
Abstract
Increasing recognition of pediatric medulloblastoma as a heterogeneous disease, with histopathological and molecular variants that have distinct biological behaviors, will impact how the disease is classified and treated. A combination of clinicopathological evaluation and assays based on molecular subgroups of disease will allow stratification of patients into risk groups and a more tailored approach to therapy. Patients with low-risk disease could be treated with de-escalated adjuvant therapy to maximize cure while reducing long-term adverse effects, and novel therapies could be sought for patients with high-risk disease. My review encompasses a brief overview of the clinical landscape, the current World Health Organization (WHO) classification of medulloblastoma, the status of molecular subgroups, and how potential stratification schemes might impact pathologists and their practice.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
This review provides a synopsis of recent developments in our understanding of the pathobiology of medulloblastoma as it relates to diagnosis and classification. In one sense it is timely. In the next couple of years, American and European clinical trials incorporating novel approaches to the therapeutic stratification of medulloblastoma will require pathologists to distinguish accurately between the classic tumor and its four variants and to report the results of assays that probe clinically relevant aspects of the tumor’s biological heterogeneity. Thus, our concept of medulloblastoma pathology will have advanced significantly from a time when the diagnosis could be made on little more than sections stained with hematoxylin and eosin and the tumor was regarded as the archetypal primitive neuroectodermal tumor (PNET), and therefore histogenetically indistinguishable from other central nervous system (CNS) embryonal tumors.
Instead, we now recognize: (i) that the tumor’s heterogeneity is reflected in its diverse histopathology and a burgeoning array of molecular abnormalities, and (ii) the clinical utility of classifying the disease according to pathological and molecular features.
Clinical setting
Progress in the treatment of childhood medulloblastoma has dramatically improved survival rates over the last three decades. Advances in surgical and anesthetic techniques and incremental refinements in adjuvant therapies over this time have resulted in cures for approximately three quarters of standard-risk patients [26, 36, 62–64, 69, 78, 84]. Cure rates are lower for infants than for older children [8, 37, 64, 77]. Unfortunately, overall improvement has been achieved at the cost of significant adverse therapeutic effects among survivors, notably cognitive problems [23, 28, 48, 57, 58, 62, 64]. Following 36 Gy of craniospinal irradiation (CSI), a child’s IQ can decrease by as much as 30 points, and a drop of 10–15 points accompanies a lower CSI dose of 24 Gy. In addition, outcome for high-risk patients, which are distinguished from standard-risk patients by having either metastatic disease at presentation or a significant amount (>1.5 cm2, MRI plane) of post-operative residual tumor, remains relatively poor, 5-year EFS being 25–40% [18, 69, 85, 93]. Thus, starting with the premise presented above—that medulloblastoma is a heterogeneous disease—current challenges are threefold:
-
(i)
For disease that is at the relatively benign end of the biological range, can adjuvant therapy be reduced to levels where adverse effects are ameliorated yet cure rates sustained, and how is this low-risk disease optimally identified?
-
(ii)
Can pathological or molecular classification identify biologically aggressive high-risk disease, beyond that presenting clinically with metastasis, and can novel approaches to adjuvant therapy be devised and cure rates improved for high-risk disease?
-
(iii)
Can small molecule drugs be designed to target specific molecular aberrations in medulloblastoma, thus effecting cures alongside reduced levels of conventional adjuvant therapies?
Addressing these challenges is not trivial. Solutions to (i) and (ii) require datasets from carefully designed clinical trials with integrated biological studies, and results may differ according to patient age. Therapeutic considerations relevant to an infant with medulloblastoma are not the same as those that apply to an adolescent with the disease; CNS maturity has been a significant factor in determining the optimal planning of adjuvant therapy, because adverse effects following radiotherapy can be particularly grave in the immature infant brain [76]. Historically, the corollary of this has been separate therapeutic trials for infants versus older children, and retrospective analyses in these cohorts of the complex interaction between clinical, pathological and molecular variables affecting outcome will necessarily be evaluated in distinct therapeutic settings. Compounding this situation is an uneven distribution of pathological and molecular disease subtypes across the range of age at presentation. For example, desmoplastic/nodular medulloblastomas account for a much higher proportion of medulloblastomas in infants than in older children, while nearly all children with medulloblastomas characterized by abnormal activation of the Wnt pathway present between the ages of 5 and 12 years [17, 19, 33].
A robust statistical analysis that accounts for all these factors in validating the predictive or prognostic utility of specific pathological or molecular features requires a large cohort of patients treated in the standardized setting of a clinical trial. Because medulloblastoma is a relatively rare disease, such trials necessarily involve multiple centers, considerable administrative organization, and a centralized approach to the evaluation of any disease marker, whether this entails central pathology review or assay(s) in a single designated laboratory [69]. Once a trial is instituted, it may take a decade to register sufficient patients for hypothesis testing, to analyze data, and to report conclusions. It is perhaps not surprising that no molecular marker has yet been chosen for therapeutic stratification in prospective trials of medulloblastoma [34]. Even though published studies of such markers now have a long history, most relied on small series of patients from a single center or cohorts of patients treated in diverse ways.
Even though the potential of enhancing the clinical utility of pathological classifications by concurrent evaluation of prognostic or predictive molecular indicators remains to be realized for medulloblastoma, studies in the last 5 years have helped to build consensus on the value and means of using molecular markers in the therapeutic stratification of childhood medulloblastoma [33, 39, 64, 69]. In addition, the production of small molecule drugs that target aberrant cell signaling processes in cancer cells, specifically those that block abnormal activation of the Shh pathway [75], will require the identification of molecular subtypes of medulloblastoma. For practical reasons, it makes sense that such assays are undertaken on the same material used for histopathological diagnosis, formalin-fixed paraffin-embedded (FFPE) tissue, putting pathologists firmly at the center of the diagnostic process.
Pathological classification
Medulloblastoma: histopathological features
In the 2007 WHO classification of CNS tumors [51], medulloblastomas are separated into the classic tumor and four variants: desmoplastic/nodular (D/N), medulloblastoma with extensive nodularity (MBEN), anaplastic medulloblastoma and large cell medulloblastoma, on the basis of their histopathological features (Fig. 1). For the first time in the history of the WHO classification, this scheme recognizes that identification of medulloblastoma variants has clinical utility [33, 39, 52]. MBENs and D/N medulloblastomas in infants have a better outcome than classic tumors, while large cell and anaplastic medulloblastomas behave aggressively.

Medulloblastoma variants. The classic medulloblastoma (a) is characterized by sheets of small cells with a high nuclear:cytoplasmic ratio and mild nuclear pleomorphism. Mitotic figures and apoptotic bodies are present among the tumor cells. Occasionally, the syncytial pattern is broken by rosettes (b) or palisades of cells. The desmoplastic/nodular medulloblastoma (c) contains nodules of differentiated neurocytic cells that express neuronal proteins and have a low growth fraction. Desmoplastic internodular regions characterized by undifferentiated embryonal cells and reticulin-positive strands of collagen are interspersed among the nodules. Another desmoplastic tumor (d), the medulloblastoma with extensive nodularity is characterized by large areas of nodule formation and restricted desmoplastic internodular regions. Internodular neurocytic cells may be present at low density and sometimes evince streaming patterns (arrow). Some classic medulloblastomas contain nodules of neurocytic cells (e), which are not circumscribed by collagen or separated by desmoplastic regions (f). These non-desmoplastic nodular tumors differ from desmoplastic tumors in three respects: age of onset, molecular cytogenetic changes, and molecular subgroup. The anaplastic medulloblastoma is characterized by marked nuclear pleomorphism, cell wrapping, a high mitotic count, and abundant apoptotic bodies (g). To qualify as an anaplastic tumor, a medulloblastoma must contain extensive regions with this phenotype. The large cell medulloblastoma (h) contains groups of large round cells with a single nucleolus. These cells rarely dominate the cytopathology; large cell tumors often contain broad areas with an anaplastic phenotype (a–e, g, h: hematoxylin & eosin, f: reticulin)
The D/N medulloblastoma and MBEN are characterized by nodules of differentiated neurocytic cells and internodular desmoplasia, which is best demonstrated by a reticulin stain (Fig. 1). The presence of nodules in desmoplastic tumors can be variable. In MBENs [29], nodules dominate the histopathology and are typically large and irregularly shaped, containing monomorphic neurocytic cells that often demonstrate linear (streaming) patterns (Fig. 1). Internodular desmoplasia is sparse, but clearly present; the MBEN is unequivocally a desmoplastic medulloblastoma. In D/N medulloblastomas, nodules of uniform neurocytic cells are usually round and scattered across desmoplastic regions in which tumor cells show more nuclear pleomorphism than is evident among intranodular cells [56]. The neurocytic differentiation shown by intranodular cells is reflected by a neuronal immunophenotype and reduced growth fraction; intranodular neurocytic cells express synaptophysin and NEU-N more strongly than internodular tumor cells, in which these proteins are weakly expressed, if at all. Ki-67 immunolabelling, which is high in internodular regions, is markedly reduced or absent among intranodular tumor cells [17]. This characteristic combination of features has helped to delineate a paucinodular D/N medulloblastoma, in which small infrequent nodules are embedded in an expanse of desmoplasia [56]. In many clinicopathological studies, the paucinodular D/N tumor, D/N medulloblastoma and MBEN are combined into a single category of desmoplastic tumors.
As D/N medulloblastoma and MBEN share fundamental histopathological features, so do anaplastic and large cell tumors. The latter contains groups of uniform large cells with round nuclei and a single nucleolus (Fig. 1). Rarely, a ‘pure’ large cell medulloblastoma is dominated by such cells, but most exhibit admixed groups of cells with a large cell or anaplastic phenotype [30, 55]. Anaplasia in medulloblastomas is basically defined as marked cytological pleomorphism. Because of the very high nuclear:cytoplasmic ratio in medulloblastomas, this effectively means nuclear pleomorphism. Cell molding, producing an irregular paving-like pattern of nuclei, and cell wrapping accompany the nuclear pleomorphism [9, 15]. By definition, the anaplastic medulloblastoma is dominated by these cytological features, and both anaplastic and large cell tumors also show increased mitotic activity and apoptosis [15, 55]. ‘Focal anaplasia’ covering a small area of the tumor is not robustly associated with outcome, and to avoid confusion the term should not be used. The rarity of the pure large cell medulloblastoma and mixture of large cell and anaplastic phenotypes in some medulloblastomas has prompted many investigators to combine the two variants into a single category of large cell/anaplastic (LC/A) tumors.
Classic medulloblastomas contain sheets of monotonous small cells with a high nuclear:cytoplasmic ratio and round nuclei (Fig. 1). Some classic tumors consist of elongated cells with oval nuclei or demonstrate moderate cytological pleomorphism [49], and limited foci of cells with an anaplastic phenotype are allowed. Rosettes or palisades of cells are present in some classic tumors, the former being regarded as a sign of differentiation, though not necessarily prognostic [17]. Rosettes of tumor cells are occasionally found in anaplastic medulloblastomas. Classic medulloblastomas may contain unequivocal nodules of neurocytic differentiation or irregular groups of neurocytic and ganglion cells [56]. Critically, the former ‘biphasic’ phenotype is not associated with internodular desmoplasia (Fig. 1); these classic, non-desmoplastic tumors with focal nodularity are distinct from D/N medulloblastomas, presenting at a different median age and harboring different molecular cytogenetic abnormalities. The ganglion cell phenotype replicates the histopathology of the CNS ganglioneuroblastoma, an embryonal tumor usually located in the cerebral hemispheres [51].
Medulloblastoma pathology: clinical associations
Classic medulloblastomas are most numerous (Table 1). In one large study of two trial cohorts that registered infants or (older) children with medulloblastoma in the UK during the 1990s, desmoplastic tumors (D/N + MBEN variants) and LC/A tumors accounted for 10.3 and 16.6%, respectively [56]. However, desmoplastic tumors occurred at a much higher frequency among infants (aged less than 3 years at presentation). Median age at presentation was correspondingly lower for desmoplastic medulloblastomas (Table 1). Other studies have also demonstrated the high frequency of desmoplastic tumors in infants [77]. A high frequency of desmoplastic tumors is also a feature of disease in adults [35].
It is now evident from several studies that desmoplastic medulloblastomas presenting in infancy have a significantly better outcome than classic or LC/A tumors in this age group. Rutkowski et al. [76, 77] have reported on this phenomenon in two cohorts, one from the German HIT-SKK’87 trial, and these findings have been reinforced by data from the SIOP CNS9204 trial [38, 56]. In the latter cohort, the outcome for infants with MBEN was no different from that for infants with D/N tumors. On the basis of these data, it seems clear that infants with desmoplastic medulloblastomas should be stratified onto a low-risk therapeutic arm in future trials; indeed, this is already a feature of the SJYC07 trial protocol at St. Jude Children’s Research Hospital.
When its distinctive phenotype was first described, the large cell medulloblastoma was recognized to have a poor prognosis [30]; indeed, it often presents with metastatic disease. Peter Burger, of Johns Hopkins Hospital, first proposed anaplasia as a histologically significant cytological feature of medulloblastomas, and it was unequivocally established as a prognostic indicator in a series of studies based on clinical trial cohorts [9, 13, 15, 55]. As an outcome indicator in multivariable analyses, it appears to be independent of metastatic disease [49, 55], though there is a trend towards an association between these variables [27].
Because the large cell medulloblastoma is rare, this and the anaplastic variant are often combined under the umbrella term ‘LC/A’ in studies that incorporate pathological phenotype into an evaluation of medulloblastoma outcome indicators, but in the individual case the tumor should be classified as large cell or anaplastic, according to the WHO scheme. The former variant is distinctive and rarely contains regions with a classic phenotype. In contrast, the latter can be difficult to separate from the classic tumor. Not all anaplastic medulloblastomas have a poor outcome, but it is important to be stringent about criteria for the diagnosis of this variant (see above), if it is to retain its clinical utility.
Molecular pathology
An extensive literature now exists on the molecular biology of medulloblastoma. This encompasses studies on the tumor’s histogenesis and cell biology [10, 14, 20, 32, 33, 53], on genetically engineered mouse (GEM) models [3, 24, 25, 40, 50, 67, 87, 88, 91, 94], and on genetic abnormalities that are potential prognostic or predictive indicators [17, 31, 36, 66, 70, 80]. Studies addressing specific aspects of the disease that are relevant to molecular classification or therapeutic stratification and likely to impact pathological practice are reviewed below.
Molecular subgroups of medulloblastoma: Shh, Wnt, and non-Shh/Wnt tumors
The rare occurrence of medulloblastoma in Gorlin syndrome (nevoid basal cell carcinoma syndrome) [21, 83], which is associated with mutations of the PTCH1 gene, focused attention on abnormalities of the Shh pathway in sporadic medulloblastomas, and led to the discovery of PTCH1 and rare SMOH and SUFU mutations in a proportion of tumors [68, 71, 72, 82, 89]. These mutations result in aberrant activation of the Shh pathway, which recreates the proliferative stimulus during CNS development of Shh released by Purkinje cells in cerebellar granule cell precursors (CGNPs). Several GEM models have been generated by manipulating the Shh pathway [41, 88, 90], supplementing data from gene expression studies in support of a molecular subgroup of medulloblastomas that is derived from CGNPs and characterized by a constitutively active Shh pathway [47, 86].

Molecular markers. Immunohistochemistry with a β-catenin antibody demonstrates combined nuclear and cytoplasmic immunoreactivity in Wnt medulloblastomas (a), in which the Wnt pathway is activated and β-catenin translocates to the nucleus. In Shh and non-Shh/Wnt tumors, immunoreactivity for β-catenin is cytoplasmic (b). Some Wnt pathway medulloblastomas show patchy β-catenin nuclear immunoreactivity, with variable nuclear staining and scattered nucleonegative cells (c). Interphase fluorescence in situ hybridization (iFISH) can be used to demonstrate: d monosomy 6 (6p, green signal; 6q, red signal), e loss of 9q22 (PTCH1 locus, green signal; 9p, red signal), f isodicentric 17q (17p, green signal; 17q, red signal), g ploidy change (tetrasomy, 4× green/red signals), and (h) MYC amplification with double minute pattern (MYC locus, green signal; 8p, red signal)

Shh and Wnt pathways. When the Shh pathway is inactive, Ptch receptors inhibit Smoothened receptors (inactive Smoothened receptors, red), preventing access of Smoothened to the primary cilium. Some smoothened receptors undergo endocytosis and are incorporated by endosomes (blue). SuFu binds Gli2 and Gli3, downstream effectors of the Shh pathway, in the cytoplasm and primary cilium. Gli2 is also targeted for degradation via the proteasomal pathway. When the Shh pathway is active, Ptch ceases to repress Smoothened (active Smoothened receptors, green), which moves into the primary cilium. Kif7 aids activation of Gli proteins in the primary cilium. SuFu no longer binds Gli2 and Gli3, and Gli2 translocates to the nucleus where it is a transcriptional activator. Gli3 (Gli3R) has repressor actions. When the Wnt pathway is inactive (inactive Frizzled receptors, red), β-catenin is phosphorylated and bound in a protein complex that processes it for degradation through the proteasomal pathway. When the Wnt pathway is active (active Frizzled receptors, green), the complex is disrupted, allowing β-catenin to translocate to the nucleus, where it acts as a transcriptional activator. SFRP can bind Wnt to prevent the binding of ligand to receptor
A similar paradigm involving Turcot syndrome and the Wnt pathway uncovered mutations of CTNNB1, APC, and AXIN1/2 in approximately 15% of medulloblastomas [12, 33, 43, 45, 46, 83, 95]. In this molecular subgroup, an active Wnt pathway produces nuclear accumulation of β-catenin [16, 42], a downstream effector of the canonical Wnt pathway (Figs. 2, 3).
Several studies in the last few years have used gene expression data from microarray analyses to investigate the range of molecular subgroups among medulloblastomas. Unsupervised hierarchical clustering methods of segregating these data produce four or five subgroups, two of which are characterized by up-regulation of genes in the Wnt or Shh pathways [47, 61, 86]. These two subgroups are clearly separated from each other and the non-Shh/Wnt subgroups on principal components analysis [61]. The non-Shh/Wnt subgroups are not obviously associated with aberrant activation of signaling pathways, although they are associated with up-regulation of specific gene classes, such as neuronal genes or photoreceptor genes [47]. In contrast to tumors in the Shh and Wnt pathway subgroups, non-Shh/Wnt subgroups are not so clearly separated from each other on principal components analysis [61].
Molecular subgroups of medulloblastoma: clinical, pathological and genetic associations
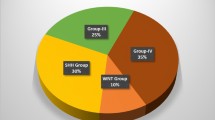
Shh, Wnt, and non-Shh/Wnt tumors account for approximately 25, 15, and 60% of medulloblastomas, respectively (Table 2). Many Shh pathway medulloblastomas have a desmoplastic phenotype and are therefore well represented among tumors presenting in infancy [47, 61, 86]. However, non-desmoplastic tumors may also have an Shh profile, particularly LC/A tumors. In contrast, Wnt pathway medulloblastomas nearly always have a classic pathology [19, 22]; LC/A tumors are rare in this subgroup, and no desmoplastic tumor has been reported to have a Wnt profile. Wnt pathway medulloblastomas do not present in infancy; they tend to present between the ages of 6 and 13 years and have a higher median age at presentation than that for all childhood medulloblastomas [19]. Non-Shh/Wnt medulloblastomas present both in infants and older children. Most non-Shh/Wnt tumors have a classic pathology, and about half of LC/A tumors are also in this subgroup. Metastatic disease at presentation characterizes some Shh pathway medulloblastomas, more often LC/A than D/N tumors, but is rare among Wnt tumors; most examples fall into the non-Shh/Wnt tumor subgroup [47].
Less than half of Shh pathway medulloblastomas have PTCH1 mutations or show copy number loss at the PTCH1 locus, 9q22, and because SMOH or SUFU mutations are uncommon other genetic aberrations related to the Shh pathway will probably be discovered in this subgroup [47, 61, 86]. Likewise, not all Wnt pathway tumors have CTNNB1 mutations, and rare APC and AXIN1/2 mutations cannot account for the remainder. Monosomy 6 is strongly associated with a Wnt profile in medulloblastomas, though the basis for this is not understood [11]. Very few other copy number abnormalities (CNAs) are found in Wnt pathway medulloblastomas; apart from monosomy 6, this subgroup of tumors is notable for its lack of CNAs across the genome [11]. Shh and Wnt tumors rarely show chromosome 17 CNAs, amplification of MYC or MYCN, or widespread ploidy change, all of which are established cytogenetic abnormalities in medulloblastomas. Instead, tumors with these abnormalities mainly fall into the non-Shh/Wnt subgroup. Other CNAs that occur predominantly in non-Shh/Wnt medulloblastomas include loss of chromosome X (in females) and loss of chromosome 8 [47].
Stratification of medulloblastoma patients: clinical, pathological and molecular factors
A molecular classification of pediatric medulloblastoma can advance approaches to therapy in two specific ways: (i) molecular outcome indicators, if independent, can be used in conjunction with clinicopathological indicators to aid identification of low-risk or high-risk disease, and (ii) tumors with aberrant activation of signaling pathways (Shh or Wnt) can be identified and targeted with pathway-specific small molecule drugs.
Many molecular markers have been proposed as outcome indicators of medulloblastoma, but generating robust data to support their utility in a clinical setting and thereby engendering confidence among clinicians to use them have both taken a relatively long time.
Wnt pathway medulloblastomas are distinct from other disease subgroups; they have an idiosyncratic epidemiology, presenting mainly in the pre-teen years, and may even have a unique histogenesis [19, 33]. They also have a favorable outcome with current therapeutic regimens; nearly all children with this tumor survive [19, 22, 86]. Stratifying children with Wnt pathway medulloblastomas to a regimen of reduced adjuvant therapy has the potential to ameliorate long-term adverse effects while maintaining cure rates. By immunohistochemistry, Wnt pathway tumors can be identified by nuclear reactivity for β-catenin. This assay captures all Wnt medulloblastomas with a CTNNB1 mutation and is regarded as the standard test for identifying this subgroup of disease [19, 22, 61]. Easy to develop for use with FFPE material, it is also a practical test for diagnostic laboratories. In forthcoming clinical trials of children older than 3 years, Wnt pathway tumors without clinical, pathological, or molecular adverse indicators could reasonably be stratified to a low-risk group.
Metastatic disease at presentation clearly designates a medulloblastoma as high-risk; this has been a cornerstone of therapeutic strategy for many years [85, 93]. LC/A phenotype is also accepted as an independent indicator of poor survival [15, 18]. A molecular marker repeatedly shown to associate with poor outcome is MYC amplification or overexpression [1, 4, 49, 66]. While these three outcome indicators are independent in most multivariable survival analyses, they also show some association [49, 79]; LC/A medulloblastomas with MYC amplification and metastatic disease at presentation are not uncommon. Less well established as indicators of adverse outcome are MYCN amplification and CNAs of chromosome 17. MYC or MYCN amplification occurs in 4–8% of medulloblastomas [49]. Like MYC, MYCN when amplified shows some association with LC/A phenotype and metastatic disease, but it remains to be proven as a prognostic marker in large trial-based cohorts [78].
Chromosome 17 CNAs are common in medulloblastomas [2, 6, 7, 17, 54, 59, 60]. Isodicentric chromosome 17q is present in approximately one-third of tumors, and chromosomal imbalance is even more frequent. Proposals have frequently been advanced for using isodicentric chromosome 17q, loss of 17p, or gain of 17q as an indicator of poor outcome in medulloblastoma stratification [31, 65, 66]. However, some studies have found no association between chromosome 17 CNAs and outcome [5, 17]. This disparity may relate to the proportion of Wnt tumors in an individual study. Wnt tumors have a good outcome, and very few have chromosome 17 CNAs [11]; the more Wnt tumors in an analyzed cohort, the more likely it is that chromosome 17 CNAs will be associated with a poor outcome. There is a clear argument for establishing any association between chromosome 17 CNAs and poor outcome among medulloblastomas that exclude Wnt tumors, in which context it might be more likely to have clinical utility.
It is important to acknowledge the complex interplay between clinical, pathological, and molecular outcome indicators when establishing which variables are best used in classification or stratification schemes. An optimal scheme may be different for infants and older children. Among infants, the critical distinction may focus on pathological variant, rather than molecular subgroup, because of the prevalence of Shh tumors among these patients (Fig. 4). Among older children, identifying the Wnt pathway medulloblastoma is clearly a critical step, but identifying other molecular subgroups may be less important than dividing non-Wnt tumors on the basis of clinical, pathological, or molecular cytogenetic factors (Fig. 5). Although a recent study claims that immunohistochemical markers of four molecular subgroups divide medulloblastomas into significantly different and clinically useful outcome groups [61], it might be difficult to abandon stratification by metastatic status or pathological variant in favor of this approach, even though many patients with metastatic disease fall into one of the reported molecular subgroups.

Potential stratification scheme for infant medulloblastoma. The scheme makes an assumption that metastatic disease at presentation does not alter the favorable outcome of infants with a desmoplastic tumor. Some data suggest this, but the hypothesis has yet to be proven. The scheme also offers the option (dotted lines) to treat Shh pathway tumors with a Shh pathway antagonist, although adverse effects on bone growth may ultimately limit its use in infants

Potential stratification scheme for non-infant children with medulloblastoma. The scheme makes an assumption (see text) that MYCN amplification does not have the same association with poor outcome as MYC amplification. Some studies suggest that tests for amplification of either gene should be used to identify high-risk tumors. The scheme also offers the option to treat Shh pathway tumors with an Shh pathway antagonist. GTR gross total resection
Several novel agents that inhibit activation of the Shh pathway and that are currently under investigation could be extremely valuable in the treatment of the Shh subgroup of medulloblastoma [73–75, 81]. GDC-0449 binds to and inhibits the Smoothened receptor and has shown some success in reducing disease load and prolonging survival. Unfortunately, there are two potential setbacks to what promises to be the first targeted treatment of medulloblastoma: (i) Shh pathway inhibition appears to disrupt bone growth [44], and so may be unsuitable for use in young children, and (ii) a SMOH mutation that disrupts the binding of GDC-0449 can abolish its effectiveness, leading to tumor recurrence [92].
In future, the classification or stratification of medulloblastoma will involve progressively more reference to molecular indices that reflect clinically relevant aspects of the tumor’s biology. It seems likely that clinical and pathological factors will be fundamental to any scheme, but pathologists will increasingly be asked to integrate the results of molecular assays into their evaluation of the medulloblastoma, whether these are prognostic or predictive markers or establish the presence of targets for small molecular drugs.
References
Aldosari N, Bigner SH, Burger PC, Becker L, Kepner JL, Friedman HS, McLendon RE (2002) MYCC and MYCN oncogene amplification in medulloblastoma. A fluorescence in situ hybridization study on paraffin sections from the Children’s Oncology Group. Arch Pathol Lab Med 126:540–544
Aldosari N, Rasheed BK, McLendon RE, Friedman HS, Bigner DD, Bigner SH (2000) Characterization of chromosome 17 abnormalities in medulloblastomas. Acta Neuropathol 99:345–351
Ayrault O, Zhao H, Zindy F, Qu C, Sherr CJ, Roussel MF (2010) Atoh1 inhibits neuronal differentiation and collaborates with Gli1 to generate medulloblastoma-initiating cells. Cancer Res 70:5618–5627
Badiali M, Pession A, Basso G, Andreini L, Rigobello L, Galassi E, Giangaspero F (1991) N-myc and c-myc oncogenes amplification in medulloblastomas. Evidence of particularly aggressive behavior of a tumor with c-myc amplification. Tumori 77:118–121
Biegel JA, Janss AJ, Raffel C, Sutton L, Rorke LB, Harper JM, Phillips PC (1997) Prognostic significance of chromosome 17p deletions in childhood primitive neuroectodermal tumors (medulloblastomas) of the central nervous system. Clin Cancer Res 3:473–478
Biegel JA, Rorke LB, Janss AJ, Sutton LN, Parmiter AH (1995) Isochromosome 17q demonstrated by interphase fluorescence in situ hybridization in primitive neuroectodermal tumors of the central nervous system. Genes Chromosomes Cancer 14:85–96
Biegel JA, Rorke LB, Packer RJ, Sutton LN, Schut L, Bonner K, Emanuel BS (1989) Isochromosome 17q in primitive neuroectodermal tumors of the central nervous system. Genes Chromosomes Cancer 1:139–147
Bouffet E (2010) Medulloblastoma in infants: the critical issues of the dilemma. Curr Oncol 17:2–3
Brown HG, Kepner JL, Perlman EJ, Friedman HS, Strother DR, Duffner PK, Kun LE, Goldthwaite PT, Burger PC (2000) “Large cell/anaplastic” medulloblastomas: a Pediatric Oncology Group Study. J Neuropathol Exp Neurol 59:857–865
Buhren J, Christoph AH, Buslei R, Albrecht S, Wiestler OD, Pietsch T (2000) Expression of the neurotrophin receptor p75NTR in medulloblastomas is correlated with distinct histological and clinical features: evidence for a medulloblastoma subtype derived from the external granule cell layer. J Neuropathol Exp Neurol 59:229–240
Clifford SC, Lusher ME, Lindsey JC, Langdon JA, Gilbertson RJ, Straughton D, Ellison DW (2006) Wnt/Wingless pathway activation and chromosome 6 loss characterize a distinct molecular sub-group of medulloblastomas associated with a favorable prognosis. Cell Cycle 5:2666–2670
Dahmen RP, Koch A, Denkhaus D, Tonn JC, Sorensen N, Berthold F, Behrens J, Birchmeier W, Wiestler OD, Pietsch T (2001) Deletions of AXIN1, a component of the WNT/wingless pathway, in sporadic medulloblastomas. Cancer Res 61:7039–7043
Eberhart CG, Burger PC (2003) Anaplasia and grading in medulloblastomas. Brain Pathol 13:376–385
Eberhart CG, Kaufman WE, Tihan T, Burger PC (2001) Apoptosis, neuronal maturation, and neurotrophin expression within medulloblastoma nodules. J Neuropathol Exp Neurol 60:462–469
Eberhart CG, Kepner JL, Goldthwaite PT, Kun LE, Duffner PK, Friedman HS, Strother DR, Burger PC (2002) Histopathologic grading of medulloblastomas: a Pediatric Oncology Group study. Cancer 94:552–560
Eberhart CG, Tihan T, Burger PC (2000) Nuclear localization and mutation of beta-catenin in medulloblastomas. J Neuropathol Exp Neurol 59:333–337
Ellison D (2002) Classifying the medulloblastoma: insights from morphology and molecular genetics. Neuropathol Appl Neurobiol 28:257–282
Ellison DW, Clifford SC, Gajjar A, Gilbertson RJ (2003) What’s new in neuro-oncology? Recent advances in medulloblastoma. Eur J Paediatr Neurol 7:53–66
Ellison DW, Onilude OE, Lindsey JC, Lusher ME, Weston CL, Taylor RE, Pearson AD, Clifford SC (2005) beta-Catenin status predicts a favorable outcome in childhood medulloblastoma. J Clin Oncol 23:7951–7957
Emmenegger BA, Wechsler-Reya RJ (2008) Stem cells and the origin and propagation of brain tumors. J Child Neurol 23:1172–1178
Evans DG, Farndon PA, Burnell LD, Gattamaneni HR, Birch JM (1991) The incidence of Gorlin syndrome in 173 consecutive cases of medulloblastoma. Br J Cancer 64:959–961
Fattet S, Haberler C, Legoix P, Varlet P, Lellouch-Tubiana A, Lair S, Manie E, Raquin MA, Bours D, Carpentier S, Barillot E, Grill J, Doz F, Puget S, Janoueix-Lerosey I, Delattre O (2009) Beta-catenin status in paediatric medulloblastomas: correlation of immunohistochemical expression with mutational status, genetic profiles, and clinical characteristics. J Pathol 218:86–94
Frange P, Alapetite C, Gaboriaud G, Bours D, Zucker JM, Zerah M, Brisse H, Chevignard M, Mosseri V, Bouffet E, Doz F (2009) From childhood to adulthood: long-term outcome of medulloblastoma patients. The Institut Curie experience (1980–2000). J Neurooncol 95:271–279
Frappart PO, Lee Y, Lamont J, McKinnon PJ (2007) BRCA2 is required for neurogenesis and suppression of medulloblastoma. EMBO J 26:2732–2742
Fults DW (2005) Modeling medulloblastoma with genetically engineered mice. Neurosurg Focus 19:E7
Gajjar A, Chintagumpala M, Ashley D, Kellie S, Kun LE, Merchant TE, Woo S, Wheeler G, Ahern V, Krasin MJ, Fouladi M, Broniscer A, Krance R, Hale GA, Stewart CF, Dauser R, Sanford RA, Fuller C, Lau C, Boyett JM, Wallace D, Gilbertson RJ (2006) Risk-adapted craniospinal radiotherapy followed by high-dose chemotherapy and stem-cell rescue in children with newly diagnosed medulloblastoma (St Jude Medulloblastoma-96): long-term results from a prospective, multicentre trial. Lancet Oncol 7:813–820
Gajjar A, Hernan R, Kocak M, Fuller C, Lee Y, McKinnon PJ, Wallace D, Lau C, Chintagumpala M, Ashley DM, Kellie SJ, Kun L, Gilbertson RJ (2004) Clinical, histopathologic, and molecular markers of prognosis: toward a new disease risk stratification system for medulloblastoma. J Clin Oncol 22:984–993
Gajjar A, Mulhern RK, Heideman RL, Sanford RA, Douglass EC, Kovnar EH, Langston JA, Jenkins JJ, Kun LE (1994) Medulloblastoma in very young children: outcome of definitive craniospinal irradiation following incomplete response to chemotherapy. J Clin Oncol 12:1212–1216
Giangaspero F, Perilongo G, Fondelli MP, Brisigotti M, Carollo C, Burnelli R, Burger PC, Garre ML (1999) Medulloblastoma with extensive nodularity: a variant with favorable prognosis. J Neurosurg 91:971–977
Giangaspero F, Rigobello L, Badiali M, Loda M, Andreini L, Basso G, Zorzi F, Montaldi A (1992) Large-cell medulloblastomas. A distinct variant with highly aggressive behavior. Am J Surg Pathol 16:687–693
Gilbertson R, Wickramasinghe C, Hernan R, Balaji V, Hunt D, Jones-Wallace D, Crolla J, Perry R, Lunec J, Pearson A, Ellison D (2001) Clinical and molecular stratification of disease risk in medulloblastoma. Br J Cancer 85:705–712
Gilbertson RJ (2004) Medulloblastoma: signalling a change in treatment. Lancet Oncol 5:209–218
Gilbertson RJ, Ellison DW (2008) The origins of medulloblastoma subtypes. Annu Rev Pathol 3:341–365
Gilbertson RJ, Gajjar A (2005) Molecular biology of medulloblastoma: will it ever make a difference to clinical management? J Neurooncol 75:273–278
Giordana MT, D’Agostino C, Pollo B, Silvani A, Ferracini R, Paiolo A, Ghiglione P, Chio A (2005) Anaplasia is rare and does not influence prognosis in adult medulloblastoma. J Neuropathol Exp Neurol 64:869–874
Gottardo NG, Gajjar A (2006) Current Therapy for Medulloblastoma. Curr Treat Options Neurol 8:319–334
Grill J, Sainte-Rose C, Jouvet A, Gentet JC, Lejars O, Frappaz D, Doz F, Rialland X, Pichon F, Bertozzi AI, Chastagner P, Couanet D, Habrand JL, Raquin MA, Le Deley MC, Kalifa C (2005) Treatment of medulloblastoma with postoperative chemotherapy alone: an SFOP prospective trial in young children. Lancet Oncol 6:573–580
Grundy RG, Wilne SH, Robinson KJ, Ironside JW, Cox T, Chong WK, Michalski A, Campbell RH, Bailey CC, Thorp N, Pizer B, Punt J, Walker DA, Ellison DW, Machin D (2010) Primary postoperative chemotherapy without radiotherapy for treatment of brain tumors other than ependymoma in children under 3 years: results of the first UKCCSG/SIOP CNS 9204 trial. Eur J Cancer 46:120–133
Gulino A, Arcella A, Giangaspero F (2008) Pathological and molecular heterogeneity of medulloblastoma. Curr Opin Oncol 20:668–675
Hallahan AR, Pritchard JI, Hansen S, Benson M, Stoeck J, Hatton BA, Russell TL, Ellenbogen RG, Bernstein ID, Beachy PA, Olson JM (2004) The SmoA1 mouse model reveals that notch signaling is critical for the growth and survival of sonic hedgehog-induced medulloblastomas. Cancer Res 64:7794–7800
Hatton BA, Villavicencio EH, Tsuchiya KD, Pritchard JI, Ditzler S, Pullar B, Hansen S, Knoblaugh SE, Lee D, Eberhart CG, Hallahan AR, Olson JM (2008) The Smo/Smo model: hedgehog-induced medulloblastoma with 90% incidence and leptomeningeal spread. Cancer Res 68:1768–1776
Henderson BR, Fagotto F (2002) The ins and outs of APC and beta-catenin nuclear transport. EMBO Rep 3:834–839
Huang H, Mahler-Araujo BM, Sankila A, Chimelli L, Yonekawa Y, Kleihues P, Ohgaki H (2000) APC mutations in sporadic medulloblastomas. Am J Pathol 156:433–437
Kimura H, Ng JM, Curran T (2008) Transient inhibition of the Hedgehog pathway in young mice causes permanent defects in bone structure. Cancer Cell 13:249–260
Koch A, Hrychyk A, Hartmann W, Waha A, Mikeska T, Schuller U, Sorensen N, Berthold F, Goodyer CG, Wiestler OD, Birchmeier W, Behrens J, Pietsch T (2007) Mutations of the Wnt antagonist AXIN2 (Conductin) result in TCF-dependent transcription in medulloblastomas. Int J Cancer 121:284–291
Koch A, Waha A, Tonn JC, Sorensen N, Berthold F, Wolter M, Reifenberger J, Hartmann W, Friedl W, Reifenberger G, Wiestler OD, Pietsch T (2001) Somatic mutations of WNT/wingless signaling pathway components in primitive neuroectodermal tumors. Int J Cancer 93:445–449
Kool M, Koster J, Bunt J, Hasselt NE, Lakeman A, van Sluis P, Troost D, Meeteren NS, Caron HN, Cloos J, Mrsic A, Ylstra B, Grajkowska W, Hartmann W, Pietsch T, Ellison D, Clifford SC, Versteeg R (2008) Integrated genomics identifies five medulloblastoma subtypes with distinct genetic profiles, pathway signatures and clinicopathological features. PLoS One 3:e3088
Kun LE, Mulhern RK, Crisco JJ (1983) Quality of life in children treated for brain tumors. Intellectual, emotional, and academic function. J Neurosurg 58:1–6
Lamont JM, McManamy CS, Pearson AD, Clifford SC, Ellison DW (2004) Combined histopathological and molecular cytogenetic stratification of medulloblastoma patients. Clin Cancer Res 10:5482–5493
Lee Y, Kawagoe R, Sasai K, Li Y, Russell HR, Curran T, McKinnon PJ (2007) Loss of suppressor-of-fused function promotes tumorigenesis. Oncogene 26:6442–6447
Louis DN (2007) WHO classification of tumors of the central nervous system. International Agency for Research on Cancer, Lyon
Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, Scheithauer BW, Kleihues P (2007) The 2007 WHO classification of tumors of the central nervous system. Acta Neuropathol 114:97–109
Marino S (2005) Medulloblastoma: developmental mechanisms out of control. Trends Mol Med 11:17–22
McCabe MG, Ichimura K, Liu L, Plant K, Backlund LM, Pearson DM, Collins VP (2006) High-resolution array-based comparative genomic hybridization of medulloblastomas and supratentorial primitive neuroectodermal tumors. J Neuropathol Exp Neurol 65:549–561
McManamy CS, Lamont JM, Taylor RE, Cole M, Pearson AD, Clifford SC, Ellison DW (2003) Morphophenotypic variation predicts clinical behavior in childhood non-desmoplastic medulloblastomas. J Neuropathol Exp Neurol 62:627–632
McManamy CS, Pears J, Weston CL, Hanzely Z, Ironside JW, Taylor RE, Grundy RG, Clifford SC, Ellison DW (2007) Nodule formation and desmoplasia in medulloblastomas-defining the nodular/desmoplastic variant and its biological behavior. Brain Pathol 17:151–164
Merchant TE, Kun LE, Krasin MJ, Wallace D, Chintagumpala MM, Woo SY, Ashley DM, Sexton M, Kellie SJ, Ahern V, Gajjar A (2008) Multi-institution prospective trial of reduced-dose craniospinal irradiation (23.4 Gy) followed by conformal posterior fossa (36 Gy) and primary site irradiation (55.8 Gy) and dose-intensive chemotherapy for average-risk medulloblastoma. Int J Radiat Oncol Biol Phys 70:782–787
Mulhern RK, Merchant TE, Gajjar A, Reddick WE, Kun LE (2004) Late neurocognitive sequelae in survivors of brain tumors in childhood. Lancet Oncol 5:399–408
Nicholson J, Wickramasinghe C, Ross F, Crolla J, Ellison D (2000) Imbalances of chromosome 17 in medulloblastomas determined by comparative genomic hybridisation and fluorescence in situ hybridisation. Mol Pathol 53:313–319
Nicholson JC, Ross FM, Kohler JA, Ellison DW (1999) Comparative genomic hybridization and histological variation in primitive neuroectodermal tumors. Br J Cancer 80:1322–1331
Northcott PA, Korshunov A, Witt H, Hielscher T, Eberhart CG, Mack S, Bouffet E, Clifford SC, Hawkins CE, French P, Rutka JT, Pfister S, Taylor MD (2010) Medulloblastoma comprises four distinct molecular variants. J Clin Oncol (in press)
Oyharcabal-Bourden V, Kalifa C, Gentet JC, Frappaz D, Edan C, Chastagner P, Sariban E, Pagnier A, Babin A, Pichon F, Neuenschwander S, Vinchon M, Bours D, Mosseri V, Le Gales C, Ruchoux M, Carrie C, Doz F (2005) Standard-risk medulloblastoma treated by adjuvant chemotherapy followed by reduced-dose craniospinal radiation therapy: a French Society of Pediatric Oncology Study. J Clin Oncol 23:4726–4734
Packer RJ, Gajjar A, Vezina G, Rorke-Adams L, Burger PC, Robertson PL, Bayer L, LaFond D, Donahue BR, Marymont MH, Muraszko K, Langston J, Sposto R (2006) Phase III study of craniospinal radiation therapy followed by adjuvant chemotherapy for newly diagnosed average-risk medulloblastoma. J Clin Oncol 24:4202–4208
Packer RJ, Vezina G (2008) Management of and prognosis with medulloblastoma: therapy at a crossroads. Arch Neurol 65:1419–1424
Pan E, Pellarin M, Holmes E, Smirnov I, Misra A, Eberhart CG, Burger PC, Biegel JA, Feuerstein BG (2005) Isochromosome 17q is a negative prognostic factor in poor-risk childhood medulloblastoma patients. Clin Cancer Res 11:4733–4740
Pfister S, Remke M, Benner A, Mendrzyk F, Toedt G, Felsberg J, Wittmann A, Devens F, Gerber NU, Joos S, Kulozik A, Reifenberger G, Rutkowski S, Wiestler OD, Radlwimmer B, Scheurlen W, Lichter P, Korshunov A (2009) Outcome prediction in pediatric medulloblastoma based on DNA copy-number aberrations of chromosomes 6q and 17q and the MYC and MYCN loci. J Clin Oncol 27:1627–1636
Piedimonte LR, Wailes IK, Weiner HL (2005) Medulloblastoma: mouse models and novel targeted therapies based on the Sonic hedgehog pathway. Neurosurg Focus 19:E8
Pietsch T, Waha A, Koch A, Kraus J, Albrecht S, Tonn J, Sorensen N, Berthold F, Henk B, Schmandt N, Wolf HK, von Deimling A, Wainwright B, Chenevix-Trench G, Wiestler OD, Wicking C (1997) Medulloblastomas of the desmoplastic variant carry mutations of the human homologue of Drosophila patched. Cancer Res 57:2085–2088
Pizer BL, Clifford SC (2009) The potential impact of tumor biology on improved clinical practice for medulloblastoma: progress towards biologically driven clinical trials. Br J Neurosurg 23:364–375
Pomeroy SL, Tamayo P, Gaasenbeek M, Sturla LM, Angelo M, McLaughlin ME, Kim JY, Goumnerova LC, Black PM, Lau C, Allen JC, Zagzag D, Olson JM, Curran T, Wetmore C, Biegel JA, Poggio T, Mukherjee S, Rifkin R, Califano A, Stolovitzky G, Louis DN, Mesirov JP, Lander ES, Golub TR (2002) Prediction of central nervous system embryonal tumor outcome based on gene expression. Nature 415:436–442
Raffel C, Jenkins RB, Frederick L, Hebrink D, Alderete B, Fults DW, James CD (1997) Sporadic medulloblastomas contain PTCH mutations. Cancer Res 57:842–845
Reifenberger J, Wolter M, Weber RG, Megahed M, Ruzicka T, Lichter P, Reifenberger G (1998) Missense mutations in SMOH in sporadic basal cell carcinomas of the skin and primitive neuroectodermal tumors of the central nervous system. Cancer Res 58:1798–1803
Romer J, Curran T (2005) Targeting medulloblastoma: small-molecule inhibitors of the Sonic Hedgehog pathway as potential cancer therapeutics. Cancer Res 65:4975–4978
Romer JT, Kimura H, Magdaleno S, Sasai K, Fuller C, Baines H, Connelly M, Stewart CF, Gould S, Rubin LL, Curran T (2004) Suppression of the Shh pathway using a small molecule inhibitor eliminates medulloblastoma in Ptc1(±)p53(−/−) mice. Cancer Cell 6:229–240
Rudin CM, Hann CL, Laterra J, Yauch RL, Callahan CA, Fu L, Holcomb T, Stinson J, Gould SE, Coleman B, LoRusso PM, Von Hoff DD, de Sauvage FJ, Low JA (2009) Treatment of medulloblastoma with hedgehog pathway inhibitor GDC-0449. N Engl J Med 361:1173–1178
Rutkowski S, Bode U, Deinlein F, Ottensmeier H, Warmuth-Metz M, Soerensen N, Graf N, Emser A, Pietsch T, Wolff JE, Kortmann RD, Kuehl J (2005) Treatment of early childhood medulloblastoma by postoperative chemotherapy alone. N Engl J Med 352:978–986
Rutkowski S, Gerber NU, von Hoff K, Gnekow A, Bode U, Graf N, Berthold F, Henze G, Wolff JE, Warmuth-Metz M, Soerensen N, Emser A, Ottensmeier H, Deinlein F, Schlegel PG, Kortmann RD, Pietsch T, Kuehl J (2009) Treatment of early childhood medulloblastoma by postoperative chemotherapy and deferred radiotherapy. Neuro Oncol 11:201–210
Rutkowski S, von Bueren A, von Hoff K, Hartmann W, Shalaby T, Deinlein F, Warmuth-Metz M, Soerensen N, Emser A, Bode U, Mittler U, Urban C, Benesch M, Kortmann RD, Schlegel PG, Kuehl J, Pietsch T, Grotzer M (2007) Prognostic relevance of clinical and biological risk factors in childhood medulloblastoma: results of patients treated in the prospective multicenter trial HIT’91. Clin Cancer Res 13:2651–2657
Stearns D, Chaudhry A, Abel TW, Burger PC, Dang CV, Eberhart CG (2006) c-myc overexpression causes anaplasia in medulloblastoma. Cancer Res 66:673–681
Tabori U, Baskin B, Shago M, Alon N, Taylor MD, Ray PN, Bouffet E, Malkin D, Hawkins C (2010) Universal poor survival in children with medulloblastoma harboring somatic TP53 mutations. J Clin Oncol 28:1345–1350
Taniguchi E, Cho MJ, Arenkiel BR, Hansen MS, Rivera OJ, McCleish AT, Qualman SJ, Guttridge DC, Scott MP, Capecchi MR, Keller C (2009) Bortezomib reverses a post-translational mechanism of tumorigenesis for patched1 haploinsufficiency in medulloblastoma. Pediatr Blood Cancer 53:136–144
Taylor MD, Liu L, Raffel C, Hui CC, Mainprize TG, Zhang X, Agatep R, Chiappa S, Gao L, Lowrance A, Hao A, Goldstein AM, Stavrou T, Scherer SW, Dura WT, Wainwright B, Squire JA, Rutka JT, Hogg D (2002) Mutations in SUFU predispose to medulloblastoma. Nat Genet 31:306–310
Taylor MD, Mainprize TG, Rutka JT (2000) Molecular insight into medulloblastoma and central nervous system primitive neuroectodermal tumor biology from hereditary syndromes: a review. Neurosurgery 47:888–901
Taylor RE, Bailey CC, Robinson K, Weston CL, Ellison D, Ironside J, Lucraft H, Gilbertson R, Tait DM, Walker DA, Pizer BL, Imeson J, Lashford LS (2003) Results of a randomized study of preradiation chemotherapy versus radiotherapy alone for nonmetastatic medulloblastoma: the International Society of Paediatric Oncology/United Kingdom Children’s Cancer Study Group PNET-3 Study. J Clin Oncol 21:1581–1591
Taylor RE, Bailey CC, Robinson KJ, Weston CL, Walker DA, Ellison D, Ironside J, Pizer BL, Lashford LS (2005) Outcome for patients with metastatic (M2–3) medulloblastoma treated with SIOP/UKCCSG PNET-3 chemotherapy. Eur J Cancer 41:727–734
Thompson MC, Fuller C, Hogg TL, Dalton J, Finkelstein D, Lau CC, Chintagumpala M, Adesina A, Ashley DM, Kellie SJ, Taylor MD, Curran T, Gajjar A, Gilbertson RJ (2006) Genomics identifies medulloblastoma subgroups that are enriched for specific genetic alterations. J Clin Oncol 24:1924–1931
Uziel T, Karginov FV, Xie S, Parker JS, Wang YD, Gajjar A, He L, Ellison D, Gilbertson RJ, Hannon G, Roussel MF (2009) The miR-17 92 cluster collaborates with the Sonic Hedgehog pathway in medulloblastoma. Proc Natl Acad Sci USA 106:2812–2817
Uziel T, Zindy F, Xie S, Lee Y, Forget A, Magdaleno S, Rehg JE, Calabrese C, Solecki D, Eberhart CG, Sherr SE, Plimmer S, Clifford SC, Hatten ME, McKinnon PJ, Gilbertson RJ, Curran T, Sherr CJ, Roussel MF (2005) The tumor suppressors Ink4c and p53 collaborate independently with Patched to suppress medulloblastoma formation. Genes Dev 19:2656–2667
Vorechovsky I, Tingby O, Hartman M, Stromberg B, Nister M, Collins VP, Toftgard R (1997) Somatic mutations in the human homologue of Drosophila patched in primitive neuroectodermal tumors. Oncogene 15:361–366
Wetmore C, Eberhart DE, Curran T (2001) Loss of p53 but not ARF accelerates medulloblastoma in mice heterozygous for patched. Cancer Res 61:513–516
Yang ZJ, Ellis T, Markant SL, Read TA, Kessler JD, Bourboulas M, Schuller U, Machold R, Fishell G, Rowitch DH, Wainwright BJ, Wechsler-Reya RJ (2008) Medulloblastoma can be initiated by deletion of Patched in lineage-restricted progenitors or stem cells. Cancer Cell 14:135–145
Yauch RL, Dijkgraaf GJ, Alicke B, Januario T, Ahn CP, Holcomb T, Pujara K, Stinson J, Callahan CA, Tang T, Bazan JF, Kan Z, Seshagiri S, Hann CL, Gould SE, Low JA, Rudin CM, de Sauvage FJ (2009) Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science 326:572–574
Zeltzer PM, Boyett JM, Finlay JL, Albright AL, Rorke LB, Milstein JM, Allen JC, Stevens KR, Stanley P, Li H, Wisoff JH, Geyer JR, McGuire-Cullen P, Stehbens JA, Shurin SB, Packer RJ (1999) Metastasis stage, adjuvant treatment, and residual tumor are prognostic factors for medulloblastoma in children: conclusions from the Children’s Cancer Group 921 randomized phase III study. J Clin Oncol 17:832–845
Zindy F, Nilsson LM, Nguyen L, Meunier C, Smeyne RJ, Rehg JE, Eberhart C, Sherr CJ, Roussel MF (2003) Hemangiosarcomas, medulloblastomas, and other tumors in Ink4c/p53-null mice. Cancer Res 63:5420–5427
Zurawel RH, Chiappa SA, Allen C, Raffel C (1998) Sporadic medulloblastomas contain oncogenic beta-catenin mutations. Cancer Res 58:896–899
Acknowledgments
I am grateful to Dr. Stefan Rutkowski (Universitätsklinikum Hamburg-Eppendorf) and Drs. Stacey Ogden and Peter McKinnon (St. Jude Children’s Research Hospital) for advice.
Conflict of interest
I have no conflict of interest to declare.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ellison, D.W. Childhood medulloblastoma: novel approaches to the classification of a heterogeneous disease. Acta Neuropathol 120, 305–316 (2010). https://doi.org/10.1007/s00401-010-0726-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00401-010-0726-6