgression between wild and selectively bred Sydney rock oysters Saccostrea glomerata. Mar Ecol Prog Ser 570:127–139
Thorpe JP, Solé-Cava AM, Watts PC (2000) Exploited marine invertebrates: genetics and fisheries. In: Solé-Cava AM, Russo CAM, Thorpe JP (eds) Marine genetics. Springer, Dordrecht, pp 165–184
Van Oosterhout C, Hutchinson WF, Wills DPM, Shipley P (2004) MICRO-CHECKER: software for identifying and correcting genotyping errors in microsatellite data. Mol Ecol Notes 4:535–538
Wang Y, Guo X (2007) Development and characterization of EST-SSR markers in the eastern oyster Crassostrea virginica. Mar Biotechnol 9:500–511
Wang Y, Wang X, Wang A, Guo X (2010) A 16-microsatellite multiplex assay for parentage assignment in the eastern oyster (Crassostrea virginica Gmelin). Aquaculture 308:S28–S33
Waples RS (1989) A generalized approach for estimating effective population size from temporal changes in allele frequency. Genetics 121:379–391
Waples RS (2006) A bias correction for estimates of effective population size based on linkage disequilibrium at unlinked gene loci. Conserv Genet 7:167–184
Waples RS (2016) Tiny estimates of the Ne/N ration in marine fishes: are they real? J Fish Biol 89:2479–2504
Waples RS (2018) Null alleles and FIS x FST correlations. J Hered 109:457–461
Waples RS, Do C (2010) Linkage disequilibrium estimates of contemporary Ne using highly variable genetic markers: a largely untapped resource for applied conservation and evolution. Evol Appl 3:244–262
Ward RD (2006) The importance of identifying spatial population structure in restocking and stock enhancement programmes. Fish Res 80:9–18
Weersing K, Toonen RJ (2009) Population genetics, larval dispersal, and connectivity in marine systems. Mar Ecol Prog Ser 393:1–12
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Zu Ermgassen PSE, Zu PS, Spalding MD et al (2012) Historical ecology with real numbers: past and present extent and biomass of an imperilled estuarine habitat. Proc R Soc B 279:3393–3400
Acknowledgements
We thank Dr. Marta Gómez-Chiarri for access to laboratory space and equipment, and Jessica Piez and Saebom Sohn for assistance in the lab. Dillon McNulty, Bray Beltran, and Jeanne Parente helped with oyster collections in the field. We also thank Mary Sullivan for help with GIS, Eric Schneider and Gary Casabona for useful discussions about shellfish restoration in RI, and two anonymous reviewers for very thoughtful and constructive comments on this manuscript. This work was funded through USDA ARS CRIS Project #803031000003, The Nature Conservancy’s GLOBE internship program, and a non-competitive grant from the USDA NRCS. Access to the University of Rhode Island Genomics and Sequencing Center, which is supported in part by the National Science Foundation under EPSCoR Cooperative Agreement # EPS-1004057, was also instrumental for the completion of this work.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Jaris, H., Brown, D.S. & Proestou, D.A. Assessing the contribution of aquaculture and restoration to wild oyster populations in a Rhode Island coastal lagoon. Conserv Genet 20, 503–516 (2019). https://doi.org/10.1007/s10592-019-01153-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-019-01153-9