
Abstract
Pediatric brain tumors are the leading cause of childhood cancer mortality with medulloblastoma (MB) representing the most frequent malignant tumor. Although standardization of therapy resulted in a 2-fold reduction in mortality in patients with MB by 2002, it became clear that further improvements in clinical outcome would require a deeper understanding of the biology of MB. Employing the four main molecular MB subgroups (Wnt, Shh, Group 3 and Group 4), a restratification into clinicogenomic risk categories quantified an unacceptable survival for the high-risk group, urging researchers to focus their efforts towards acquiring a greater biological understanding of these children. Advancing in parallel with the molecular characterization and understanding of pediatric MB is the clinicogenomic correlations giving rise to recommendations for neurosurgical care. While unique observations that distinct radiological patterns can be identified to inform the MB molecular subgroup preoperatively, current neurosurgical practice remains maximal safe surgical resection followed by risk-adapted provision of adjuvant therapy in the context of a clinical trial.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
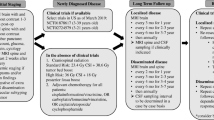
Pediatric brain tumors are the leading cause of childhood cancer mortality with medulloblastoma (MB) representing the most frequent malignant tumor [1]. Our understanding of the molecular basis of pediatric MB has increased significantly since its first description by two eminent dually trained neurosurgeons and neuropathologists, Drs. Percival Bailey and Harvey Cushing in 1925 [2]. Since then, multiple risk stratifications were developed in efforts to guide and standardize clinical practice. Clinically, high-risk disease is identified by an age less than 3 years and metastasis at diagnosis or postoperative residual disease greater than 1.5 cm2 [3]. Over the last 30 years, a multimodal approach to disease management employing a standardized risk stratification system has made significant improvements in outcome survival (OS) for these children; however, ample potential for improvements remain [4]. Traditional treatment for children age 3 years and older involves maximal safe resection, adjuvant craniospinal irradiation, and chemotherapy with vincristine, cisplatin, cyclophosphamide, and lomustine [5]. In children under the age of 3 years, the balance between the devastating neurodevelopmental and neurocognitive side effects of radiation therapy and disease control prompted the neurooncological community to learn from the early experiences of postoperative irradiation of the developing brain [6]. In these young children, a radiation sparing, high-dose chemotherapy protocol is administered with radiation reserved for salvage therapy. While childhood MB survivorship witnessed a significant improvement nearing 80% for standard-risk patients, the treatment toxicity and long-term sequelae of the current regime considerably impact their quality of life, further emphasizing the need for a deeper understanding of the biology of MB (Fig. 1) [7,8,9,10,11,12,13].

Clinical risk stratification for adjuvant therapy in pediatric MB [3]. *CSF, cerebrospinal fluid; CSI, craniospinal irradiation
2 Molecular era
At the turn of the millennium, high-throughput methods for studying the genome and transcriptome became available facilitating early studies to subdivide histologically similar leukemias into clinically and molecularly distinct groups [14]. A subsequent study by Pomeroy et al. utilized transcriptomics to study primitive neuroectodermal tumors (PNETs) of the CNS and demonstrated that 2 histologically similar tumors, MB and supratentorial PNET were molecularly distinguishable [15]. Further, MB with classic histopathology were biologically distinct from nodular desmoplastic histology providing the initial evidence for what is now regarded as the sonic hedgehog (Shh) MB subrgroup [15]. Subsequent high-throughput characterization of a larger series of primary MB tumor samples identified definitive clusters that appeared to be distinct from one another [16,17,18,19]. These findings prompted a gathering of an international expert panel at a consensus conference in Boston in 2010. This panel of experts determined that the data supported the existence of four main MB subgroups (Wnt, Shh, Group 3 (G3) and Group 4 (G4)) based on multiple genomic platforms with distinct demographics and clinical features [20]. The Wingless (Wnt) and Shh pathways were characterized by upregulation of genes in the canonical Wnt or Shh pathways respectively. These 2 subgroups were separated from each other and other subgroups on principal components analysis and were associated with improved clinical outcomes when compared with G3 and G4 subgroups, which were less well-characterized and clinically correlated with a greater likelihood of metastatic disease and poor clinical outcome [17,18,19,20,21,22,23] (Table 1). Following an international consensus, the WHO provided an update on the classification of tumors of the central nervous system (CNS) to include the molecular parameters in addition to histology to define many tumor entities. For MB, the genetically defined subcategories included the Wnt activated, Shh activated and TP53 mutant, Shh activated and TP53 wild type, and non-Wnt/non-Shh, Group 3 (G3) and Group 4 (G4) [24].
After review of 5 years of data within the published and unpublished literature since the pediatric MB molecular consensus, Ramaswamy et al. updated the clinical risk stratification integrating the breadth of evidence for currently available biomarkers and clinical outcome. Risk groups were defined based on current survival rates: low risk (> 90% survival), average (standard) risk (75–90% survival), high risk (50–75% survival), and very high risk (< 50% survival). The Wnt subgroup and nonmetastatic G4 MB with chromosome 11 loss or whole chromosome 17 gain were recognized as low risk and may qualify for reduced therapy. High-risk patients encompassed patients with metastatic Shh, G4 tumors, and MYCN-amplified Shh MB. And, G3 with metastasis or Shh with TP53 mutations were identified as very high risk (Table 2) [25]. For this reason, Ramaswamy urges researchers to consider investigating alternative treatments for the high- and very high-risk patients for whom there is currently no alternative targeted treatments outwith the local standard or risk-adapted protocols available at local and centers running clinical trials respectively.
3 Recurrent disease
Recurrent pediatric MB is a therapeutically challenging disease secondary to its near universal resistance to contemporary therapeutic strategies [26,27,28,29,30,31]. While risk-based therapy has resulted in relatively stable 5-year progression-free survival (PFS) and overall survival (OS) rates, there still remains a significant group of children in whom the tumor progresses despite novel approaches to salvage therapy [29]. In general, the PFS of MB has been observed to generally decrease between 1 and 3 years after diagnosis and stabilizes between 3 and 5 years [26]. Owing to the difficulty in treating recurrent disease, the OS mirrors PFS rates with decreases observed after year 3 and 4 after diagnosis and stabilized by year 5. Of note, while contrary to clinical intuition, metastatic disease at presentation has not been associated with disease recurrence [26, 28, 29]. One explanation for this observation may be the differences in risk stratification and upfront treatment provided in metastatic disease.
Molecularly, Ramaswamy et al. observed that despite significant genetic alterations, MB maintains its molecular subgroup at recurrence [32]. One explanation is the high degree of clonal selection that may occur where a dominant clone that is resistant to the intensive upfront chemoradiotherapy propagate to dominate the recurrent tumor. One very important consideration in the management of lesions suspicious for a recurrent MB of childhood is differentiating it from secondary tumors. Ramaswamy et al. emphasize the importance of confirming the diagnosis of a later MB recurrence as radiation-induced high-grade gliomas have been observed [33, 34]. Once the histology is confirmed as an MB recurrence, various strategies for therapeutic salvage have been observed in the literature. In pediatric-refractory or relapsed brain tumors, a recent single-center review observed a poor prognosis in recurrent/treatment refractory pediatric MB despite standardization of upfront therapy and provision of multimodal salvage therapy [35]. Two risk-adapted clinical trials aimed at young children, SJYC07 and ACNS1221, unfortunately did not observe an improvement in overall event-free survival leading investigators to re-evaluate our therapeutic strategies for recurrent disease [36, 37].
A current ongoing clinical trial looking at molecularly-driven doublet therapies for children and young adults with recurrent brain tumors includes recurrent childhood MB (i.e., SJDAWN). SJDAWN is underway at St. Jude's Children's Research Hospital; this study proposed a novel approach to evaluate combinatory therapies of new agents based on tumor type and molecular features. In this study, investigators hypothesized that the 2 predictably active drugs (i.e., doublet) will increase the efficacy of each agent. In MB, G3/4 MB were enrolled into stratum A (i.e., ribociclib and gemcitabine), Wnt/Shh MB were enrolled into stratum B (i.e., ribociclib and trametinib), and skeletally mature Shh MB children with chromosome 9q loss or PTCH1 mutation and off Smoothened (SMO) inhibitor for > 6 months were enrolled into stratum C (ribociclib and sonidegib) [38]. Ribociclib is a cyclin D1/CDK4 and CDK6 inhibitor, gemcitabine is a nucleic acid synthesis inhibitor, trametinib is a mitogen-activated, extracellular signal-regulated kinase (MEK) inhibitor, and sonidegib is a Shh pathway (SMO) inhibitor. The rationale for combining a cell cycle inhibitor with a more targeted molecularly targeted agent is the synergistic effects on tumor propagation and maintenance. The results of this trial are eagerly awaited. All in all, as we make greater gains in the understanding of the biology of recurrent MB to inform combinatory targeted therapies, the ultimate goal of the pediatric neuro-oncological community is to transform recurrent MB in to an entity that is no longer universally fatal
4 Radiogenomics of pediatric medulloblastoma
4.1 Location
As the molecular subgrouping of MB was further integrated as a clinical standard of care, parallels between preoperative imaging, intraoperative findings, and the postoperative molecular diagnosis were retrospectively sought. Perreault et al. found 100% concordance between tumor location on MRI imaging either using 1.5T and 3T machine and surgical identification of tumor in their large cohort (N = 99) of pediatric MB. Furthermore, the tumor location was highly predictive of molecular subgroups. Wnt tumors occurred along the cerebellar peduncle or cerebellopontine angle (CPA), cerebellar hemispheric location was characteristic of Shh tumors, and G3 and G4 MB were primarily midline and occupied the 4th ventricle [39, 40]. Logistic regression analysis within their discovery cohort revealed that tumor location was a significant predictor of MB subgroups. Further computational analysis confirmed G3 and G4 MB predominated within the midline fourth ventricle (Fig. 2).

Locations of molecularly distinct pediatric MB subtypes. a. Wnt tumors with tendency to CPA. b Shh tumors with predilection to the cerebellar hemisphere. c–d G3 and G4 MB with tendency towards the midline with avid enhancement in G3 MB
4.2 Enhancement pattern
The enhancement patterns of pediatric MB have also been characterized and Perreault et al. indicated unique to G3 MB, an ill-defined tumor margin is a feature not commonly present in the other 3 subgroups. When observed in a non-G3 MB, it was universally in the 3/14 Shh MB. In fact, minimal or no enhancement was characteristic of G4 MB. In contrast, the majority of G3 MB displayed contrast enhancement (Fig. 2), a feature that radiologically distinguished midline fourth ventricular G3 tumors from G4 MB tumors from G4 MB [41]. Mata-Mbemba et al. further delineated metastatic imaging patterns characteristic of specific molecular subgroups that may aid in the radiological prediction of a molecular subgrouping of MB prior to histopathological and genomic analysis. These authors found that the presence of spinal metastases in G3 MB (p < 0.01) and ependymal metastasis in the third ventricular infundibular recess with a mismatch pattern (i.e., diffusion restricting but minimal contrast enhancing) was significantly associated with G4 MB (p < 0.02) [40]. These enhancement patterns can help to differentiate molecular subgrouping when the imaging features of the primary tumor converge.
Although the unique observations that distinct radiological patterns can be identified to inform the MB molecular subgroup preoperatively, current practice remains maximal safe surgical resection followed by risk-adapted provision of adjuvant therapy. With greater resolution and understanding of the molecular basis of MB, a time where preoperative imaging features reliably predicts molecular subgrouping to influence intraoperative decision making (after intraoperative histopathological diagnosis) as well as providing adjuvant intraoperative targeted therapy may become a reality.
5 Implications of molecular subgrouping to the neuro-oncology multidisciplinary team
Until a multidisciplinary pediatric neuro-oncology team can reliably predict the diagnosis in addition to molecular subgrouping of pediatric MB (i.e., radiogenomics, liquid biopsy [42]), we continue to manage these children with maximal safe resection followed by adjuvant therapy. Multiple clinical trials have been proposed and executed employing the molecular subgroupings of pediatric MB, exploring tumor vulnerabilities, which has resulted in both successes and identification of areas for improvement for future trials [36]. One consistent message is that childhood MB should be treated in the context of a clinical trial whenever possible.
Wnt MB have the most favorable clinical outcome. Phoenix et al. provide one explanation for the improved PFS and OS observed in Wnt MB describing paracrine signals driven by mutant β-catenin induce an aberrant fenestrated vasculature that permits the accumulation of high levels of intratumoral adjuvant chemotherapy thereby facilitating the observed robust therapeutic response [43]. These findings and the general clinical observations that Wnt MB experiences the best prognosis have led to clinical efforts to spare this subgroup of patients from the devastating neurocognitive effects of radiation therapy in the clinical trial setting (e.g., SIOP-PNET5-MB, SJMB12, ACNS1422) [44,45,46,47,48]. Moxon-Emre et al. in their retrospective review of childhood MB treated between 1993 and 2013 found children in all subgroups declined in intellectual outcomes after surgery and adjuvant chemoradiotherapy, with a reduced dose of craniospinal irradiation (CSI) in addition to a tumor bed boost preserving intellectual functioning in carefully selected children diagnosed with Wnt and G4 MB [44]. In contrast however, G3 MB patients treated with reduced-dose CSI and tumor bed boost were not associated with preserved intellectual functioning and may be secondary to its location and relationship to the 4th ventricle [39]. As expected, treated hydrocephalus and cerebellar mutism were significant covariates that affected all measures of intellectual functioning reemphasizing the importance of prompt identification of posterior fossa tumors in children [49] and intraoperative decision-making [50].
The current therapeutic priority is to identify effective novel therapies to target the very high-risk metastatic and MYC-amplified MB [51]. As MYC is a highly stable protein, there is a consensus that it has the potential to be an effective target for MYC-amplified cancers. Unfortunately, effective means to target MYC are not yet available [52,53,54,55,56] and the search for alternative mediators of the aggressive phenotype of G3 MB continues. Bouffet describes the evolution of the management of high-risk MB over the last few decades highlighting important confounders in previous clinical trials [57]. The clear difference between high- and average-risk protocols is the dose of radiation to the neuraxis. The trials summarized in this review were unfortunately difficult to interpret as most of the studies included 2 phenotypically different populations that we now understand to be distinct pathophysiological entities (i.e., metastatic MB and incomplete resections). With the identification of molecular subgroups with clinical and prognostic significance, the dichotomy between standard- and high-risk patients and results from these high-risk MB trials become increasingly difficult to interpret.
Transitioning from a phenotypic (metastatic disease, incomplete resection) to molecular risk stratification (e.g., metastatic G3 MB, Shh with TP53 mutation) has led to new clinical trial strategies to employ molecular risk-adapted therapy. SJYC07 was a multicenter phase 2 trial with a risk-adapted protocol according to molecular subtype for young children with MB [36]. This study enrolled children younger than 3 years with a newly diagnosed MB. Patients were stratified to low-, intermediate-, and high-risk treatment groups with all patients receiving identical induction chemotherapy, with high-risk patients receiving an additional 5 doses of vinblastine. Induction followed by risk-adapted consolidation therapy where low-risk patients received cyclophosphamide, etoposide, and carboplatin while intermediate risk patients received 54Gy focal radiation to the tumor bed, and high-risk patients receiving targeted topotecan and cyclophosphamide. All patients received identical maintenance therapy with cyclophosphamide, topotecan, and erlotinib. Accrual into the low-risk group was suspended after an interim analysis observing the 1-year PFS was below the stopping rule boundary. Five-year PFS was 31.3% overall and 55.3%, 24.6%, and 16.7% in the low-, intermediate-, and high-risk groups respectively [36]. While the risk-adapted approach did not improve PFS in young children with MB, methylation subgroup analysis identified SHH subgroup (51.5%) had an improved PFS compared with G3 MB (8.3%). Furthermore, two distinct methylation subtypes within the infant SHH were identified namely iSHH-I (5-year PFS = 27.8%) and iSHH-II (5-year PFS 75.4%) where the latter subgroup had improved PFS in the absence of radiation, intraventricular chemotherapy, or high-dose chemotherapy compared with the iSHH-I subtype. iSHH-I cohort were enriched in SUFU alterations and chromosome 2 gains, and devoid of SMO mutations, a consideration for enrollment into clinical trials targeting the Shh pathway.
While there are some advocates of de-escalation therapy in non-Wnt low-risk MB (i.e., young children with desmoplastic MB with Shh activation), there are significant anxieties associated with its safety owing to the general feeling within the neurooncological community that the first treatment regimen provided to these children is the best chance of cure or disease control, though there is evidence of successful salvage in the literature [35]. This emphasizes the importance of greater biological understanding to inform future clinical trial design.
6 Implications of molecular subgrouping to the neurosurgeon
Despite the seemingly arbitrary cutoff of 1.5 cm2 residual tumor volume constituting a distinct factor in pediatric MB to upgrade a patient from standard to high-risk adjuvant treatment, pediatric neurosurgeons continue to advocate for quality of life versus OS. It is difficult to predict whether the integration of predictive factors in radiogenomics of molecular subgroups will alter the practice of some pediatric neurosurgeons to more aggressively resect a tumor with imaging features suggestive of a G3 MB versus a Wnt MB. In fact, irrespective of the subgrouping, the mainstay of surgical management for childhood MB remains safe maximal resection.
This assertion however was further substantiated in 2016, when Thompson et al. reported the result of their large (N = 787) retrospective cohort of histopathologically diagnosed MB from 35 international institutions [58]. Extent of resection was categorized as gross total resection (GTR) (no residual tumor), near-total resection (NTR) (< 1.5 cm2 residual tumor), and subtotal resection (> 1.5 cm2 residual tumor). The prognostic benefit of increased extent of resection for patient with MB was further amplified after molecular subgrouping was taken into account. Specifically, no benefit in OS was observed for GTR compared with NTR which suggests leaving behind a small tumor residual to spare the child of devastating post-surgical deficits is an intraoperative consideration [58]. This recommendation is validated further with the observation that pediatric MB is associated with a high preponderance to the development of post-operative syndromes such as cerebellar mutism secondary to its multiple high risk anatomical features (i.e., invasion of the brainstem floor, cerebellar peduncle) of the tumor entity [33, 39]. Moreover, while a second surgery for progressing post-operative residual is feasible with a recent International Society of Pediatric Oncology (SIOP) study showing second surgery to be significantly associated with prolonged survival in recurrent/progressive disease; it can be a technically more challenging operation with greater risk of complication. In this study, Sabel et al. observed the survival benefit of surgery after relapse compared with children who were salvaged with radiation and high-dose chemotherapy alone, validating the role for second surgery, not simply in the context of histopathological confirmation but with the goal of safe maximal second resection. [29]. All in all, safe maximal surgical resection for primary and recurrent/treatment refractory tumors plays an integral role in the management of the childhood MB.
7 Summary
The standardized management of childhood MB has reduced the mortality of standard-risk children; however, a greater understanding of not only higher-risk patients but also all MB is imperative for successful management. Recent research has re-stratified these children into molecular data-informed risk categories identifying biological aberrations and suggested priorities and novel strategies for therapeutic drug discovery. Radiogenomics has also gained momentum with consistencies observed between molecular subgroup diagnosis and imaging features but is in its infancy in terms of clinical utility. Despite the molecular data guided changes in adjuvant medical treatment strategies in childhood MB, the neurosurgical management of maximal safe surgical resection has remained a mainstay.
References
Ellison, D. W., et al. (2011). Medulloblastoma: clinicopathological correlates of SHH, WNT, and non-SHH/WNT molecular subgroups. Acta Neuropathologica, 121(3), 381–396.
Bailey, P., Cushings H. (1925). Medulloblastoma cerebelli:a common type of midcerebellar glioma of childhood. Archives of Neurology and Psychiatry, 14, 192–224.
Polkinghorn, W. R., & Tarbell, N. J. (2007). Medulloblastoma: tumorigenesis, current clinical paradigm, and efforts to improve risk stratification. Nature Clinical Practice. Oncology, 4(5), 295–304.
Ellison, D. (2002). Classifying the medulloblastoma: insights from morphology and molecular genetics. Neuropathology and Applied Neurobiology, 28(4), 257–282.
Packer, R. J. (2007). Craniospinal radiation therapy followed by adjuvant chemotherapy for newly diagnosed average-risk medulloblastoma. Current Neurology and Neuroscience Reports, 7(2), 130–132.
Packer, R. J., Sutton, L. N., Atkins, T. E., Radcliffe, J., Bunin, G. R., D’Angio, G., Siegel, K. R., & Schut, L. (1989). A prospective study of cognitive function in children receiving whole-brain radiotherapy and chemotherapy: 2-year results. Journal of Neurosurgery, 70(5), 707–713.
Bull, K. S., et al. (2014). Child-related characteristics predicting subsequent health-related quality of life in 8- to 14-year-old children with and without cerebellar tumors: a prospective longitudinal study. Neurooncol Pract, 1(3), 114–122.
Ellison, D. W. (2010). Childhood medulloblastoma: novel approaches to the classification of a heterogeneous disease. Acta Neuropathologica, 120(3), 305–316.
Ris, M. D., Packer, R., Goldwein, J., Jones-Wallace, D., & Boyett, J. M. (2001). Intellectual outcome after reduced-dose radiation therapy plus adjuvant chemotherapy for medulloblastoma: a Children’s Cancer Group study. Journal of Clinical Oncology, 19(15), 3470–3476.
Palmer, S. L., et al. (2003). Predicting intellectual outcome among children treated with 35-40 Gy craniospinal irradiation for medulloblastoma. Neuropsychology, 17(4), 548–555.
Palmer, S. L., et al. (2001). Patterns of intellectual development among survivors of pediatric medulloblastoma: a longitudinal analysis. Journal of Clinical Oncology, 19(8), 2302–2308.
Mulhern, R. K., Merchant, T. E., Gajjar, A., Reddick, W. E., & Kun, L. E. (2004). Late neurocognitive sequelae in survivors of brain tumours in childhood. The Lancet Oncology, 5(7), 399–408.
Yoo, H. J., Kim, H., Park, H. J., Kim, D. S., Ra, Y. S., & Shin, H. Y. (2016). Neurocognitive function and health-related quality of life in pediatric Korean survivors of medulloblastoma. Journal of Korean Medical Science, 31(11), 1726–1734.
Golub, T. R., et al. (1999). Molecular classification of cancer: class discovery and class prediction by gene expression monitoring. Science, 286(5439), 531–537.
Pomeroy, S. L., et al. (2002). Prediction of central nervous system embryonal tumour outcome based on gene expression. Nature, 415(6870), 436–442.
Thompson, M. C., Fuller, C., Hogg, T. L., Dalton, J., Finkelstein, D., Lau, C. C., Chintagumpala, M., Adesina, A., Ashley, D. M., Kellie, S. J., Taylor, M. D., Curran, T., Gajjar, A., & Gilbertson, R. J. (2006). Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. Journal of Clinical Oncology, 24(12), 1924–1931.
Kool, M., Koster, J., Bunt, J., Hasselt, N. E., Lakeman, A., van Sluis, P., Troost, D., Meeteren, N. S., Caron, H. N., Cloos, J., Mrsić, A., Ylstra, B., Grajkowska, W., Hartmann, W., Pietsch, T., Ellison, D., Clifford, S. C., & Versteeg, R. (2008). Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS One, 3(8), e3088.
Northcott, P. A., Korshunov, A., Witt, H., Hielscher, T., Eberhart, C. G., Mack, S., Bouffet, E., Clifford, S. C., Hawkins, C. E., French, P., Rutka, J. T., Pfister, S., & Taylor, M. D. (2011). Medulloblastoma comprises four distinct molecular variants. Journal of Clinical Oncology, 29(11), 1408–1414.
Cho, Y. J., Tsherniak, A., Tamayo, P., Santagata, S., Ligon, A., Greulich, H., Berhoukim, R., Amani, V., Goumnerova, L., Eberhart, C. G., Lau, C. C., Olson, J. M., Gilbertson, R. J., Gajjar, A., Delattre, O., Kool, M., Ligon, K., Meyerson, M., Mesirov, J. P., & Pomeroy, S. L. (2011). Integrative genomic analysis of medulloblastoma identifies a molecular subgroup that drives poor clinical outcome. Journal of Clinical Oncology, 29(11), 1424–1430.
Taylor, M. D., Northcott, P. A., Korshunov, A., Remke, M., Cho, Y. J., Clifford, S. C., Eberhart, C. G., Parsons, D. W., Rutkowski, S., Gajjar, A., Ellison, D. W., Lichter, P., Gilbertson, R. J., Pomeroy, S. L., Kool, M., & Pfister, S. M. (2012). Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathologica, 123(4), 465–472.
Kool, M., Korshunov, A., Remke, M., Jones, D. T., Schlanstein, M., Northcott, P. A., Cho, Y. J., Koster, J., Schouten-van Meeteren, A., van Vuurden, D., Clifford, S. C., Pietsch, T., von Bueren, A., Rutkowski, S., McCabe, M., Collins, V. P., Bäcklund, M. L., Haberler, C., Bourdeaut, F., Delattre, O., Doz, F., Ellison, D. W., Gilbertson, R. J., Pomeroy, S. L., Taylor, M. D., Lichter, P., & Pfister, S. M. (2012). Molecular subgroups of medulloblastoma: an international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, group 3, and group 4 medulloblastomas. Acta Neuropathologica, 123(4), 473–484.
Clifford, S. C., Lusher, M. E., Lindsey, J. C., Langdon, J. A., Gilbertson, R. J., Straughton, D., & Ellison, D. W. (2006). Wnt/wingless pathway activation and chromosome 6 loss characterize a distinct molecular sub-group of medulloblastomas associated with a favorable prognosis. Cell Cycle, 5(22), 2666–2670.
Ellison, D. W., et al. (2005). Beta-catenin status predicts a favorable outcome in childhood medulloblastoma: the United Kingdom Children’s Cancer Study Group Brain Tumour Committee. Journal of Clinical Oncology, 23(31), 7951–7957.
Louis, D. N., Perry, A., Reifenberger, G., von Deimling, A., Figarella-Branger, D., Cavenee, W. K., Ohgaki, H., Wiestler, O. D., Kleihues, P., & Ellison, D. W. (2016). The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathologica, 131(6), 803–820.
Ramaswamy, V., Remke M, Bouffet E., Bailey S., Clifford SC., et al. (2016). Risk stratification of childhood medulloblastoma in the molecular era: the current consensus. Acta Neuropathologica, 131(6), 821–31.
Johnston, D. L., et al. (2018). Survival following tumor recurrence in children with medulloblastoma. Journal of Pediatric Hematology/Oncology.
Johnston, D. L., Keene, D., Kostova, M., Lafay-Cousin, L., Fryer, C., Scheinemann, K., Carret, A. S., Fleming, A., Percy, V., Afzal, S., Wilson, B., Bowes, L., Zelcer, S., Mpofu, C., Silva, M., Larouche, V., Brossard, J., Strother, D., & Bouffet, E. (2015). Survival of children with medulloblastoma in Canada diagnosed between 1990 and 2009 inclusive. Journal of Neuro-Oncology, 124(2), 247–253.
Koschmann, C., et al. (2016). Survival after relapse of medulloblastoma. Journal of Pediatric Hematology/Oncology, 38(4), 269–273.
Sabel, M., Fleischhack, G., Tippelt, S., Gustafsson, G., Doz, F., Kortmann, R., Massimino, M., Navajas, A., von Hoff, K., Rutkowski, S., Warmuth-Metz, M., Clifford, S. C., Pietsch, T., Pizer, B., Lannering, B., & SIOP-E Brain Tumour Group. (2016). Relapse patterns and outcome after relapse in standard risk medulloblastoma: a report from the HIT-SIOP-PNET4 study. Journal of Neuro-Oncology, 129(3), 515–524.
Bowers, D. C., Gargan, L., Weprin, B. E., Mulne, A. F., Elterman, R. D., Munoz, L., Giller, C. A., & Winick, N. J. (2007). Impact of site of tumor recurrence upon survival for children with recurrent or progressive medulloblastoma. Journal of Neurosurgery, 107(1 Suppl), 5–10.
Perreault, S., Lober, R. M., Carret, A. S., Zhang, G., Hershon, L., Décarie, J. C., Yeom, K., Vogel, H., Fisher, P. G., & Partap, S. (2013). Relapse patterns in pediatric embryonal central nervous system tumors. Journal of Neuro-Oncology, 115(2), 209–215.
Cavalli, F. M. G., Remke, M., Rampasek, L., Peacock, J., Shih, D. J. H., Luu, B., Garzia, L., Torchia, J., Nor, C., Morrissy, A. S., Agnihotri, S., Thompson, Y. Y., Kuzan-Fischer, C. M., Farooq, H., Isaev, K., Daniels, C., Cho, B. K., Kim, S. K., Wang, K. C., Lee, J. Y., Grajkowska, W. A., Perek-Polnik, M., Vasiljevic, A., Faure-Conter, C., Jouvet, A., Giannini, C., Nageswara Rao, A. A., Li, K. K. W., Ng, H. K., Eberhart, C. G., Pollack, I. F., Hamilton, R. L., Gillespie, G. Y., Olson, J. M., Leary, S., Weiss, W. A., Lach, B., Chambless, L. B., Thompson, R. C., Cooper, M. K., Vibhakar, R., Hauser, P., van Veelen, M., Kros, J. M., French, P. J., Ra, Y. S., Kumabe, T., López-Aguilar, E., Zitterbart, K., Sterba, J., Finocchiaro, G., Massimino, M., van Meir, E., Osuka, S., Shofuda, T., Klekner, A., Zollo, M., Leonard, J. R., Rubin, J. B., Jabado, N., Albrecht, S., Mora, J., van Meter, T., Jung, S., Moore, A. S., Hallahan, A. R., Chan, J. A., Tirapelli, D. P. C., Carlotti, C. G., Fouladi, M., Pimentel, J., Faria, C. C., Saad, A. G., Massimi, L., Liau, L. M., Wheeler, H., Nakamura, H., Elbabaa, S. K., Perezpeña-Diazconti, M., Chico Ponce de León, F., Robinson, S., Zapotocky, M., Lassaletta, A., Huang, A., Hawkins, C. E., Tabori, U., Bouffet, E., Bartels, U., Dirks, P. B., Rutka, J. T., Bader, G. D., Reimand, J., Goldenberg, A., Ramaswamy, V., & Taylor, M. D. (2017). Intertumoral heterogeneity within medulloblastoma subgroups. Cancer Cell, 31(6), 737–754 e6.
Ramaswamy, V., Remke, M., Bouffet, E., Faria, C. C., Perreault, S., Cho, Y. J., Shih, D. J., Luu, B., Dubuc, A. M., Northcott, P. A., Schüller, U., Gururangan, S., McLendon, R., Bigner, D., Fouladi, M., Ligon, K. L., Pomeroy, S. L., Dunn, S., Triscott, J., Jabado, N., Fontebasso, A., Jones, D. T., Kool, M., Karajannis, M. A., Gardner, S. L., Zagzag, D., Nunes, S., Pimentel, J., Mora, J., Lipp, E., Walter, A. W., Ryzhova, M., Zheludkova, O., Kumirova, E., Alshami, J., Croul, S. E., Rutka, J. T., Hawkins, C., Tabori, U., Codispoti, K. E., Packer, R. J., Pfister, S. M., Korshunov, A., & Taylor, M. D. (2013). Recurrence patterns across medulloblastoma subgroups: an integrated clinical and molecular analysis. The Lancet Oncology, 14(12), 1200–1207.
Packer, R. J., Zhou, T., Holmes, E., Vezina, G., & Gajjar, A. (2013). Survival and secondary tumors in children with medulloblastoma receiving radiotherapy and adjuvant chemotherapy: results of Children’s Oncology Group trial A9961. Neuro-Oncology, 15(1), 97–103.
Kameda-Smith, M. M., Wang, A., Abdulhadi, N., Voth, R., Sergeant, A., Maharaj, A., Bakhshinyan, D., Adile, A. A., Pai, A. M., Ajani, O., Yarascavitch, B., Alyman, M. C., Duckworth, J., Samaan, M. C., Farrokhyar, F., Singh, S. K., Fleming, A., & Pediatric Brain Tumour Study Group. (2019). Salvage therapy for childhood medulloblastoma: a single center experience. The Canadian Journal of Neurological Sciences, 46(4), 403–414.
Robinson, G. W., et al. (2018). Risk-adapted therapy for young children with medulloblastoma (SJYC07): therapeutic and molecular outcomes from a multicentre, phase 2 trial. The Lancet Oncology, 19(6), 768–784.
Lafay-Cousin, L., Bouffet, E., Onar-Thomas, A., Billups, C. A., Hawkins, C., Eberhart, C., Horbinski, C., Robinson, G. W., Strother, D. R., Heier, L., Souweidane, M. M., Fouladi, M., Gajjar, A., & Children Oncology Group. (2017). ACNS1221: A phase II study for the treatment of non metastatic desmoplastic medulloblastoma in children less than 4 years of age-a report of the Children Oncology Group. Journal of Clinical Oncology,35(15_suppl), 10505–10505.
ClinicalTrials.gov. Bethesda (MD): National Library of Medicine (US). Identifier: NCT03434262, SJDWAN: St. Jude Children's Research Hospital Phase 1 study evaluating molecularly-driven doublet therapies for children and young adults with recurrent brain tumours. Available from: https://clinicaltrials.gov/ct2/show/NCT03434262?term;SJDAWN&draw;2&rank;1. Accessed 31 Oct 2019.
Perreault, S., Ramaswamy, V., Achrol, A. S., Chao, K., Liu, T. T., Shih, D., Remke, M., Schubert, S., Bouffet, E., Fisher, P. G., Partap, S., Vogel, H., Taylor, M. D., Cho, Y. J., & Yeom, K. W. (2014). MRI surrogates for molecular subgroups of medulloblastoma. AJNR. American Journal of Neuroradiology, 35(7), 1263–1269.
Mata-Mbemba, D., Zapotocky, M., Laughlin, S., Taylor, M. D., Ramaswamy, V., & Raybaud, C. (2018). MRI characteristics of primary tumors and metastatic lesions in molecular subgroups of pediatric medulloblastoma: a single-center study. AJNR. American Journal of Neuroradiology, 39(5), 949–955.
Lastowska, M., et al. (2018). Medulloblastoma with transitional features between group 3 and group 4 is associated with good prognosis. Journal of Neuro-Oncology, 138(2), 231–240.
Bonner, E. R., et al. (2018). Liquid biopsy for pediatric central nervous system tumors. NPJ Precis Oncol, 2, 29.
Phoenix, T. N., et al. (2016). Medulloblastoma genotype dictates blood brain barrier phenotype. Cancer Cell, 29(4), 508–522.
Moxon-Emre, I., Bouffet, E., Taylor, M. D., Laperriere, N., Scantlebury, N., Law, N., Spiegler, B. J., Malkin, D., Janzen, L., & Mabbott, D. (2014). Impact of craniospinal dose, boost volume, and neurologic complications on intellectual outcome in patients with medulloblastoma. Journal of Clinical Oncology, 32(17), 1760–1768.
ClinicalTrials.gov, A study assessing the feasibility of a surgery and chemotherapy-only in children with Wnt positive medulloblastoma. 2018, ethesda (MD): National Library of Medicine (US).
ClinicalTrials.gov, Reduced craniospinal radiation therapy and chemotherapy in treating younger patients with newly diagnosed WNT-driven medulloblastoma. 2018, National Library of Medicine (US): Bethesda (MD).
ClinicalTrials.gov, A clinical and molecular risk-directed therapy for newly diagnosed medulloblastoma. 2013: Bethesda (MD): National Library of Medicine (US).
ClinicalTrials.gov, International Society of Paediatric Oncology (SIOP) PNET 5 Medulloblastoma. 2014, ethesda (MD): National Library of Medicine (US).
Kameda-Smith, M. M., et al. (2013). Time to diagnosis of paediatric posterior fossa tumours: an 11-year west of Scotland experience 2000-2011. British Journal of Neurosurgery, 27(3), 364–369.
Sergeant, A., et al. (2017). Analysis of surgical and MRI factors associated with cerebellar mutism. Journal of Neuro-Oncology, 133(3), 539–552.
Gottardo, N. G., Hansford, J. R., McGlade, J., Alvaro, F., Ashley, D. M., Bailey, S., Baker, D. L., Bourdeaut, F., Cho, Y. J., Clay, M., Clifford, S. C., Cohn, R. J., Cole, C. H., Dallas, P. B., Downie, P., Doz, F., Ellison, D. W., Endersby, R., Fisher, P. G., Hassall, T., Heath, J. A., Hii, H. L., Jones, D. T., Junckerstorff, R., Kellie, S., Kool, M., Kotecha, R. S., Lichter, P., Laughton, S. J., Lee, S., McCowage, G., Northcott, P. A., Olson, J. M., Packer, R. J., Pfister, S. M., Pietsch, T., Pizer, B., Pomeroy, S. L., Remke, M., Robinson, G. W., Rutkowski, S., Schoep, T., Shelat, A. A., Stewart, C. F., Sullivan, M., Taylor, M. D., Wainwright, B., Walwyn, T., Weiss, W. A., Williamson, D., & Gajjar, A. (2014). Medulloblastoma down under 2013: a report from the third annual meeting of the International Medulloblastoma Working Group. Acta Neuropathologica, 127(2), 189–201.
ClinicalTrials.gov, A dose exploration study with MK-8628 in participants with selected advanced solid tumors (MK-8628-006). 2018, Bethesda (MD): National Library of Medicine (US).
ClinicalTrials.gov, A dose-finding study of MK-8628, a small molecule inhibitor of the bromodomain and extra-terminal (BET) proteins, in adults with selected advanced solid tumors (MK-8628-003). 2018, National Library of Medicine (US). Bethesda (MD).
Graff, J. N., Higano, C. S., Hahn, N. M., Taylor, M. H., Zhang, B., Zhou, X., Venkatakrishnan, K., Leonard, E. J., & Sarantopoulos, J. (2016). Open-label, multicenter, phase 1 study of alisertib (MLN8237), an aurora a kinase inhibitor, with docetaxel in patients with solid tumors. Cancer, 122(16), 2524–2533.
Schoffski, P., et al. (2011). Phase I, open-label, multicentre, dose-escalation, pharmacokinetic and pharmacodynamic trial of the oral aurora kinase inhibitor PF-03814735 in advanced solid tumours. European Journal of Cancer, 47(15), 2256–2264.
Seymour, J. F., et al. (2014). A phase 2 study of MK-0457 in patients with BCR-ABL T315I mutant chronic myelogenous leukemia and Philadelphia chromosome-positive acute lymphoblastic leukemia. Blood Cancer Journal, 4, e238.
Bouffet, E. (2019). Management of high-risk medulloblastoma. Neurochirurgie.
Thompson, E. M., et al. (2016). Prognostic value of medulloblastoma extent of resection after accounting for molecular subgroup: a retrospective integrated clinical and molecular analysis. The Lancet Oncology.
Acknowledgments
I would like to acknowledge all of the children, families, and international society of researchers who have helped advance the understanding of pediatric MB.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kameda-Smith, M.M. Pediatric medulloblastoma in the molecular era: what are the surgical implications?. Cancer Metastasis Rev 39, 235–243 (2020). https://doi.org/10.1007/s10555-020-09865-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10555-020-09865-y