
Abstract
The current survival outcome for children with medulloblastoma is a remarkable testimony for the multidisciplinary approach in the management of pediatric brain tumors. Children older than 3 years of age who have a less than 1.5 cm2 residual tumor after resection with no evidence of dissemination (M0) are classified as having standard-risk disease, while those with a larger tumor bed residual or with tumor dissemination are classified as having high-risk disease. Treatment for these patients includes craniospinal irradiation followed by a boost to the posterior fossa or posterior fossa tumor bed as well as cisplatin-based chemotherapy. For children younger than 3 years, surgery followed by chemotherapy is the most common treatment approach with or without primary site radiotherapy (RT). Currently, four molecular subtypes of medulloblastoma with prognostic implications have been identified. Current protocols are examining de-escalation of treatment for some children with Wnt-pathway tumors with more aggressive therapy for Group 3 and 4 subtypes.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others


1 Epidemiology
Medulloblastoma is an embryonal tumor of the cerebellum and accounts for about 20% of brain and 40% of all posterior fossa tumors in children. In the United States, there are approximately 400 cases per year, with a peak age of incidence between 5 and 6 years (Gurney et al. 1996). More than 70% are seen in children younger than 10 years while about 20% of cases are seen in those younger than 2 years of age. Medulloblastoma is more common in males and accounts for two-thirds of all cases.
2 Predisposing Factors
A previous report showed that 6.4% of patients with medulloblastoma have an associated genetic syndrome or congenital anomaly (Evans et al. 1993). Gorlin syndrome or basal cell nevus syndrome, a rare autosomal dominant condition characterized by multiple basal cell carcinomas, odontogenic keratocysts, and ocular abnormalities, has been associated with desmoplastic medulloblastoma. Type 2 Turcot syndrome has been associated with medulloblastoma and familial adenomatous polyposis (Paraf et al. 1997).
3 Presenting Symptoms
Children can present with a constellation of signs and symptoms related to cerebellar involvement, hydrocephalus (from obstruction of the fourth ventricle), invasion of the brainstem (direct extension), and tumor dissemination. Gait abnormalities, truncal unsteadiness, and difficulty with fine motor coordination are often seen. Patients with hydrocephalus may present with morning emesis and headaches, while those with brainstem involvement can present with double vision. Patients with spinal dissemination can present with back pain, and rarely cord compression can be a presenting sign.
4 Radiographic and Pathologic Findings
4.1 Radiology
The classic location of a medulloblastoma is midline and involves the vermis. However, in older children and adolescents, medulloblastoma may present as a well-lateralized lesion or as a tumor in the cerebellopontine angle. Some have suggested that the different molecular subtypes of medulloblastoma may have tumors whose origins are in certain locations in the posterior fossa. In one report, the wingless (Wnt) tumors were likely to be midline and infiltrative of the brainstem while the sonic hedgehog (SHH) pathway tumors were more likely to be in the cerebellar hemisphere (Gibson et al. 2010). Others have not seen any correlation with molecular subtype and tumor location (Teo et al. 2013).
4.2 Pathology
The different subtypes of medulloblastoma have traditionally included the classic, nodular desmoplastic, and large cell anaplastic histologies. The nodular desmoplastic histology has been reported to be the most common subtype in infants and has a more favorable outcome. The large cell, anaplastic subtype has the worst prognosis, while the classic subtype, which is the most common, has an intermediate prognosis. More recently, medulloblastoma has been subdivided into four molecular subtypes: Wnt, SHH, Group 3, and Group 4 (Northcott et al. 2011). Medulloblastoma activated through the Wnt signaling pathway has the best outcome with more than 90% survival; it comprises only 10% of all medulloblastoma. It is typically seen in older children and young adults and tends to be the classic histology. Most of these tumors have mutations in the CTNNB1 gene. Wnt medulloblastoma is associated with complete or partial loss of chromosome 6, while the other subtypes are most frequently associated with alterations in chromosome 17. The SHH subtype has a bimodal peak in infancy and in late childhood and early adulthood. It comprises 30% of all medulloblastoma and has an intermediate prognosis. The PTCH1 tumor suppressor gene is most commonly mutated but other genes such as SMO and SUFU may also be mutated. The desmoplastic histology tends to be of SHH subtype. Group 3 tumors have the worst outcome and comprise 25% of all medulloblastoma. They have the highest incidence of tumor dissemination at initial diagnosis and typically occur in infants and young children. Boys are affected more than girls. Recurrent amplification of MYC has been identified. Group 4 tumors comprise the remaining medulloblastomas and also have an intermediate prognosis. They tend to occur in boys and seen in children of all ages. Isochromosome 17q has been described more often in Group 4 tumors. Figure 6.1 shows the different molecular subtypes and associated demographic, clinical, and genetic features (Taylor et al. 2012). Significant differences in patterns of failure within the molecular subtypes are evident with local relapse within the posterior fossa most frequent for patients with SHH while metastatic relapse seems to be more frequent in the Group 3 and 4 tumors (Ramaswamy et al. 2013).

Molecular subgroups of medulloblastoma. Four molecular subgroups have been identified in children. The patient demographic and tumor characteristics are presented (Taylor et al. 2012)
5 Workup
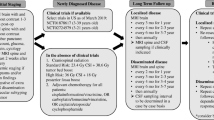
Children with medulloblastoma typically will have a magnetic resonance imaging (MRI) scan of the brain prior to resection and diagnosis. Postoperatively, patients undergo a MRI of the brain within 24–48 h after surgery to determine the amount of residual tumor. About 2 weeks after surgery, children with medulloblastoma should have a lumbar tap to rule out malignant cells in the cerebrospinal fluid and a MRI of the entire spine to evaluate for leptomeningeal dissemination. In one study looking at detection of malignant cells, the lumbar tap was more sensitive than ventricular shunt taps (Gajjar et al. 1999). Both lumbar tap and MRI of spine are complementary; if either exam is not done, leptomeningeal dissemination can be missed in about 15% of cases (Fouladi et al. 1999). Bone scan and bone marrow biopsy are only performed nowadays in the rare case of bone pain or abnormal blood count in the setting of leptomeningeal dissemination. Extraneural metastasis is uncommon at initial diagnosis and seen in ≤2% of cases (Mazloom et al. 2010).
The modified Chang staging system is used to note the absence or presence and degree of leptomeningeal spread (Table 6.1) (Chang et al. 1969). In general, about two-thirds of patients will have M0 disease. M1 disease is designated in the presence of malignant cytology from a lumbar tap without MRI evidence of gross tumor spread. Patients with M2 and M3 disease traditionally have worse outcome compared to M1 disease (Harisiadis and Chang 1977).
Prior to the age of molecular subtyping of medulloblastoma, the two most important prognostic factors have been the amount of residual in the tumor bed and the M status (Zeltzer et al. 1999). Patients with disease limited to the primary site with less than 1.5 cm2 residual and no dissemination (M0) are classified as having standard- or average-risk disease. Patients with either more than 1.5 cm2 residual or leptomeningeal or extraneural spread (M1–M4) are classified as having high-risk disease.
6 Acute Management
Hydrocephalus leading to increased intracranial pressure can be managed by an extraventricular drain, ventriculostomy, or a shunt. In general, most patients with posterior fossa tumors will have an intraoperative extraventricular drain (EVD) if there is significant pre-resection hydrocephalus. The EVD is weaned postoperatively and removed if tolerated. If persistent hydrocephalus remains with inability to wean EVD, a CSF shunt or endoscopic third ventriculostomy is performed (Lin and Riva-Cambrin 2015). The most common type of shunt used is the ventriculoperitoneal shunt. Possible complications of a shunt include malfunction and obstruction, infection, and the rare case of extraneural metastasis.
7 Treatment
7.1 Surgery
In addition to diversion of CSF to manage hydrocephalus, resection of the primary tumor is an important component of treatment. Maximal safe resection is performed with little or no residual as possible. As stated above, a tumor residual of more than 1.5 cm2 has been associated with a worse survival outcome (Zeltzer et al. 1999). Possible complications of surgery include aseptic meningitis, cervical spine instability, and posterior fossa syndrome. The posterior fossa or cerebellar mutism syndrome is classically seen at 1 or 2 days after surgery and can be accompanied by personality changes, emotional lability, decreased initiation of voluntary movements, and disturbance of ingestion (Pollack 1997). Although it can be associated with surgery for nonneoplastic and neoplastic conditions, children with medulloblastoma are the most commonly affected, and mutism usually lasts for 2–4 months with residual dysarthria. Approximately 8–25% of children with medulloblastoma develop posterior fossa syndrome (Doxey et al. 1999). The underlying cause is still not known, but involvement of the dentate nucleus, bilateral interruption of the dentothalamocortical pathway, and injury to median structures of the cerebellum have been postulated as a possible explanation for this condition. There is some evidence that the incidence of posterior fossa syndrome is increasing, proportional to the increase in more aggressive surgery (Korah et al. 2010).
7.2 Radiotherapy
Medulloblastoma is a radiosensitive tumor. The mean inactivation dose and surviving fraction after 2 Gy have been reported to be 1.43 Gy and 27% (Deschavanne and Fertil 1996). RT is a very important component of treatment in the curative management of these patients. Radiotherapy dose, volume, and sequencing of RT and chemotherapy have all been implicated as factors which can influence outcome and are discussed below (Castro-Vita et al. 1980; Garton et al. 1990; Tarbell et al. 1991; Jenkin 1969; Paulino 1997). RT technique is likewise important (Carrie et al. 1992, 1999). It has been previously shown that inadequate coverage of the cribriform plate can lead to a higher risk of subfrontal recurrences (Jereb et al. 1984). Protraction of RT treatment duration for more than 45–50 days has been associated with worse posterior fossa control and survival (Paulino et al. 2003; del Charco et al. 1998; Taylor et al. 2004).
7.2.1 Radiotherapy Volume
Historically, the greatest advance in the curative management of medulloblastoma has been the use of craniospinal irradiation (CSI). The landmark Paterson and Farr study established that CSI is the appropriate RT treatment for medulloblastoma (Paterson and Farr 1953). The use of less extensive RT fields (posterior fossa only, posterior fossa and spinal RT) has been associated with worse outcome (Castro-Vita et al. 1980; Jenkin 1969). In the era of chemotherapy, the French M4 protocol utilized chemotherapy to decrease the RT volume to just the posterior fossa and spine (Bouffet et al. 1992). Of 16 children treated, only 3 (18%) were alive and disease-free at a mean follow-up of 6 years. The most common site of relapse was in the supratentorial brain, the site which was not irradiated, accounting for 69% of relapses. Currently, the only children not treated with CSI are the very young patients (<3 years old) where CSI may result in severe neurocognitive and musculoskeletal sequelae (Ashley et al. 2012; Duffner et al. 1993).
After CSI, additional radiation is given to the posterior fossa where the original tumor is located. Historically, the entire posterior fossa (PF) received the boost RT dose. Treatment fields were largely based on using bony landmarks in the skull. Because of advances in imaging and RT treatment delivery and concerns regarding toxicity of RT, some investigators have boosted only the tumor bed and any residual tumor with a safety margin. Current available data indicate that isolated non-tumor bed PF failures occur in <5% of patients when only the tumor bed receives the boost RT dose (Merchant et al. 2008; Paulino et al. 2011; Douglas et al. 2004; Wolden et al. 2003). A study from Toronto suggests that this approach improves neurocognitive outcome compared to treatment of the entire posterior fossa (Moxon-Emre et al. 2014). The recently closed Children’s Oncology Group (COG) ACNS0031 protocol randomized standard-risk patients to PF boost vs. tumor bed boost (tumor bed with a 1.5 cm safety margin for clinical target volume or CTV); the preliminary results of this phase III study showed that the entire posterior fossa does not need to receive the boost dose (Michalski et al. 2016).
7.2.2 Radiotherapy Dose
Prior to the era of routine use of chemotherapy, the standard CSI dose was approximately 36 Gy in 20 fractions. In a Children’s Cancer Group study, Packer and colleagues reported a 79% 5-year progression-free survival in standard-risk medulloblastoma with a 23.4 Gy CSI dose followed by a boost with vincristine, lomustine, and cisplatin (Packer et al. 1999). A subsequent COG study randomizing standard-risk patients to two different chemotherapy regimens confirmed the efficacy of 23.4 Gy CSI dose (Packer et al. 2006). Currently, the standard CSI dose for standard-risk patients is 23.4 Gy in 13 fractions followed by a RT boost in conjunction with chemotherapy. When delivering 23.4 Gy CSI for standard-risk patients, RT is started within 30 days of the surgery. The SIOP II study showed an inferior survival outcome for 23.4 Gy CSI patients when RT was delayed, by giving initial chemotherapy; however, one of the criticisms of the study is the possible use of a less efficient chemotherapy regimen (Bailey et al. 1995). The SFOP M7 protocol delivered pre-RT chemotherapy with a reduced supratentorial dose of 27 Gy; RT was delivered at 7 weeks after surgery (Gentet et al. 1995). The 5-year EFS was 74% for patients with macroscopically complete or subtotal excision and M0 disease. The MSFOP 93 nonrandomized protocol delivered pre-RT chemotherapy with RT delivered at the latest 15 weeks after surgery (Oyharcabal-Bourden et al. 2005). RT dose was 25 Gy CSI and 55 Gy PF RT and showed a 5-year recurrence-free survival rate of 64.8%. Although the latter 2 studies suggest that RT can be delayed, the best outcomes that have been reported using a reduced CSI dose with chemotherapy have delivered upfront RT (Packer et al. 1999, 2006).
Some groups have recommended a 36 Gy CSI dose for standard-risk anaplastic medulloblastoma because of their inferior prognosis compared to other histologic subtypes. Currently, the COG ACNS0332 guidelines recommend the use of 36 Gy CSI even for M0 anaplastic medulloblastoma, and this subgroup is currently excluded from the ongoing European PNET5 protocol for standard-risk patients (Chintagumpala et al. 2016).
Studies using a 23.4 Gy CSI dose have shown neurocognitive deficits especially in children younger than 7 years of age (Ris et al. 2001, 2013). Pilot studies from Children’s Hospital of Philadelphia and Indiana University have conflicting results on efficacy and neurocognitive outcome in young children treated with 18 Gy CSI (Goldwein et al. 1996; Jakacki et al. 2004). The COG ACS0031 protocol randomized standard-risk children 3–7 years old to either 18 Gy or 23.4 Gy CSI dose followed by a boost with chemotherapy; the preliminary results inidicate an inferior event-free survival outcome for those treated with 18 Gy CSI (Michalski et al. 2016).
For patients with high-risk disease, CSI doses have typically ranged from 36 to 39.6 Gy in 20 to 22 fractions followed by a posterior fossa/tumor bed boost. Results from the POG 9031 study indicate that the use of this dose regimen with chemotherapy was associated with a favorable 5-year event-free survival (Tarbell et al. 2013). POG 9031 looked at the sequencing of chemotherapy and RT with patients either receiving 3 cycles of cisplatin and etoposide before RT or after RT. Both arms of the study received consolidative vincristine and cyclophosphamide. The 5-year event-free survival (EFS) rates were 66% for chemotherapy first arm and 70% for RT first arm (p = 0.54). The HIT ’91 study showed no difference in EFS or OS among M2 and M3 patients receiving maintenance chemotherapy (RT first) and sandwich chemotherapy (chemotherapy first) (Kortmann 2014; Kortmann et al. 2000). The CSI dose given in this study was 35.2 Gy in 22 fractions with a boost to the posterior fossa for a total dose of 55.2 Gy. In patients with M0 or M1 disease, patients who had RT first had a better EFS and OS. The SIOP/UKCCSG PNET3 tested pre-RT chemotherapy including vincristine, etoposide, carboplatin, and cyclophosphamide followed by 35 Gy CSI and 55 Gy total PF dose for M2–M3 patients with a 5-year PFS of 35% (Taylor et al. 2005).
Some studies have looked at using altered fractionation RT to increase dose for better tumor control. The HIT-SIOP PNET 4 Trial randomized patients with standard-risk medulloblastoma to 23.4 Gy CSI and 54 Gy posterior fossa (1.8 Gy/fraction) over 30 fractions in 6 weeks to 36 Gy CSI, 60 Gy to posterior fossa and 68 Gy to tumor bed (1.0 Gy/fraction given twice daily) over 68 fractions in 6.8 weeks. Both arms included vincristine during RT followed by post-RT 8 cycles of vincristine, lomustine, and cisplatin (Lannering et al. 2012). The 5-year EFS and OS rates were the same in both arms. The PNET 4 showed that hyperfractionation was associated with a better executive function with a trend for better verbal outcomes in the children less than 8 years at the time of RT (Kennedy et al. 2014; Camara-Costa et al. 2015). A study from Mumbai utilizing hyperfractionated RT showed no significant decline in all tested domains of cognitive function in 20 children with a 2-year follow-up (Gupta et al. 2012). Regarding the high-risk group, the Milan strategy for disseminated medulloblastoma included pre-RT chemotherapy, a hyperfractionated accelerated radiotherapy (HART) regimen of 39 Gy CSI (1.3 Gy/fraction twice daily) followed a posterior fossa boost to a total dose of 60 Gy (1.5 Gy/fraction twice daily) and 2 myeloablative courses for persistent disseminated disease before HART (Gandola et al. 2009). The 5-year event-free and overall survival rates were 70 and 73%, comparable to results of the POG 9031 protocol discussed above. To date, the results of altered fractionation RT to increase dose have not shown any significant survival benefit over conventional fractionation.
Table 6.2 outlines some of the major trials performed in standard- and high-risk childhood medulloblastoma (Packer et al. 2006; Bailey et al. 1995; Tarbell et al. 2013; Lannering et al. 2012; von Hoff et al. 2009; Taylor et al. 2003; Gajjar et al. 2006).
7.3 Chemotherapy
Most patients with medulloblastoma will receive chemotherapy. In infants, chemotherapy is used to delay the institution of RT; in older children, chemotherapy is used to lower the CSI dose in standard-risk patients and improve survival outcome in high-risk patients. Earlier randomized trials have not demonstrated an improvement in survival outcome in patients treated with chemotherapy; however on subset analysis, there is a suggestion that patient with high-risk tumors may have a benefit for the addition of chemotherapy (Krischer et al. 1991; Evans et al. 1990; Tait et al. 1990).
A latter study, SIOP/UKCCSG PNET-3, randomized patients to 35 Gy CSI with 20 Gy posterior fossa boost or chemotherapy followed by the same RT (Taylor et al. 2003). Chemotherapy consisted of vincristine, etoposide, carboplatin, and cyclophosphamide. The 3- and 5-year EFS were 78.5% and 74.2% vs. 64.8% and 59.8% for chemo + RT and RT alone, respectively. There was no statistically significant difference in 3- or 5-year OS.
Although currently, the use of chemotherapy in standard-risk medulloblastoma is considered “standard of care” practice, there is some evidence that RT alone given in a hyperfractionated regimen may give the same survival outcome with acceptable neurotoxicity. The French M-SFOP 98 protocol delivered hyperfractionated RT to 36 Gy in 36 fractions followed by a conformal tumor bed boost to a total dose of 68 Gy in 68 fractions (1 Gy BID) (Carrie et al. 2009). The 3-year progression-free and OS rates were 81% and 89%, respectively. Of the 48 patients <19 years of age, 22 were evaluable and there was no decrease in IQ scores during the first 2 years of follow-up.
8 Infant Medulloblastoma
Unfortunately about 20% of medulloblastoma occur in the very young. The survival outcomes of these children have traditionally been poor because of the omission or delay in delivering CSI. Radiotherapy to the craniospinal axis in the very young has been associated with severe cognitive dysfunction and musculoskeletal abnormalities (Johnston et al. 2009; Walter et al. 1999). For many years, the standard approach was to perform resection followed by chemotherapy; CSI was delayed until the child had turned 3 years. The Baby POG study showed a 1- and 2-year progression-free survival (PFS) of 42% and 34% with this approach (Duffner et al. 1993). One of the major advances in infant medulloblastoma has been the discovery that the completely resected desmoplastic subtype without evidence of leptomeningeal spread (M0) has a favorable outcome (Rutkowski et al. 2005; von Bueren et al. 2011). The HIT-SKK ’92 trial delivered chemotherapy after resection to 43 children younger than 3 years of age; the regimen included intravenous cyclophosphamide, vincristine, methotrexate, carboplatin and etoposide and intraventricular methotrexate (Rutkowski et al. 2005). RT was not part of the treatment regimen. The 5-year PFS for patients with completely resected, M0 tumors and desmoplastic tumors were 82% and 85%, respectively. The HIT-SKK ’2000 trial showed the excellent outcome of this approach for tumors with desmoplastic histology and medulloblastoma with extensive nodularity (von Bueren et al. 2011). This study also showed that local recurrence was the predominant pattern of failure for infants with M0 disease. Building on this knowledge, the COG P9934 study routinely delivered local RT to the primary site in M0 patients (Ashley et al. 2012). The results showed that the local control and EFS were better with conformal RT compared to a previous infant medulloblastoma study, POG9233, which delivered only adjuvant chemotherapy (Geyer et al. 2005). However, an analysis of children with non-desmoplastic histology did not reveal any improvement in EFS with conformal RT (23% vs. 14%, p = 0.92). The BB SFOP French protocol enrolled 79 patients (15 with metastases) who were treated with postoperative chemotherapy, with RT reserved at progression. The 5-year OS was 73% for no local residual, 41% for patients with local residual disease, and 13% for metastatic patients; the 5-year PFS was 41% for no local residual compared to 0% for those with subtotal resection (Grill et al. 2005). At present, the use of local RT remains controversial and is done at the discretion of the treating physicians.
9 Target Delineation and Technique
Target delineation and technique for craniospinal axis irradiation will be discussed in another chapter. In general, there is a theoretical advantage of using protons for children because of the lack of the exit dose when treating the spinal field. There should be lower dose to the thyroid gland, heart, lungs, abdominal organs, and gonads with the use of protons when compared to photons (Johnstone et al. 2013). Acute toxicity such as nausea and vomiting, decrease in blood counts, intervention for management of esophagitis and weight loss are less with proton compared to photon-treated adult medulloblastoma patients (Brown et al. 2013). There are also several modeling studies which indicate that proton beam therapy may reduce the risk of radiation-induced secondary tumors (Miralbell et al. 2002). Many of these patients are currently being treated in the supine position as this is more comfortable for the patient, provides better airway access during anesthesia, and is more reproducible (Verma et al. 2015).
For target delineation of the posterior fossa, the entire posterior fossa is contoured and considered as the clinical target volume (CTV). A 0.3–0.5 mm margin is added depending on institutional practice for the planning target volume (PTV). For tumor bed delineation, the preoperative and postoperative MRI of the patient needs to be reviewed. The tumor cavity is outlined in addition to any residual tumor. The preoperative MRI can help guide which areas need to be included based on where tumor had contact with normal brain tissue. An additional margin of 1–1.5 cm to the contoured cavity and residual tumor is added, respecting boundaries of normal anatomic structures such as bone. Examples of contours for posterior fossa boost and tumor bed boost are presented in Fig. 6.2. As shown above, the bilateral cochlea is also contoured. For standard- and high-risk patients, we try and limit the mean dose to the cochlea to <37 Gy and <43 Gy, respectively (Paulino et al. 2010).

A patient with standard-risk medulloblastoma. Note the coverage of the cribriform plate as part of the planning target volume or PTV (dark blue) for craniospinal irradiation. The upper 6 slices show a posterior fossa boost while the lower 9 slices show a tumor bed boost. For the posterior fossa boost, the clinical target volume (CTV) is the entire posterior fossa delineated in red and PTV is the orange line. For the tumor bed boost the gross tumor volume (GTV) is the purple shaded area, CTV is orange line and PTV is purple line. (Buchsbaum and Paulino 2015)
For the boost portion of treatment, both protons and intensity modulated radiotherapy (IMRT) have been used. One advantage of the tumor bed over the posterior fossa boost is a lower cochlear dose. Long-term studies using an intensity modulated radiation therapy (IMRT) boost to the tumor bed indicate that only 25% of patients have Grade 3–4 ototoxicity, compared to about 65% Grade 3–4 ototoxicity in the era of parallel-opposed lateral fields, treating the entire posterior fossa (Paulino et al. 2010; Huang et al. 2002). Protons also have a theoretical advantage of reducing dose to the hippocampus, hypothalamus, and pituitary which may translate to better cognitive and endocrine function compared to photon treatments. For standard-risk medulloblastoma, there does not seem to be a difference in progression-free or OS in patients treated with protons vs. photons (Eaton et al. 2016a). There is also some evidence that proton patients have a reduced risk of hypothyroidism, sex hormone deficiency, requirement for any endocrine replacement therapy, and height impairment (Eaton et al. 2016b).
10 Follow-Up and Outcomes
10.1 Follow-Up Guidelines
In general, follow-up is scheduled every 3 months for the first year after completion of therapy, every 6 months for the next 2 years, and annually after 3 years. A history and physical examination is performed in addition to MRI of the brain and spine during these visits. It is uncommon to get recurrences after 7 years of therapy so patients usually are followed up for possible complications of treatment (Belza et al. 1991). The patient will also require endocrine evaluation every 6 months for the first 3 years and annually thereafter. Audiograms are often performed yearly after therapy and may depend on the patient’s symptomatology and auditory findings. The topic of surveillance for treated medulloblastoma patients has been questioned as salvage is poor for these patients (Torres et al. 1994; Shaw et al. 1997). Some have argued that patients who recur will be symptomatic.
11 Future Directions
Current studies are underway to determine whether treatment of children with medulloblastoma can be tailored according to molecular subtypes. For example, children with M0 classic medulloblastoma with a Wnt molecular signature may need less treatment such as RT omission or lowering of RT dose, whereas for Group 3 tumors, treatments may be more intensive with use of systemic or targeted agents. For Group 3 and 4 tumors, further delineation of biological targets is needed for better outcomes. Long-term follow-up of patients treated with protons is necessary to determine efficacy and long-term complications when compared to children treated with photons (Wolden 2013).
References
Ashley DM et al (2012) Induction chemotherapy and conformal radiation therapy for very young children with nonmetastatic medulloblastoma: Children’s Oncology Group study P9934. J Clin Oncol 30(26):3181–3186
Bailey CC et al (1995) Prospective randomised trial of chemotherapy given before radiotherapy in childhood medulloblastoma. International Society of Paediatric Oncology (SIOP) and the (German) Society of Paediatric Oncology (GPO): SIOP II. Med Pediatr Oncol 25(3):166–178
Belza MG et al (1991) Medulloblastoma: freedom from relapse longer than 8 years—a therapeutic cure? J Neurosurg 75(4):575–582
Bouffet E et al (1992) M4 protocol for cerebellar medulloblastoma: supratentorial radiotherapy may not be avoided. Int J Radiat Oncol Biol Phys 24(1):79–85
Brown AP et al (2013) Proton beam craniospinal irradiation reduces acute toxicity for adults with medulloblastoma. Int J Radiat Oncol Biol Phys 86(2):277–284
Buchsbaum J, Paulino AC (2015) Pediatric brain tumors. In: Riaz N, Lee NY, Lu JJ (eds) Target volume delineation for conformal and intensity-modulated radiation therapy. Springer, Cham, pp 495–514
Camara-Costa H et al (2015) Neuropsychological outcome of children treated for standard risk medulloblastoma in the PNET4 European randomized controlled trial of Hyperfractionated versus standard radiation therapy and maintenance chemotherapy. Int J Radiat Oncol Biol Phys 92(5):978–985
Carrie C et al (1992) Quality control of radiotherapeutic treatment of medulloblastoma in a multicentric study: the contribution of radiotherapy technique to tumour relapse. The French Medulloblastoma Group. Radiother Oncol 24(2):77–81
Carrie C et al (1999) Impact of targeting deviations on outcome in medulloblastoma: study of the French Society of Pediatric Oncology (SFOP). Int J Radiat Oncol Biol Phys 45(2):435–439
Carrie C et al (2009) Online quality control, hyperfractionated radiotherapy alone and reduced boost volume for standard risk medulloblastoma: long-term results of MSFOP 98. J Clin Oncol 27(11):1879–1883
Castro-Vita H et al (1980) Medulloblastomas. Rev Interam Radiol 5(3):77–82
Chang CH, Housepian EM, Herbert C Jr (1969) An operative staging system and a megavoltage radiotherapeutic technic for cerebellar medulloblastomas. Radiology 93(6):1351–1359
Chintagumpala M, Paulino A, Panigrahy A, Hawkins C, Jae A, Parsons DW (2016) Embryonal and pineal region tumors. In: Poplack DG, Pizzo PA (eds) Principles and practice of pediatric oncology. Wolters Kluwer, Philadelphia, pp 671–699
del Charco JO et al (1998) Medulloblastoma: time-dose relationship based on a 30-year review. Int J Radiat Oncol Biol Phys 42(1):147–154
Deschavanne PJ, Fertil B (1996) A review of human cell radiosensitivity in vitro. Int J Radiat Oncol Biol Phys 34(1):251–266
Douglas JG et al (2004) Concurrent chemotherapy and reduced-dose cranial spinal irradiation followed by conformal posterior fossa tumor bed boost for average-risk medulloblastoma: efficacy and patterns of failure. Int J Radiat Oncol Biol Phys 58(4):1161–1164
Doxey D et al (1999) Posterior fossa syndrome: identifiable risk factors and irreversible complications. Pediatr Neurosurg 31(3):131–136
Duffner PK et al (1993) Postoperative chemotherapy and delayed radiation in children less than three years of age with malignant brain tumors. N Engl J Med 328(24):1725–1731
Eaton BR et al (2016a) Clinical outcomes among children with standard-risk medulloblastoma treated with proton and photon radiation therapy: a comparison of disease control and overall survival. Int J Radiat Oncol Biol Phys 94(1):133–138
Eaton BR et al (2016b) Endocrine outcomes with proton and photon radiotherapy for standard risk medulloblastoma. Neuro-Oncology 18(6):881–887
Evans AE et al (1990) The treatment of medulloblastoma. Results of a prospective randomized trial of radiation therapy with and without CCNU, vincristine, and prednisone. J Neurosurg 72(4):572–582
Evans G et al (1993) Congenital anomalies and genetic syndromes in 173 cases of medulloblastoma. Med Pediatr Oncol 21(6):433–434
Fouladi M et al (1999) Comparison of CSF cytology and spinal magnetic resonance imaging in the detection of leptomeningeal disease in pediatric medulloblastoma or primitive neuroectodermal tumor. J Clin Oncol 17(10):3234–3237
Gajjar A et al (1999) Comparison of lumbar and shunt cerebrospinal fluid specimens for cytologic detection of leptomeningeal disease in pediatric patients with brain tumors. J Clin Oncol 17(6):1825–1828
Gajjar A et al (2006) Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma-96): long-term results from a prospective, multicentre trial. Lancet Oncol 7(10):813–820
Gandola L et al (2009) Hyperfractionated accelerated radiotherapy in the Milan strategy for metastatic medulloblastoma. J Clin Oncol 27(4):566–571
Garton GR et al (1990) Medulloblastoma—prognostic factors and outcome of treatment: review of the Mayo Clinic experience. Mayo Clin Proc 65(8):1077–1086
Gentet JC et al (1995) Preirradiation chemotherapy including “eight drugs in 1 day” regimen and high-dose methotrexate in childhood medulloblastoma: results of the M7 French Cooperative study. J Neurosurg 82(4):608–614
Geyer JR et al (2005) Multiagent chemotherapy and deferred radiotherapy in infants with malignant brain tumors: a report from the Children’s Cancer Group. J Clin Oncol 23(30):7621–7631
Gibson P et al (2010) Subtypes of medulloblastoma have distinct developmental origins. Nature 468(7327):1095–1099
Goldwein JW et al (1996) Updated results of a pilot study of low dose craniospinal irradiation plus chemotherapy for children under five with cerebellar primitive neuroectodermal tumors (medulloblastoma). Int J Radiat Oncol Biol Phys 34(4):899–904
Grill J et al (2005) Treatment of medulloblastoma with postoperative chemotherapy alone: an SFOP prospective trial in young children. Lancet Oncol 6(8):573–580
Gupta T et al (2012) Early clinical outcomes demonstrate preserved cognitive function in children with average-risk medulloblastoma when treated with hyperfractionated radiation therapy. Int J Radiat Oncol Biol Phys 83(5):1534–1540
Gurney JG et al (1996) Trends in cancer incidence among children in the U.S. Cancer 78(3):532–541
Harisiadis L, Chang CH (1977) Medulloblastoma in children: a correlation between staging and results of treatment. Int J Radiat Oncol Biol Phys 2(9–10):833–841
Huang E et al (2002) Intensity-modulated radiation therapy for pediatric medulloblastoma: early report on the reduction of ototoxicity. Int J Radiat Oncol Biol Phys 52(3):599–605
Jakacki RI et al (2004) A pilot study of preirradiation chemotherapy and 1800 cGy craniospinal irradiation in young children with medulloblastoma. Int J Radiat Oncol Biol Phys 60(2):531–536
Jenkin RD (1969) Medulloblastoma in childhood: radiation therapy. Can Med Assoc J 100(2):51–53
Jereb B et al (1984) Radiation for medulloblastoma adjusted to prevent recurrence to the cribriform plate region. Cancer 54(3):602–604
Johnston DL et al (2009) Medulloblastoma in children under the age of three years: a retrospective Canadian review. J Neuro-Oncol 94(1):51–56
Johnstone PA et al (2013) Pediatric CSI: are protons the only ethical approach? Int J Radiat Oncol Biol Phys 87(2):228–230
Kennedy C et al (2014) Quality of survival and growth in children and young adults in the PNET4 European controlled trial of hyperfractionated versus conventional radiation therapy for standard-risk medulloblastoma. Int J Radiat Oncol Biol Phys 88(2):292–300
Korah MP et al (2010) Incidence, risks, and sequelae of posterior fossa syndrome in pediatric medulloblastoma. Int J Radiat Oncol Biol Phys 77(1):106–112
Kortmann RD (2014) The chemotherapy before or after radiation therapy does not influence survival of children with high-risk medulloblastomas: results of the multicenter and randomized study of the Pediatric Oncology Group (POG 9031). Strahlenther Onkol 190(1):106–108
Kortmann RD et al (2000) Postoperative neoadjuvant chemotherapy before radiotherapy as compared to immediate radiotherapy followed by maintenance chemotherapy in the treatment of medulloblastoma in childhood: results of the German prospective randomized trial HIT ‘91. Int J Radiat Oncol Biol Phys 46(2):269–279
Krischer JP et al (1991) Nitrogen mustard, vincristine, procarbazine, and prednisone as adjuvant chemotherapy in the treatment of medulloblastoma. A Pediatric Oncology Group study. J Neurosurg 74(6):905–909
Lannering B et al (2012) Hyperfractionated versus conventional radiotherapy followed by chemotherapy in standard-risk medulloblastoma: results from the randomized multicenter HIT-SIOP PNET 4 trial. J Clin Oncol 30(26):3187–3193
Lin CT, Riva-Cambrin JK (2015) Management of posterior fossa tumors and hydrocephalus in children: a review. Childs Nerv Syst 31(10):1781–1789
Mazloom A, Zangeneh AH, Paulino AC (2010) Prognostic factors after extraneural metastasis of medulloblastoma. Int J Radiat Oncol Biol Phys 78(1):72–78
Merchant TE et al (2008) Multi-institution prospective trial of reduced-dose craniospinal irradiation (23.4 Gy) followed by conformal posterior fossa (36 Gy) and primary site irradiation (55.8 Gy) and dose-intensive chemotherapy for average-risk medulloblastoma. Int J Radiat Oncol Biol Phys 70(3):782–787
Michalski JM et al (2016) Results of COG ACNS0331: A phase III trial of involved-field radiotherapy (IFRT) and low dose craniospinal irradiation (LD-CSI) with chemotherapy in average-risk medulloblastoma: a report from the children’s oncology group. Int J Radiat Oncol Biol Phys 96(5):937–938
Miralbell R et al (2002) Potential reduction of the incidence of radiation-induced second cancers by using proton beams in the treatment of pediatric tumors. Int J Radiat Oncol Biol Phys 54(3):824–829
Moxon-Emre I et al (2014) Impact of craniospinal dose, boost volume, and neurologic complications on intellectual outcome in patients with medulloblastoma. J Clin Oncol 32(17):1760–1768
Northcott PA et al (2011) Medulloblastoma comprises four distinct molecular variants. J Clin Oncol 29(11):1408–1414
Oyharcabal-Bourden V et al (2005) Standard-risk medulloblastoma treated by adjuvant chemotherapy followed by reduced-dose craniospinal radiation therapy: a French Society of Pediatric Oncology Study. J Clin Oncol 23(21):4726–4734
Packer RJ et al (1999) Treatment of children with medulloblastomas with reduced-dose craniospinal radiation therapy and adjuvant chemotherapy: a Children’s Cancer Group study. J Clin Oncol 17(7):2127–2136
Packer RJ et al (2006) Phase III study of craniospinal radiation therapy followed by adjuvant chemotherapy for newly diagnosed average-risk medulloblastoma. J Clin Oncol 24(25):4202–4208
Paraf F, Jothy S, Van Meir EG (1997) Brain tumor-polyposis syndrome: two genetic diseases? J Clin Oncol 15(7):2744–2758
Paterson E, Farr RF (1953) Cerebellar medulloblastoma: treatment by irradiation of the whole central nervous system. Acta Radiol 39(4):323–336
Paulino AC (1997) Radiotherapeutic management of medulloblastoma. Oncology (Williston Park) 11(6):813–823; discussion 823, 827–8, 831.
Paulino AC et al (2003) Protracted radiotherapy treatment duration in medulloblastoma. Am J Clin Oncol 26(1):55–59
Paulino AC et al (2010) Ototoxicity after intensity-modulated radiation therapy and cisplatin-based chemotherapy in children with medulloblastoma. Int J Radiat Oncol Biol Phys 78(5):1445–1450
Paulino AC et al (2011) Local control after craniospinal irradiation, intensity-modulated radiotherapy boost, and chemotherapy in childhood medulloblastoma. Cancer 117(3):635–641
Pollack IF (1997) Posterior fossa syndrome. Int Rev Neurobiol 41:411–432
Ramaswamy V et al (2013) Recurrence patterns across medulloblastoma subgroups: an integrated clinical and molecular analysis. Lancet Oncol 14(12):1200–1207
Ris MD et al (2001) Intellectual outcome after reduced-dose radiation therapy plus adjuvant chemotherapy for medulloblastoma: a Children’s Cancer Group study. J Clin Oncol 19(15):3470–3476
Ris MD et al (2013) Intellectual and academic outcome following two chemotherapy regimens and radiotherapy for average-risk medulloblastoma: COG A9961. Pediatr Blood Cancer 60(8):1350–1357
Rutkowski S et al (2005) Treatment of early childhood medulloblastoma by postoperative chemotherapy alone. N Engl J Med 352(10):978–986
Shaw DW et al (1997) Asymptomatic recurrence detection with surveillance scanning in children with medulloblastoma. J Clin Oncol 15(5):1811–1813
Tait DM et al (1990) Adjuvant chemotherapy for medulloblastoma: the first multi-centre control trial of the International Society of Paediatric Oncology (SIOP I). Eur J Cancer 26(4):464–469
Tarbell NJ et al (1991) The change in patterns of relapse in medulloblastoma. Cancer 68(7):1600–1604
Tarbell NJ et al (2013) High-risk medulloblastoma: a pediatric oncology group randomized trial of chemotherapy before or after radiation therapy (POG 9031). J Clin Oncol 31(23):2936–2941
Taylor RE et al (2003) Results of a randomized study of preradiation chemotherapy versus radiotherapy alone for nonmetastatic medulloblastoma: the International Society of Paediatric Oncology/United Kingdom Children’s Cancer Study Group PNET-3 study. J Clin Oncol 21(8):1581–1591
Taylor RE et al (2004) Impact of radiotherapy parameters on outcome in the International Society of Paediatric Oncology/United Kingdom Children’s Cancer Study Group PNET-3 study of preradiotherapy chemotherapy for M0-M1 medulloblastoma. Int J Radiat Oncol Biol Phys 58(4):1184–1193
Taylor RE et al (2005) Outcome for patients with metastatic (M2-3) medulloblastoma treated with SIOP/UKCCSG PNET-3 chemotherapy. Eur J Cancer 41(5):727–734
Taylor MD et al (2012) Molecular subgroups of medulloblastoma: the current consensus. Acta Neuropathol 123(4):465–472
Teo WY et al (2013) Implications of tumor location on subtypes of medulloblastoma. Pediatr Blood Cancer 60(9):1408–1410
Torres CF et al (1994) Surveillance scanning of children with medulloblastoma. N Engl J Med 330(13):892–895
Verma J et al (2015) Comparison of supine and prone craniospinal irradiation in children with medulloblastoma. Pract Radiat Oncol 5(2):93–98
von Bueren AO et al (2011) Treatment of young children with localized medulloblastoma by chemotherapy alone: results of the prospective, multicenter trial HIT 2000 confirming the prognostic impact of histology. Neuro-Oncology 13(6):669–679
von Hoff K et al (2009) Long-term outcome and clinical prognostic factors in children with medulloblastoma treated in the prospective randomised multicentre trial HIT’91. Eur J Cancer 45(7):1209–1217
Walter AW et al (1999) Survival and neurodevelopmental outcome of young children with medulloblastoma at St Jude Children’s Research Hospital. J Clin Oncol 17(12):3720–3728
Wolden SL (2013) Protons for craniospinal radiation: are clinical data important? Int J Radiat Oncol Biol Phys 87(2):231–232
Wolden SL et al (2003) Patterns of failure using a conformal radiation therapy tumor bed boost for medulloblastoma. J Clin Oncol 21(16):3079–3083
Zeltzer PM et al (1999) Metastasis stage, adjuvant treatment, and residual tumor are prognostic factors for medulloblastoma in children: conclusions from the Children’s Cancer Group 921 randomized phase III study. J Clin Oncol 17(3):832–845
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG
About this chapter

Cite this chapter
Paulino, A.C., Carrie, C. (2018). Medulloblastoma. In: Mahajan, A., Paulino, A. (eds) Radiation Oncology for Pediatric CNS Tumors. Springer, Cham. https://doi.org/10.1007/978-3-319-55430-3_6
Download citation
DOI: https://doi.org/10.1007/978-3-319-55430-3_6
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-55428-0
Online ISBN: 978-3-319-55430-3
eBook Packages: MedicineMedicine (R0)