
Abstract
The native European flat oyster Ostrea edulis is listed in the OSPAR Convention for the Protection of the Marine Environment of the North-East Atlantic (species and habitat protection) and in the UK Biodiversity Action Plan. Once extremely abundant in the nineteenth century, European stocks of O. edulis have declined during the twentieth century to rare, small, localised populations due to overexploitation, habitat degradation and, most recently, the parasitic disease bonamiosis. Selective breeding programmes for resistance to bonamiosis have been initiated in France and Ireland. High genetic diversity and bonamiosis-resistance would be important features of any sustainable restoration programmes for O. edulis. Oysters were sampled across Europe from four hatchery sources, four pond-cultured sources and four wild, but managed fisheries and were genotyped at five microsatellite loci. Hatchery-produced populations from small numbers of broodstock showed a significant loss of genetic diversity relative to wild populations and pedigree reconstruction revealed that they were each composed of a single large full-sib family and several small full-sib families. This extremely low effective population size highlights the variance in reproductive success among the potential breeders. Pond-cultured oysters were intermediate in genetic diversity and effective population size between hatchery and wild populations. Controlled hatchery production allows the development of bonamiosis-resistant strains, but at the expense of genetic diversity. Large scale pond culture on the other hand can provide a good level of genetic diversity. A mixture of these two approaches is required to ensure a healthy and sustainable restoration programme for O. edulis in Europe.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
An increasing number of exploited marine species are threatened through overharvesting, habitat loss or degradation, and/or diseases (Gaffney 2006) and, as a consequence, restoration programmes are being developed. Any restoration programme should be coupled with habitat restoration/rehabilitation (McCay et al. 2003) and should attempt to restore populations that have the highest possible genetic diversity. Without sufficient genetic diversity and suitably restored habitat the long term sustainability of restored populations remains at risk.
There are three main strategies for molluscan shellfish population restoration attempts: adult transplant, juvenile seeding and larval release. Shellfish restoration projects generally focus on the second of these strategies and involve the release of hatchery-produced progenies for restocking and enhancing stocks of natural populations (Gaffney 2006; Laing et al. 2006). Gaffney (2006) reviewed the three major genetic concerns relevant to shellfish restoration projects: (i) identifying source broodstock (after population genetics study at the geographical distribution scale of the species), (ii) maintaining the maximum genetic variability possible and reducing inbreeding of hatchery stocks used for restoration purpose and (iii) assessing the potential impact on the effective population size in wild populations of restocking with hatchery-propagated spat. Restocking with genetically improved hatchery strains could potentially have positive effects on wild populations particularly in terms of growth rate and disease resistance (Gaffney 2006). Several strategies were tested in Florida to restore and enhance populations of hard clam Mercenaria spp. (Arnold et al. 2002) and bay scallop Argopecten irradians (Arnold et al. 2005), following habitat degradation and loss. Hatchery-produced bay scallops were deployed in cages, but no significant contribution from the restoration stock to the wild population could be detected with mitochondrial DNA markers (Wilbur et al. 2005). On the other hand trials of bay scallop larval release produced an increase in the scallops in the larval enclosure relative to the control enclosure (Arnold 2008).
Oyster larvae settle out of the plankton and cement themselves to hard substrates, preferentially other oyster shells. This natural process, when not disturbed by a fishery, leads to oyster reefs that can support high biodiversity in their associated community. The importance of the restoration of oyster reef habitat has been emphasised by Mann (2000) and considerable investment has been made into the restoration of reefs of the eastern cupped oyster, Crassostrea virginica, in Chesapeake Bay, USA (Brumbaugh et al. 2000). In two separate studies, four million (Milbury et al. 2004) and 0.75 million (Hare et al. 2006) C. virginica hatchery-produced spat with improved disease tolerance were planted out in 1997 and 2002, respectively, on natural oyster beds in different areas of the Chesapeake Bay. The hatchery stocks could be distinguished from the wild oysters by diagnostic single nucleotide polymorphisms (SNPs) in their mitochondrial DNA, but in neither attempt was a high enhancement success detected (Milbury et al. 2004; Hare et al. 2006). Such studies on attempts at bivalve restoration from hatchery production demonstrate the importance of an evidential basis to conservation outcomes (Pullin and Knight 2009).
The native European flat oyster Ostrea edulis is listed in the OSPAR Convention for the Protection of the Marine Environment of the North-East Atlantic (species and habitat protection) and in the UK Biodiversity Action Plan and Laing et al. (2005) carried out a feasibility study for its restoration.
Ostrea edulis is a sessile, filter-feeding bivalve mollusc with a distribution ranging from Norway to Morocco in the Atlantic Ocean, in the Mediterranean Sea and extending into the Black Sea. This species was once of huge economic importance in Europe but there were massive declines in abundance in the late 1800s and early 1900s probably due to overfishing, habitat deterioration and unidentified diseases. Following a plateau of low level fisheries in Europe into the 1960s a further drastic decline in oyster numbers occurred due to two parasitic diseases, marteiliosis (Marteilia refringens), mainly affecting estuarine populations, and the more serious bonamiosis (Bonamia ostreae) that causes heavy mortalities in both intertidal and subtidal areas.
Selective breeding programmes for resistance to bonamiosis have been initiated in France (Naciri-Graven et al. 1998) and Ireland (Culloty et al. 2004) and have demonstrated an important potential for genetic gain through selective breeding. Additionally, significant differences in growth, mortality and susceptibility to bonamiosis and other diseases were observed between geographic origins and between families (da Silva et al. 2005). More recently, quantitative trait loci (QTLs) linked to bonamiosis resistance have been mapped (Lallias et al. 2009) and the search for candidate genes involved in the resistance to bonamiosis has been initiated (Morga et al., unpublished results). Such studies highlight the possibilities of restoring flat oyster populations by restocking with hatchery-produced bonamiosis-resistant seed.
Before implementing such restoration programmes in O. edulis, it is important to assess the potential impact of hatchery-propagated stocks on the genetic variability and the effective population size of wild populations (Gaffney 2006). Several studies have reported the loss of genetic diversity and a reduced effective population size in hatchery populations of shellfish, particularly oysters (Gosling 1982; Hedgecock and Sly 1990; Gaffney et al. 1992; Hedgecock et al. 1992; Saavedra and Guerra 1996; Saavedra 1997; Launey et al. 2001; Boudry et al. 2002; Sobolewska and Beaumont 2005; Appleyard and Ward 2006; Taris et al. 2007; Lind et al. 2009). So, while hatchery production is an effective method to produce large numbers of seed for restoration, and controlled conditions allow the development of disease resistant strains, its major drawback is the loss of genetic diversity.
The earliest method for artificial oyster production was the use of large ponds that were seeded with hundreds of adult oysters and the resultant spat collected on shells or tiles placed in the pond. Ponds were originally crude un-lined basins 2–3 m deep and up to 10 ha in area. Natural ponds or “polls” were exploited in parts of Norway that enabled enhanced spat production by making use of the greenhouse effect of a layer of surface fresh water (Kirkland et al. 1983). Ponds fell out of favour in the 1960s when hatchery culture proved to be more controlled and more reliable, but recently smaller replicated butyl-lined ponds have been revived and have proved to be a reliable source of oyster spat production (Gathorne-Hardy and Hugh-Jones 2004; Laing et al. 2005). However, there are no published data on the potential loss of genetic diversity inherent in pond production.
Traditionally, natural recruitment from wild populations was encouraged by the laying of tiles or shells to collect oyster spat. Young oysters were then scraped from the tiles and re-seeded into the fishery. This enhancement of natural recruitment by providing abundant settlement substrate would be expected to maintain existing genetic variability.
In the present study we have used O. edulis supplied from hatchery culture, pond culture and from natural recruitment to compare the genetic diversity at highly variable microsatellite loci generated by these three types of seed sources. Such information is critical to any proposed restoration programme for this species in Europe.
Materials and methods
Sampling
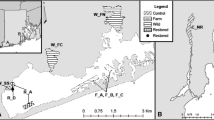
Twelve O. edulis populations were sampled (Fig. 1). Between 30 and 48 oysters per population were analysed.

Sample sites of O. edulis. WQ Quiberon (Brittany, France), WLR Loch Ryan (Scotland), WRF Ria Formosa (Portugal), WGr Grevelingen (the Netherlands), PRo Rossmore (Ireland), PVe Venø, Struer (Denmark), PBo Boemlo (Norway), PVa Vaagstranda (Norway), HLK Loch Kishorn (UK, commercial hatchery), HO Orkney (UK, commercial hatchery), HBOR Brittany (France, research hatchery) and HMED Mediterranean (France, research hatchery)
The wild or managed populations were sampled in Loch Ryan (Scotland, WLR) in 2006; in Quiberon (France, WQ) and Grevelingen (the Netherlands, WGr) in 2007; and in Ria Formosa (Portugal, WRF) in 2008. Quiberon, a large bay in southern Brittany, is a major site of flat oyster spat collection and on-growing. Grevelingen is a landlocked population, in a shallow (2–10 m) marine lake. Over-exploitation of its stocks lead to successive importations of foreign spat (MacKenzie et al. 1997). Since 1964, no commercial production and no foreign importations have been reported for Grevelingen oysters (Drinkwaard 1999). Ria Formosa is a shallow coastal lagoon located in Algarve (southern Portugal). The clam Ruditapes decussatus and the cupped oysters Crassostrea gigas and Crassostrea angulata are farmed in this site (Chícharo and Chícharo 2001) but flat oysters are not sufficiently abundant for commercial exploitation (Batista, personal communication). The Loch Ryan population represents a long-managed wild stock with regular natural spatfall and is the largest fishery for native oysters in Scotland (Hugh-Jones 2003). Oysters from France, Holland or Essex were laid in Loch Ryan in 1880s and in 1960s (Beaumont et al. 2006).
The pond-produced populations were sampled in Boemlo (Norway, PBo), Vaagstranda (Norway, PVa), Venø (Denmark, PVe) in 2007 and in Rossmore (Ireland, PRo) in 1999. In Norway two types of ponds (heliothermic polls) are traditionally used for cultivation (Kirkland et al. 1983; MacKenzie et al. 1997). The “breed-polls”, 5–10 m deep, 1–5 ha in area, exhibit strong salinity stratification, have restricted water exchange with the outside fjord (tidal exchanges controlled by a gate) and are used for spawning and collecting spat. The “spat-polls”, larger (up to 20 m deep and 40 ha in area), exchange water with the outside fjord and are used as fattening grounds (MacKenzie et al. 1997). The Boemlo population (Boemlo Skjell Ltd) was sampled from a “breed-poll” of 2 ha, 5–6 m deep, containing around 2,500 oysters, the broodstock originating in the 1980s from a mixture of different populations around Norway (Magnesen, personal communication). The Vaagstranda population (Arctic Oysters Ltd) was sampled from a “spat-poll” of 40 ha, 10–12 m deep, containing 100,000 oysters which originated from an introduction from Holland in 1930 (Magnesen, personal communication). The Danish Venø population was produced in 2004 in outdoor ponds (about 0.07 ha, 1.7 m deep) containing 400 oysters originated from Limfjord (Nissum Bredning) (Ommaney, personal communication). The Rossmore population (Cork Harbour) was produced in the context of selective breeding programme for resistance to bonamiosis: oysters were bred in spatting ponds from older oysters that survived the epizooty or were resistant to B. ostreae and that have been selectively bred for several generations (Sobolewska and Beaumont 2005). The Rossmore pond production was based on 22 butyl-lined ponds (0.04 ha, 2 m deep). These ponds were stocked with up to 700–800 oysters (Laing et al. 2005; Gathorne-Hardy and Hugh-Jones 2004).
The hatchery-produced populations were derived either from a commercial hatchery (Seasalter Shellfish Ltd, Whitstable, England) or from a research hatchery (Ifremer, Argenton, France). The Loch Kishorn population (Scotland, HLK), sampled in 2000, originated from Seasalter hatchery (original seed) (Sobolewska and Beaumont 2005). The Orkney population (Scotland, HO), sampled in 2000, was three generations removed from original Seasalter seed (Sobolewska and Beaumont 2005). The HBOR population resulted from the mixing of two spawning events (11/06/2007 and 27/06/2007) from 58 B. ostreae-resistant broodstock originating from the French selective breeding programme (Naciri-Graven et al. 1998). The HMED population resulted from the mixing of two spawning events (09/06/2007 and 22/06/2007) from 95 wild Mediterranean oysters from Thau lagoon.
DNA extraction and amplification of microsatellite loci
Genomic DNA was extracted from gill tissue, using a standard chloroform/isoamylalcohol method (Sambrook et al. 1989) and purified with the Wizard DNA Clean Up System (Promega). DNA quantification was performed using a spectrophotometer (BioPhotometer, Eppendorf). Five microsatellite markers were amplified following the authors’ instructions: OeduJ12, OeduT5 (Launey et al. 2002); Oedu.B11 (Naciri et al. 1995); Oedu.HA7 (Sobolewska et al. 2001) and Oe3/44 (Morgan et al. 2000). OeduT5 and Oedu.HA7 are distributed in the same linkage group, OeduJ12, Oedu.B11 and Oe3/44 being distributed on three different linkage groups (Lallias et al. 2007).
Genetic analysis
Genetic diversity within each of the twelve populations was measured as the number of alleles per locus (N a), the observed heterozygosity (H o) and unbiased expected heterozygosity (H e) (Nei 1978) under Hardy–Weinberg equilibrium. Allelic richness (A) (correcting frequency for unequal sample sizes) (El Mousadik and Petit 1996) was estimated per locus and per sample. All analyses were performed with FSTAT ver. 2.9 (Goudet 1995). Allelic richness and expected heterozygosity were compared between the three groups (wild, pond, hatchery) using a one-way ANOVA marker by marker followed by Tukey’s pairwise comparisons, or a Kruskall–Wallis test when variances were not equal (PAST software, Hammer et al. 2001). Wright’s (1931) F-statistics were computed according to Weir and Cockerham’s (1984) estimators, using FSTAT. Deviations from Hardy–Weinberg equilibrium (F is) were computed in each sampled population and genetic differentiation between populations was estimated using Wright’s fixation index F st. The significance of departure from zero of F is (or F st) values was assessed by randomizing alleles within samples (or genotypes among samples), based on 2,000 randomizations and after Bonferroni adjustment.
Linkage disequilibrium (LD) analysis
Linkage disequilibrium (LD) was assessed by permutation tests (1,000 permutations) with GENETIX 4.1 software (Belkhir et al. 1996–2001), for each pair of markers in each population.
Estimation of effective breeding sizes
Effective breeding sizes (N b) were estimated for the 12 populations sampled using three different methods. The heterozygote excess method was implemented in Colony v2.0 (Wang 2009). The LD method (Hill 1981) was implemented in LDNE program (Waples and Do 2008). For this method, the lowest allele frequency used (P crit value) was 0.02, as recommended by Waples and Do (2009). Sibship-based estimates were obtained using Colony v2.0 (Wang 2009), assuming a polygamous breeding system for males and females, and using the full likelihood model with medium precision and no prior information. Also, Spearman rank correlations (r s) were calculated between N b estimates and allelic richness.
Pedigree reconstruction analysis
Pedigree reconstruction on the four hatchery-produced populations was performed with PEDIGREE 2.2 (Herbinger et al. 2006), which partitions individuals into family groups (full-sibs (FS) or half-sibs (HS)) based on molecular marker data in the absence of parental information. Four parameters are chosen by the user: number of iterations of the Markov Chain, FS constraint (to choose between a FS partition and a kin group partition), temperature of the Markov Chain and weight (W) used in computing the partition score.
The four hatchery datasets were analysed with the FS partition algorithm in order to detect the presence of FS families. To generate the best (with the highest score) FS partition with W = 1, we performed four runs with one million iterations and temperature of 10, followed by four runs with one million iterations and temperature of 30 and used the best FS partition W = 1 as a start-up partition file to check that no better partition could be found. This procedure was repeated with W = 5 and W = 10. The three best FS partitions obtained with an increasing weight were then compared with the COMPARE function of PEDIGREE 2.2.
Results
LD
No significant LD was observed for the four wild populations and PVa. Significant LD was observed for PBo (one out of ten pairs), PRo (five out of ten pairs), PVe and HO (six out of ten pairs), HBOR (eight out of ten pairs), HLK and HMED (all ten pairs of loci).
Comparison of genetic diversity between wild, pond and hatchery populations
Allelic richness ranged from 6.14 (HBOR) to 19.08 (WLR) (Table 1). Mean allelic richness (averaged over five loci and four populations) was 7.57 for the hatchery-derived populations, 14.07 for the pond-produced populations and 18.43 for the wild populations. One-way ANOVA performed for each marker revealed that, at each locus, hatchery-derived populations exhibited a significantly lower mean allelic richness than the pond-derived and the wild populations (P < 0.05). For three out of five loci (Oedu.B11, Oedu.HA7 and OeduT5), pond-derived populations exhibited a significantly lower (P < 0.05) allelic richness than the wild populations. Mean observed heterozygosity varied between 0.756 (PBo) and 0.856 (HBOR) and mean expected heterozygosity ranged from 0.680 (HMED) to 0.915 (WLR) (Table 1). Mean expected heterozygosity (averaged over five loci and four populations) was 0.743, 0.857 and 0.900 for hatchery-produced, pond-produced, and wild populations, respectively. For four out of five loci (all but Oe3/44), one-way ANOVA revealed that hatchery derived populations exhibited significantly lower expected heterozygosities than pond derived populations and wild populations (P < 0.05 for each pairwise comparison).
Wright’s (1931) F is calculated for all five loci in each population showed significant overall heterozygote deficiencies for four populations (WQ, WGr, WLR and PBo) after Bonferroni adjustment (P < 0.05), mainly due to locus Oedu.B11 (Table 1). Null alleles were suspected at that locus. After the removal of locus Oedu.B11 from the analyses, no significant deficiencies of heterozygotes were observed. Significant heterozygote excesses were observed for two of the hatchery populations (HBOR and HMED) (P < 0.05 after Bonferroni adjustment) (Table 1) for all markers. Finally, the four hatchery populations exhibited a significant overall excess of heterozygotes (P < 0.05) after the removal of locus Oedu.B11 (Table 1).
Genetic differentiation among populations
Fst values for pairwise comparison among populations from O. edulis are given in Table 2. Among the wild populations, Fst values were low, ranging from 0.006 to 0.044. Only the Grevelingen population was significantly differentiated from the Quiberon and the Ria Formosa populations. Pond and hatchery-produced populations were significantly differentiated from each other and from the wild populations (Fst values ranging from 0.006 to 0.254). HMED was the population exhibiting the highest Fst values (from 0.126 to 0.254) in the pairwise comparison with the other populations.
Effective breeding sizes
Nb estimates for the 12 O. edulis populations are shown in Table 3. The heterozygote excess method only generated Nb estimates for two hatchery populations, HBOR (7) and HMED (7), without 95% confidence intervals. With the LD method, very high effective breeding sizes were obtained for the four wild populations and two Norwegian pond-produced populations (PBo and PVa) (from 138 [74–536] to ∞ [448–∞]). Estimated Nb were far smaller for PVe: 33 [26–45] and PRo: 49 [37–70]. For the hatchery-produced populations, relatively small Nb were reported for HO: 19 [14–25]; and very small Nb for HBOR: 6 [3–10], HLK: 4 [3–8] and HMED: 2 [2–3]. By contrast, the sibship assignment (SA)-based method produced Nb estimates and finite 95% confidence intervals for the 12 sampled populations. Both LD and SA methods gave similar Nb estimates for six populations: the two small pond-produced and the four hatchery-produced populations. However, for the two large pond-produced and the four wild populations, the SA method produced far smaller Nb estimates than the LD method. Figure 2 shows significant positive correlations between the two Nb estimates and allelic richness in the 12 flat oyster populations (rs (LD) = 0.869, P < 0.001; rs (SA) = 0.931, P < 0.001).

Nb estimates and allelic richness for 12 O. edulis populations. LDNb estimates based on the linkage disequilibrium method, SANb estimates based on the SA method. Upper X axis for SA estimates, lower X axis for LD estimates. Straight line regression for LD estimates of Nb, dotted line regression for SA estimates of Nb
Pedigree reconstruction analysis
For the HMED population, the best FS partition (score 3924.03, W = 5) identified eight groups (Fig. 3). We were able to reconstruct the parental genotypes of the large FS family (30 offspring) and calculate segregation distortion amongst the progeny (Table 4).

Distributions of individuals from the four hatchery-produced populations into FS groups using the program PEDIGREE 2.2 (Herbinger et al. 2006). Each vertical bar represents a FS group. The output of the program has been modified for the populations HBOR and HLK due to evidence of null alleles (see “Pedigree reconstruction analysis” and Table 5)
The best FS partition (score 1576.25, W = 5) revealed eight groups in the HBOR population (Fig. 3). The parental genotypes of the two largest FS families could be reconstructed and were identical at four out of five loci. Differences between them occurred only at the Oedu.B11 locus. The parental Oedu.B11 genotypes of the first family were 122/122 × 122/166 but were 128/128 × 128/166 for the second family. Four different genotypes were present among the progeny of the two first families: 128/166, 128/128, 122/166 and 122/122. Therefore, this was compatible with the segregation of a null allele (Table 5). We concluded that the two largest FS families (18 and 12 offspring, respectively) in this HBOR population were in fact a single FS family of 30 offspring (Fig. 3; Table 4).
For the HLK population, the best FS partition (score 1796.43; W = 10) identified seven groups (Fig. 3). After reconstructing the parental genotypes of the largest FS family containing 14 offspring, it was noted that a separate group of four offspring could be placed within that FS family if an Oedu.B11 null allele was present in one parent (Table 5). Therefore, we concluded that the largest FS family in the HLK sample consisted of 18 offspring (Fig. 3; Table 4).
Finally, the HO population revealed (score 3759.14, W = 5) 16 groups (Fig. 3). By changing W from 5 to 10 some groups with one or two offspring merged with other small FS groups. Parental genotypes could not be reconstructed for any FS family. Therefore, the HO population appeared to be composed of several small FS families, in contrast to the HMED, HBOR and HLK populations.
Discussion
Loss of genetic diversity
Our results highlight the loss of genetic diversity in hatchery populations in comparison with pond-produced and wild populations, both in terms of a reduction in allelic richness and in the expected heterozygosity. Pond-produced populations were quite effective in maintaining the genetic diversity but small scale ponds still showed a significant reduction in allelic richness relative to wild populations. These results are not surprising because reduced genetic diversity has been reported in hatchery or aquaculture populations of finfish (Bouza et al. 1997; Lundrigan et al. 2005; Liu et al. 2005; Machado-Schiaffino et al. 2007), algae (Guillemin et al. 2008) and shellfish (Gaffney et al. 1992; Hedgecock et al. 1992; Saavedra 1997; Taris et al. 2006; Hara and Sekino 2007; Lind et al. 2009). However, we are able to demonstrate that alternative managed reproduction methods using large scale ponds can retain high genetic diversity in the flat oyster and that this information is of significant value for restoration of this species.
High variance in reproductive success and small effective breeding size
Nb estimates were obtained based on different methods (Table 3). As reported by Beebee (2009) and Wang (2009), the heterozygote excess method performed very badly and gave meaningful estimates for only two (out of 12) populations. The LD and SA methods gave consistent Nb estimates for six populations (the four hatchery and the two small pond populations), bolstering confidence in our results. However, the SA method gave far smaller Nb estimates (below 70), compared with the LD method (between 138 and infinity), for the two large pond and the four wild populations. The low number of markers used in this study, as well as a sample size much smaller than the actual effective population size could explain the discrepancies in the Nb estimates. Indeed, in such a situation, the SA method becomes biased and yields confidence intervals that are too narrow (Wang 2009). Also, the SA method assumes sampling from a single cohort; violation of this assumption could have an impact on Nb estimates. The LD-based Nb estimates, although less precise, seem therefore to give more realistic estimates in our study. Despite the slight discrepancies in the Nb estimates, allelic richness analyses and Nb estimates gave consistent results: significant positive correlations between LD-based or SA-based Nb estimates and allelic richness were observed (Fig. 2). This is in agreement with the neutral theory that predicts a positive, albeit nonlinear, relationship between ln Ne and genetic diversity (Soulé 1976). Such a linear correlation has previously been shown by Beebee (2009) in toad populations.
Our results revealed that the four hatchery populations had much reduced effective breeding sizes compared with wild and pond populations (Table 3). Also, the occurrence of heterozygote excesses in the hatchery populations (Table 1) could be a consequence of low N b (Luikart and Cornuet 1999). HLK and HO populations both originated from Seasalter hatchery, the HLK oysters being first generation while the HO oysters are three generations removed from Seasalter imports. Interestingly HO was associated with a higher N b than HLK. Following a hatchery bottleneck, the LD is expected to decay over time (assuming random mating of hatchery-produced oysters after the bottleneck), with a consequent increase in N b estimate. However, hatchery bottlenecks are likely to vary in their strength even within the same hatchery. Therefore, it would be best to make temporal comparisons of N b estimates to assess the within-hatchery variance, before drawing conclusions about the long term consequences of hatchery practices.
The negative impacts on genetic diversity of hatchery practices such as mass spawning, culling of slow-growing larvae (Taris et al. 2006) or communal rearing of different families have been widely documented. In particular, the impact in terms of a reduced N b has been previously reported for many shellfish species (e.g. Hedgecock et al. 1992), and oysters in particular: the pearl oyster Pinctada maxima (Lind et al. 2009); the cupped oysters C. gigas and C. virginica (e.g. Hedgecock and Sly 1990; Gaffney et al. 1992; Appleyard and Ward 2006) and the flat oyster O. edulis (e.g. Saavedra and Guerra 1996; Saavedra 1997; Launey et al. 2001). Some remediation of the problem can be achieved by pooling the progeny of multiple spawning events (Gaffney et al. 1992) or by performing controlled spawnings (Lind et al. 2009). However, flat oysters are sequential hermaphrodites and an individual’s sex at any time cannot be determined non-destructively. A further complication is that males produce spermatozeugmata and females brood their eggs and early larvae up to approximately 10 days after fertilisation (O’Foighil 1989) adding to the difficulty of making controlled multiple crosses or mass matings. We know that two hatchery populations used in our study (HBOR and HMED) involved the pooling of two spawning events but they still had a very low effective breeding size (below six or 12 according to the LD and SA methods, respectively). Even at the small pond scale (Venø, Rossmore), where 400–800 broodstock were used, there was a reduced N b although more genetic diversity was retained than the hatchery-produced populations. Therefore, simply increasing the number of breeders does not necessarily increase the effective breeding size. It is only when we move to the Norwegian ponds of larger dimensions that LD-based N b approaches that of the wild populations. In such ponds, the oyster population remains undisturbed from year to year, allowing spatial dynamics such as clumping to develop among breeders, which may affect N b. This does not happen in small ponds (such as PRo) because they are emptied and cleaned each year.
Pedigree reconstruction of the four hatchery-produced populations revealed that three of those populations (HMED, HBOR, and HLK) were composed of a large FS family and a few small FS families. Pedigree reconstruction and N b estimation gave congruent results: HMED is the hatchery population with the lowest N b and with the highest skew in family contribution, while HO is the hatchery population with the highest N b and the highest number of contributing families as well as a more even contribution of families (Fig. 3; Table 3). This also strongly highlights the variance in reproductive success among the potential breeders, something that has also been demonstrated for salmonids (Herbinger et al. 2006). High relatedness among progenies, as demonstrated by our pedigree reconstruction analyses, could have serious implications for the long-term management of hatchery stocks, rapidly leading to inbreeding depression (Bierne et al. 1998; Naciri-Graven et al. 2000; Taris et al. 2007) due to a high genetic load in oysters (Launey and Hedgecock 2001). Recent studies reported high variance of reproductive success in oysters both in the wild (Li and Hedgecock 1998; Hedgecock et al. 2007) and under aquaculture conditions (Hedgecock et al. 1992; Boudry et al. 2002; Taris et al. 2007; Lallias et al., unpublished results). Variance in reproductive success seems a fundamental factor influencing the N b. Therefore, further studies should be implemented to improve hatchery practices, in order to reduce variance in family contributions.
Implications for restoration
Hatchery-produced populations of oysters are associated with a significant loss of allelic diversity and heterozygosity. Moreover, almost no genetic differentiation was observed across the wild populations (as shown by Launey et al. 2002; Sobolewska and Beaumont 2005; Beaumont et al. 2006), whereas hatchery practices led the hatchery-produced populations to be highly differentiated from the wild populations (Table 2). Unless hatcheries significantly change their methods of production, the restoration of wild populations of O. edulis by hatchery-produced stocks could be detrimental to the conservation of oysters and the long-term sustainability of their fisheries. Further studies are needed to estimate census numbers and N e of European oyster populations to better estimate the potential impacts and benefits of supportive breeding (Ryman and Laikre 1991; Gaffney 2006). There is little sense in mounting a major restoration programme for flat oysters if it will not lead to increased population sizes of wide genetic diversity. Genetic diversity is essential for long term sustainability. Large scale pond-production systems could represent a valuable alternative to hatcheries for restocking flat oyster populations because they seem more efficient in the maintaining of genetic diversity.
Restoration of flat oyster populations in Europe is complicated by the existence of bonamiosis which can cause very high mortalities in flat oyster populations. At the present time, some areas in northern Europe are bonamiosis-free while the disease is common elsewhere. Restoration of oysters in bonamiosis-free regions could therefore take advantage of the potential high genetic diversity provided by large scale pond production. In bonamiosis areas the better strategy might be to attempt restoration using bonamiosis-resistant strains from hatchery (Naciri-Graven et al. 1998) or small pond-culture (Culloty et al. 2004) but also to improve hatchery and small pond production methods to gain the highest genetic diversity possible.
References
Appleyard SA, Ward RD (2006) Genetic diversity and effective population size in mass selection lines of Pacific oyster (Crassostrea gigas). Aquaculture 254:148–159
Arnold WS (2008) Application of larval release for restocking and stock enhancement of coastal marine bivalve populations. Rev Fish Sci 16:65–71
Arnold WS, Marelli DC, Parker M, Hoffman P, Frischer M, Scarpa J (2002) Enhancing hard clam (Mercenaria spp.) population density in the Indian River Lagoon, Florida: a comparison of strategies to maintain the commercial fishery. J Shellfish Res 21:659–672
Arnold WS, Blake NJ, Harrison MM, Marelli DC, Parker ML, Peters SC, Sweat DE (2005) Restoration of bay scallop (Argopecten irradians (Lamarck)) populations in Florida coastal waters: planting techniques and the growth, mortality and reproductive development of planted scallops. J Shellfish Res 24:883–904
Beaumont AR, Trebano Garcia M, Honig S, Low P (2006) Genetics of Scottish populations of the native oyster, Ostrea edulis: gene flow, human intervention and conservation. Aquat Living Resour 19:389–402
Beebee TJC (2009) A comparison of single-sample effective size estimators using empirical toad (Bufo calamita) population data: genetic compensation and population size-genetic diversity correlations. Mol Ecol 18:4790–4797
Belkhir K, Borsa P, Chikhi L, Raufaste N, Bonhomme F (1996–2001) GENETIX 4.02, logiciel sous Windows TM pour la génétique des populations. Laboratoire Génome, Populations, Interactions, CNRS UMR 5000, Université de Montpellier II, Montpellier
Bierne N, Launey S, Naciri-Graven Y, Bonhomme F (1998) Early effect of inbreeding as revealed by microsatellite analyses on Ostrea edulis larvae. Genetics 148:1893–1906
Boudry P, Collet B, Cornette F, Hervouet V, Bonhomme F (2002) High variance in reproductive success of the Pacific oyster (Crassostrea gigas, Thunberg) revealed by microsatellite-based parentage analysis of multifactorial crosses. Aquaculture 204:283–296
Bouza C, Sanchez L, Martinez P (1997) Gene diversity analysis in natural populations and cultured stocks of turbot (Scophthalmus maximus L.). Anim Genet 28:28–36
Brumbaugh RD, Sorabella LA, Garcia CO, Goldsborough WJ, Wesson JA (2000) Making a case for community-based oyster restoration: an example from Hampton Roads, Virginia, U.S.A. J Shellfish Res 19:467–472
Chícharo L, Chícharo MA (2001) Effects of environmental conditions on planktonic abundances, benthic recruitment and growth rates of the bivalve mollusc Ruditapes decussatus in a Portuguese coastal lagoon. Fish Res 53:235–250
Culloty SC, Cronin MA, Mulcahy MF (2004) Potential resistance of a number of populations of the oyster Ostrea edulis to the parasite Bonamia ostreae. Aquaculture 237:41–58
da Silva PM, Fuentes J, Villalba A (2005) Growth, mortality and disease susceptibility of oyster Ostrea edulis families obtained from brood stocks of different geographical origins, through on-growing in the Ría de Arousa (Galicia, NW Spain). Mar Biol 147:965–977
Drinkwaard A (1999) Introductions and developments of oysters in the North Sea area: a review. Helgol Meeresunters 52:301–308
El Mousadik A, Petit RJ (1996) High level of genetic differentiation for allelic richness among populations of the argan tree [Argania spinosa (L.) Skeels] endemic to Morocco. Theor Appl Genet 92:832–839
Foighil DO (1989) Role of spermatozeugmata in the spawning ecology of the brooding oyster Ostrea edulis. Gamete Res 24:219–228
Gaffney PM (2006) The role of genetics in shellfish restoration. Aquat Living Resour 19:277–282
Gaffney PM, Davis CV, Hawes RO (1992) Assessment of drift and selection in hatchery populations of oysters (Crassostrea virginica). Aquaculture 105:1–20
Gathorne-Hardy A, Hugh-Jones T (2004) Spat collection in native oyster ponds. Shellfish News 17:6–9
Gosling EM (1982) Genetic variability in hatchery-produced Pacific oysters (Crassostrea gigas Thunberg). Aquaculture 26:273–287
Goudet J (1995) FSTAT (vers. 1.2): a computer program to calculate F-statistics. J Hered 86:485–486
Guillemin ML, Faugeron S, Destombe C, Viard F, Correa JA, Valero M (2008) Genetic variation in wild and cultivated populations of the haploid-diploid red alga Gracilaria chilensis: how farming practices favor asexual reproduction and heterozygosity. Evolution 62:1500–1519
Hammer Ø, Harper DAT, Ryan PD (2001) PAST: Palaeontological statistics software package for education and data analysis. Palaeontol Electronica 4:9
Hara M, Sekino M (2007) Genetic differences between hatchery stocks and natural populations in Pacific abalone (Haliotis discus) estimated using microsatellite DNA markers. Mar Biotechnol 9:74–81
Hare MP, Allen SK, Bloomer P, Camara MD, Carnegie RB, Murfree J, Luckenbach M, Meritt D, Morrison C, Paynter K, Reece KS, Rose CG (2006) A genetic test for recruitment enhancement in Chesapeake Bay oysters, Crassostrea virginica, after population supplementation with a disease tolerant strain. Conserv Genet 7:717–734
Hedgecock D, Sly F (1990) Genetic drift and effective population sizes of hatchery-propagated stocks of the Pacific oyster, Crassostrea gigas. Aquaculture 88:21–38
Hedgecock D, Chow V, Waples RS (1992) Effective population numbers of shellfish broodstocks estimated from temporal variance in allelic frequencies. Aquaculture 108:215–232
Hedgecock D, Launey S, Pudovkin AI, Naciri Y, Lapègue S, Bonhomme F (2007) Small effective number of parents (Nb) inferred for a naturally spawned cohort of juvenile European flat oysters Ostrea edulis. Mar Biol 150:1173–1182
Herbinger CM, O’Reilly PT, Verspoor E (2006) Unravelling first-generation pedigrees in wild endangered salmon populations using molecular genetic markers. Mol Ecol 15:2261–2275
Hill WG (1981) Estimation of effective population size from data on linkage disequilibrium. Genet Res 38:209–216
Hugh-Jones T (2003) The Loch Ryan native oyster fishery. Shellfish News 15:17–18
Kirkland DW, Platt Bradbury J, Dean WE (1983) The heliothermic lake—a direct method of collecting and storing solar energy. Arch Hydrobiol 65:1–60
Laing I, Walker P, Areal F (2005) A feasibility study of the native oyster (Ostrea edulis) stock regeneration in the United Kingdom (CARD Project Report FC1016). Available via DIALOG, http://www.defra.gov.uk/science/project_data/DocumentLibrary/FC1016/FC1016_2543_FRP.pdf
Laing I, Walker P, Areal F (2006) Return of the native—is European oyster (Ostrea edulis) stock restoration in the UK feasible? Aquat Living Resour 19:283–287
Lallias D, Beaumont AR, Haley CS, Boudry P, Heurtebise S, Lapègue S (2007) A first-generation genetic linkage map of the European flat oyster Ostrea edulis (L.) based on AFLP and microsatellite markers. Anim Genet 38:560–568
Lallias D, Gomez-Raya L, Haley C, Arzul I, Heurtebise S, Beaumont A, Boudry P, Lapègue S (2009) Combining two-stage testing and interval mapping strategies to detect QTL for resistance to bonamiosis in the European flat oyster Ostrea edulis. Mar Biotechnol 11:570–584
Launey S, Hedgecock D (2001) High genetic load in the Pacific oyster Crassostrea gigas. Genetics 159:255–265
Launey S, Barre M, Gerard A, Naciri-Graven Y (2001) Population bottleneck and effective size in Bonamia ostreae-resistant populations of Ostrea edulis as inferred by microsatellite markers. Genet Res 78:259–270
Launey S, Ledu C, Boudry P, Bonhomme F, Naciri-Graven Y (2002) Geographic structure in the European flat oyster (Ostrea edulis L.) as revealed by microsatellite polymorphism. J Hered 93:331–351
Li G, Hedgecock D (1998) Genetic heterogeneity, detected by PCR-SSCP, among samples of larval Pacific oysters (Crassostrea gigas) supports the hypothesis of large variance in reproductive success. Can J Fish Aquat Sci 55:1025–1033
Lind CE, Evans BS, Knauer J, Taylor JJU, Jerry DR (2009) Decreased genetic diversity and a reduced effective population size in cultured silver-lipped pearl oysters (Pinctada maxima). Aquaculture 286:12–19
Liu Y, Chen S, Li B (2005) Assessing the genetic structure of three Japanese flounder (Paralichthys olivaceus) stocks by microsatellite markers. Aquaculture 243:103–111
Luikart G, Cornuet JM (1999) Estimating the effective number of breeders from heterozygote excess in progeny. Genetics 151:1211–1216
Lundrigan TA, Reist JD, Ferguson MM (2005) Microsatellite genetic variation within and among Arctic charr (Salvelinus alpinus) from aquaculture and natural populations in North America. Aquaculture 244:63–75
Machado-Schiaffino G, Dopico E, Garcia-Vazquez E (2007) Genetic variation losses in Atlantic salmon stocks created for supportive breeding. Aquaculture 264:59–65
MacKenzie CL, Burrel VG, Rosefield A, Hobart WL (1997) The history, present condition, and future of the molluscan fisheries of north and central America and Europe. National Marine Fisheries Service, Washington, DC
Mann R (2000) Restoring the oyster reef communities in the Chesapeake bay: a commentary. J Shellfish Res 19:335–339
McCay DPF, Peterson CH, DeAlteris JT, Catena J (2003) Restoration that targets function as opposed to structure: replacing lost bivalve production and filtration. Mar Ecol Prog Ser 264:197–212
Milbury C, Meritt D, Newell R, Gaffney P (2004) Mitochondrial DNA markers allow monitoring of oyster stock enhancement in the Chesapeake Bay. Mar Biol 145:351–359
Morgan TS, Rogers AD, Iyengar A (2000) Novel microsatellite markers for the European oyster Ostrea edulis. Mol Ecol 9:495–497
Naciri Y, Vigouroux Y, Dallas J, Desmarais E, Delsert C, Bonhomme F (1995) Identification and inheritance of (GA/TC)n and (AC/GT)n repeats in the European flat oyster Ostrea edulis (L.). Mol Mar Biol Biotechnol 4:83–89
Naciri-Graven Y, Martin A-G, Baud J-P, Renault T, Gerard A (1998) Selecting the flat oyster Ostrea edulis (L.) for survival when infected with the parasite Bonamia ostreae. J Exp Mar Biol Ecol 224:91–107
Naciri-Graven Y, Launey S, Lebayon N, Gerard A, Baud JP (2000) Influence of parentage upon growth in Ostrea edulis: evidence for inbreeding depression. Genet Res 76:159–168
Nei M (1978) Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89:583–590
Pullin AS, Knight TM (2009) Doing more good than harm—building an evidence-base for conservation and environmental management. Biol Conserv 142:931–934
Ryman N, Laikre L (1991) Effects of supportive breeding on the genetically effective population size. Conserv Biol 5:325–329
Saavedra C (1997) Low effective sizes in hatchery populations of the European oyster (Ostrea edulis): implications for the management of genetic resources. J Shellfish Res 16:441–446
Saavedra C, Guerra A (1996) Allozyme heterozygosity, founder effect and fitness traits in a cultivated population of the European oyster, Ostrea edulis. Aquaculture 139:203–224
Sambrook J, Fritsch EF, Maniatis T (1989) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, Cold Spring Harbour
Sobolewska H, Beaumont AR (2005) Genetic variation at microsatellite loci in northern populations of the European flat oyster (Ostrea edulis). J Mar Biol Assoc UK 85:955–960
Sobolewska H, Beaumont AR, Hamilton A (2001) Dinucleotide microsatellites isolated from the European flat oyster, Ostrea edulis. Mol Ecol Notes 1:79–80
Soulé ME (1976) Allozyme variation, its determinants in space and time. In: Ayala FJ (ed) Molecular evolution. Sinauer Associates, Sunderland, pp 46–59
Taris N, Ernande B, McCombie H, Boudry P (2006) Phenotypic and genetic consequences of size selection at the larval stage in the Pacific oyster (Crassostrea gigas). J Exp Mar Biol Ecol 333:147–158
Taris N, Batista FM, Boudry P (2007) Evidence of response to unintentional selection for faster development and inbreeding depression in Crassostrea gigas larvae. Aquaculture 272:S69–S79
Wang J (2009) A new method for estimating effective population sizes from a single sample of multilocus genotypes. Mol Ecol 18:2148–2164
Waples RS, Do C (2008) LDNE: a program for estimating effective population size from data on linkage disequilibrium. Mol Ecol Resour 8:753–756
Waples RS, Do C (2009) Linkage disequilibrium estimates of contemporary N e using highly variable genetic markers: a largely untapped resource for applied conservation and evolution. Evol Appl. doi:10.1111/j.1752-4571.2009.00104.x
Weir BS, Cockerham CC (1984) Estimating F-statistics for the analysis of population structure. Evolution 38:1358–1370
Wilbur AE, Seyoum S, Bert TM, Arnold WS (2005) A genetic assessment of bay scallop (Argopecten irradians) restoration efforts in Florida’s Gulf of Mexico Coastal Waters (USA). Conserv Genet 6:111–122
Wright S (1931) Evolution in Mendelian populations. Genetics 16:97–159
Acknowledgements
We would like to thank Thorolf Magnesen for supplying the two Norwegian oyster populations, Alexandra Leitão for the Ria Formosa population, and René Robert for the French hatchery HBOR and HMED samples. We thank two anonymous reviewers for useful comments and suggestions. This research has been financed by the European Regional Development Fund INTERREG IIIB Priority C, project 201—AAAG2 (Atlantic Arc Aquaculture Group 2).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Lallias, D., Boudry, P., Lapègue, S. et al. Strategies for the retention of high genetic variability in European flat oyster (Ostrea edulis) restoration programmes. Conserv Genet 11, 1899–1910 (2010). https://doi.org/10.1007/s10592-010-0081-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10592-010-0081-0