
Abstract
The spatial and seasonal dynamics of the halophilic prokaryotic community was investigated in five ponds from Sfax solar saltern (Tunisia), covering a salinity gradient ranging from 20 to 36%. Fluorescence in situ hybridization indicated that, above 24% salinity, the prokaryotic community shifted from bacterial to archaeal dominance with a remarkable increase in the proportion of detected cells. Denaturing gradient gel electrophoresis (DGGE) profiles were rather similar in all the samples analyzed, except in the lowest salinity pond (around 20% salt) where several specific archaeal and bacterial phylotypes were detected. In spite of previous studies on these salterns, DGGE analysis unveiled the presence of microorganisms not previously described in these ponds, such as Archaea related to Natronomonas or bacteria related to Alkalimnicola, as well as many new sequences of Bacteroidetes. Some phylotypes, such as those related to Haloquadratum or to some Bacteroidetes, displayed a strong dependence of salinity and/or magnesium concentrations, which in the case of Haloquadratum could be related to the presence of ecotypes. Seasonal variability in the prokaryotic community composition was focused on two ponds with the lowest (20%) and the highest salinity (36%). In contrast to the crystallized pond, where comparable profiles between autumn 2007 and summer 2008 were obtained, the non-crystallized pond showed pronounced seasonal changes and a sharp succession of “species” during the year. Canonical correspondence analysis of biological and physicochemical parameters indicated that temperature was a strong factor structuring the prokaryotic community in the non-crystallizer pond, that had salinities ranging from 20 to 23.8% during the year.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The prokaryotic community structure of different coastal solar salterns across the world has been characterized by using both culture dependent and independent methods (Burns et al. 2004; Maturrano et al. 2006). FISH and molecular analyses revealed that Bacteria and Archaea are the two components of the microbial community in such extreme systems (Benlloch et al. 1996; Antón et al. 1999, 2000; Rodríguez-Valera et al. 1999; Litchfield and Gillevet 2002; Øvreås et al. 2003). In the crystallized ponds with the highest salinity (around 35%), a very low prokaryotic diversity was depicted by using 16S rRNA gene clone libraries, with Archaea being the dominant fraction (Rodríguez-Valera et al. 1999; Antón et al. 2000, 2008). While organisms related to Haloquadratum walsbyi and Halorubrum sp. were the most abundant Archaea (Benlloch et al. 2002; Burns et al. 2004), different Salinibacter phylotypes as well as other members of Bacteroidetes, dominated the bacterial assemblage that accounted for 5–30% of microbial community (Antón et al. 2002, 2008; Øvreås et al. 2003). In contrast, at lower salinity (around 20%) ponds, a rather diverse assemblage of Bacteria and Archaea was observed (Casamayor et al. 2002; Øvreås et al. 2003).
Although considerable data are available regarding the microbial diversity in hypersaline environments (Sorensen et al. 2005; Walsh et al. 2005; Dong et al. 2006; Jiang et al. 2006; Clementino et al. 2008; Tsiamis et al. 2008; Pagaling et al. 2009), fewer papers have addressed the seasonal variability and the factors that influence the prokaryotic community dynamics (Koizumi et al. 2004; Henriques et al. 2006). However, differences in prokaryotic community distribution and composition may be found at a very small scale as a result of salinity, nutrient concentration and organic matter composition gradients (Oren 2002, 2008; Koizumi et al. 2004).
Two culture-independent studies on the microbial community of Sfax solar saltern have been conducted using 16S rRNA gene clone libraries (Baati et al. 2008; Trigui et al. 2011). These studies indicated that Sfax salterns harbour a prokaryotic diversity higher than that of other salterns previously studied. However, the above mentioned studies included only two and three sampling sites, respectively, and did not assess the seasonal variability. Here, in order to analyze the microbial community dynamics in that system, we have investigated the seasonal and spatial variability of Bacteria and Archaea phylotypes present in five hypersaline ponds along the salinity gradient in Sfax salterns.
Materials and methods
Sample collection and physico-chemical analysis
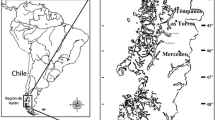
Five ponds (M1, TS18, PM2, R2, and S6) from Sfax solar salterns (34°39′N and 10°42′E) with salinities ranging from 20 to 36% were sampled in March 2008. Samples were labelled as M1MR08, TS18MR08, PM2MR08, R2MR08, and S6MR08 and used for the study of the spatial variability of the prokaryotic community. Samples were also taken from ponds M1 and TS18 in November 2007 (autumn; samples M1N07 and TS18N07), May 2008 (late spring; samples M1MY08 and TS18MY08) and August 2008 (summer; samples M1AU08 and TS18AU08) and were used for ascertaining seasonal variability. For every sample, 1 l of water was collected 20 cm below the surface in sterile bottles previously rinsed with water from the sampled pond. Salinity was determined in situ with a hand refractometer (Atago). Subsamples were sent to the research technical facilities at the University of Alicante (Spain) for chemical analysis.
DAPI-total cell counts, FISH and catalyzed reported deposition-FISH experiments
Water samples were fixed as previously described for fixation of extremely halophilic microorganisms by Antón et al. (1999). Total cell counts were determined by polycarbonate-membrane filtration after staining with DAPI (4′,6-diadimino-2-phenylindole) as described before (Snaidr et al. 1997). In situ hybridizations were performed at 46°C for 90 min as described in Snaidr et al. (1997). The group-specific probes to detect Archaea and Bacteria were respectively ARC915 and EUB338 (Amann et al. 1990; 1995, respectively). For catalyzed reported deposition (CARD)-FISH experiments, horseradish peroxidase (HRP) labeled oligonucleotide probes and tyramide signal amplification were used as described in detail (Pernthaler et al. 2004). The procedure of this approach consists of four main stages: embedding, permealization and inactivation of peroxidases, hybridization and tyramide signal amplification (Pernthaler et al. 2004). The slides were examined with an Axioplan microscope (Leica DMLA) and stained cells were counted in more than 20 different microscopic fields to estimate the cell concentration in the samples.
DNA extraction
Microbial biomass was collected by filtration of 10–20 ml of every sample with a 0.22 μm pore size GV filter (Durapore, Millipore) and DNA was extracted as described in detail in a previous work (Mutlu et al. 2008) with the exception that nucleic acids were precipitated with 0.1 vol of sodium acetate and 0.6 vol of isopropanol and centrifuged at 13,000 rpm, for 30 min, at 4°C, and washed with 70% ethanol. Genomic DNA extracts were stored at −80°C until used.
PCR amplification of bacterial and archaeal 16S rRNA genes
The extracted genomic DNA was used for PCR amplifications of bacterial and archaeal 16S rRNA genes by using the specific primers 341F-GC (GC-clamp: 5′-CGCCCGCCGCGCGCGGCGGGCGGGGCGGGGGCACGGGGGG-3′) and 344 F-GC, respectively, and the reverse universal primer 907R (Muyzer et al. 1993, 1996; Schäfer et al. 2001). Each PCR mixture contained 5 μl of 10× PCR reaction buffer (Invitrogen), 2.5 μl of 50 mM MgCl2, 1 μl of a 10 mM dNTP mixture, 1 μl of 10 μM (each) primer, 1 units of Taq polymerase, 1 μl of template DNA and sterile MilliQ water up to 50 μl. The PCR program for Bacteria was: 94°C for 5 min, 65°C for 1 min, 72°C for 3 min and 9 touchdown cycles of: 94°C for 1 min, 65°C (with a decreasing of 1°C in each cycle) for 1 min, 72°C for 3 min, followed by 20 cycles of: 94°C for 1 min, 55°C for 1 min, 72°C for 3 min. The PCR program for Archaea was: 94°C for 5 min and 30 cycles of: 94°C for 30 s, 56°C for 45 s, 72°C for 2 min. In both cases, during the final cycle the length of extension step was increased to 30 min to minimize double band formation (Janse et al. 2004).
DGGE analysis of bacterial and archaeal diversity
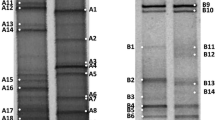
DGGE was performed by using the D-Code System (Bio-Rad, Hercules, CA). PCR products were loaded onto 6% (w/v) polyacrylamide (acrylamide:bis-acrylamide gel stock solution 37.5:1; Bio-Rad), with 45 to 65% (Archaea) and 40 to 60% (Bacteria) denaturing gradient (where 100% of denaturant consists of 7 M urea and 40% formamide) in 1× TAE buffer (40 mM Tris, pH 8.0; 20 mM acetic acid; 1 mM EDTA) and subjected to 16–18 h of electrophoresis at 60°C and 70 V. DGGE gels were stained for 30 min with SYBR Green, visualized under UV light and photographed with a Typhoon 9410 (Amersham Biosciences) system. The images were analysed to estimate the bacterial and archaeal richness (Moeseneder et al. 1999).
For sequencing and identification of DGGE fragments, bands were excised with sterile razor blades from the DGGE gels and soaked overnight into 20 μl of MilliQ water. Two microliters (100 ng approximately) of each band were then re-amplified, with the same primer set (without the GC clamp). PCR products were purified with a GFX PCR DNA and gel band purification kit (GE Healthcare) and 250 ng were sequenced with primer 907R in an ABI PRISM 3100 Genetic Analyzer (Applied Biosystems, Foster City, CA). Sequences were aligned to affiliated 16S rRNA gene sequences obtained from GenBank nucleotide database using the BLAST (Basic Local Alignment Search Tool) software at the National Centre of Biotechnology Information website (http://www.ncbi.nlm.nih.gov/).
Data analysis
Physicochemical data (salinity, pH, temperature, sodium, potassium, magnesium, calcium, chlorides and sulphate concentrations) were taken as the independent variables while biological parameters (archaeal and bacterial diversity, numbers of Archaea and Bacteria and DAPI counts) were taken as dependent variables. First, a principal component analysis (PCA) was carried out using the informatics package SPSS17.0 to study the correlation among the independent variables. Then, ordination methods were used to analyze the variation of the sequences found in the different ponds according to the physicochemical variables using canonical correspondence analysis (CCA). The resulting ordination biplot approximated the weighted average of each species (in this case, distribution of phylotypes) with respect to each of the environmental variables, which were represented as arrows. The length of these arrows indicated the relative importance of that environmental factor in explaining variation in bacterial and archaeal profiles, while the angle between arrows indicated the degree to which they were correlated (Jongman et al. 1995). A Monte Carlo test with 4,000 permutations was carried out to make assure the significance of the canonical axes. These analyses were carried out using the program package CANOCO 4.5. Paleontological Statistics Software Package (PAST) was used to compute Shannon index and estimate prokaryotic diversity by using each DGGE band as an equivalent of a “phylotype”. Relationships between archaeal and bacterial DGGE band patterns for the analyzed samples was performed by calculating a similarity dendrogram as previously described (Martínez-García et al. 2010).
Results and discussion
Physicochemical characterization of the samples
We analyzed eight samples from ponds M1 and TS18 collected in autumn 2007, late winter, late spring and summer 2008, as well as three additional samples taken in PM2, R2, and S6 ponds in late winter 2008. For every pond, a total of nine physicochemical parameters (temperature, salinity, pH, and ionic composition) were determined as shown in Table 1. The salinity of the samples ranged from 20 to 36% as determined in situ by a hand refractometer.
PCA of these nine parameters indicated that three components were needed to explain 95.1% of the total variance among the samples. The first component C1 had a very strong contribution of the salinity and magnesium concentration while C2 was mainly related with sodium concentration and C3 with temperature (Supplementary Table 1). According to these three components, the different samples were grouped as shown in Supplementary Fig. 1. With the exception of the sample pond S6 (slightly more saline than the rest but with very high magnesium concentration), all the samples taken in March grouped together and apart from the rest, while samples taken in August and May were not clearly separated and formed a non homogenous cluster. Thus, salinity was not the only factor relevant in defining differences/similarities among samples.
FISH and DAPI counts
We analyzed the prokaryotic diversity in five different ponds in March (2008), along the salinity gradient. Samples showed salinities between 20 and 36% (Table 1) and DAPI total cell values of up to 8 × 107 cells ml−1, in the range of cell densities previously measured in this system (Elloumi et al. 2009; Trigui et al. 2011) or in other coastal salterns or salt lakes (Antón et al. 2000; Burns et al. 2004; Mutlu et al. 2008). Composition of the prokaryotic community in the lowest salinity M1 sample could not be measured by standard FISH. For this reason, CARD-FISH was performed for that sample indicating that Bacteria constituted at least 13% of the prokaryotes while Archaea remained undetectable in all except the August sample, which also had the highest detection rate among M1 samples. The failure of FISH detection in most M1 samples (expect for M1 AU 08) is most likely due to a low level of metabolic activity of the community (Amann et al. 1995; Pernthaler et al. 2004). By combining flow cytometry cell sorting of SYBR Green labelled cells and phylogenetic analysis, two populations of cells with high (HNA) and low (LNA) nucleic acid content were detected in M1 and TS (Trigui et al. 2011). In agreement with our FISH results, LNA fraction, composed of cells that are either inactivated, dormant or have very low levels of activity (Gasol et al. 1999; Lebaron et al. 2002) was more abundant in M1 than HNA. Indeed, sequences related to the LNA fraction described by Trigui et al. (2011) were retrieved from M1, as discussed in the following section. For the rest of the ponds, FISH showed that prokaryotic detection percentages were above 44%, with Archaea always outnumbering Bacteria, in good agreement with data previously reported for other crystallizers (Antón et al. 1999; Rosselló-Mora et al. 2003; Maturrano et al. 2006).
DGGE analysis of prokaryotic diversity
Bacterial and archaeal assemblages were analyzed by amplification of a fragment of the 16S rRNA genes from the community followed by DGGE. DGGE patterns (Fig. 1) were rather homogeneous in all the samples analyzed, except in the case of M1. However, similarities were higher among archaeal patterns that displayed less changes both with time and along the salinity gradient. Clustering analyses of the DGGE profiles (Fig. 2) showed two main clusters (M1 samples and the rest) both for Bacteria and Archaea patterns. In a previous work on Sfax solar salterns, Baati et al. (2008) analyzed 16S rRNA gene libraries from three different kinds of ponds: M2, TS38 and S5 (that would be similar to our ponds M1, TS18 and S6, respectively). These authors also found that M2, the lowest salinity pond, was different from the rest, since no sequence present in M2 was retrieved from the rest of the ponds. In our case, M1 was also the most dissimilar pond and the only one with pond-specific bands. However, many bands present in M1 were also present in the rest of the ponds analyzed in this work, as explained below.


Dendrogram showing the relationships between archaeal a and bacterial b DGGE band patterns for every analyzed pond. Samples are labelled as in Table 1
In order to identify the different members of the prokaryotic community, DGGE bands (of approximately 550 bp) were excised from the gel and sequenced. All DGGE bands (Fig. 1) with the same position in the gel showed the same sequence (data not shown). CCA was used to correlate changes in bacterial and archaeal phylotype (i.e. DGGE band sequence) distribution in the different ponds with their physicochemical characteristics. Sequencing results from individual DGGE bands are shown in Tables 2 and 3 while Fig. 3 show graphically the changes in phylotype composition (i.e. DGGE band) for the analyzed samples according to the pond and the sampling season, respectively. With the exception of M1, that had seven bacterial and seven archaeal specific bands, most of the bands appeared in all the analyzed ponds. It is noteworthy (Fig. 3) that all these M1 specific sequences have a similar distribution in the graph according to CCA1 axis, so they correspond to samples of similar salinities although different temperatures (as can be seen by projecting them over the vectors representing these environmental parameters), as discussed below. Finally, there are pairs of archaeal/bacterial sequences that always appear together either at a given time of the year (1A/16B) or along different seasons (24A/12B). These pairs of phylotypes display the same dependence of physicochemical parameters, as shown by their similar position in the CCA space, and most likely have the same growth requirements.

CCA biplot of the axis 1 and 2 for the DGGE sequences and environmental parameters. Sequences are represented according to the ponds where they were obtained a and the months in which they were detected b sequences located in the same point of the represented spaced are indicated in the rectangles. Every sequence is represented by a circle that is divided into different sections corresponding to the proportions of the sequence found in different pond (a) and different months (b)
For Bacteria, 13 bands were excised from the gel and sequenced. Nine of them were related to Bacteroidetes, two to γ-Proteobacteria, one to α-Proteobacteria and one to Cyanobacteria. Seven bands corresponded to sequences not previously retrieved from Sfax salterns, in spite of the availability of three recent papers on the microbial diversity of this system (Baati et al. 2008; Trigui et al. 2011), although some of them were very closely related to sequences from other Mediterranean coastal salterns. Among the sequences detected here similar to previously reported Sfax sequences, two of them (7B and 9B) seemed to be specific of M ponds, since we detected them only in M1 (Figs. 2, 3) while Baati et al. (2008) detected them in M2. However, band 8B was labelled as M2 specific in Baati et al. (2008) while we found it in all the ponds. In addition, a band (12B) specific of M1 pond was very closely related to a sequence found in high magnesium bitterns from Sfax solar salterns (Baati et al. unspublished) with Alkalilimnicola sp. as the closest cultured relative (97% similarity). Very recently (Ghai et al. 2011) found that Alkalilimnicola representatives accounted for around 16% of the prokaryotic community inhabiting a saltern pond of 19% salinity. These sequences had not been previously found in the works by Baati et al. 2008 and Trigui et al. 2011 mentioned above. Only three bacterial sequences had more than 97% similarity to 16S rRNA genes from cultured strains (12B, 15B and 20B) and the rest had only distant relationships with known genera, underscoring the extend of unknown diversity present in natural environments. To this regard, it is worth mentioning the recovery of a sequence (13B) only distantly related to the α-proteobacterium Anaplasma.
Bacteroidetes was the most frequently retrieved bacterial group, with a high representation of new sequences not previously found in nature. Indeed, it seems that the description of a group of phylogenetically related halophiles is emerging within the Bacteroidetes formed by a couple of cultured genera (i.e. Salinibacter and Salisaeta) together with sequences recovered from hypersaline environments all over the world (Antón et al. 2008).
For Archaea, all the 19 bands analyzed corresponded to members of the Halobacteriales (Euryarchaeota). With the exception of bands 1A and 1B, all were closely related either to cultured haloarchaea or to sequences previously retrieved from hypersaline environments. Five of them were related to Sfax sequences obtained in Baati et al. (2008) or Trigui et al. (2011). Sequences 4A and 7A were observed in all the analyzed ponds while their homologous sequences had been previously found in M2 and S6, respectively. Three sequences (25A, 26A and 27A) were specific of pond M1 and corresponded to the low nucleic acid content found by Trigui et al. (2011) in this pond in October 2009. Among the archaeal sequences related to cultured strains, it is worth mentioning that four bands related to H. walsbyi, which has been repeatedly reported as one of the most abundant prokaryotes in coastal solar salterns, included Sfax salterns (Benlloch et al. 1996, 2002; Burns et al. 2004; Maturrano et al. 2006; Mutlu et al. 2008). Haloquadratum bands 7A and 11A are present along the whole year in all the samples, while bands 6A and 7A do not appear in M1, most likely indicating the existence of ecotypes within this species adapted to different environmental conditions. In addition, two sequences (29A and 30A) were related to the alkaliphilic haloarcheaon Natronomonas which had not been previously detected in Sfax salterns but have also been found in other neutral hypersaline environments (Benlloch et al. 2002, Burns et al. 2004; Ghai et al. 2011).
In summary, sequence analyses showed that most archaeal and bacterial phylotypes corresponded to members of Euryarchaeota and Bacteroidetes, respectively, which was in agreement with previous studies of Sfax salterns carried out using rRNA gene clone libraries (Baati et al. 2008; Trigui et al. 2011), as well as analysis of hypersaline environments from over the world (Mutlu et al. 2008; Burns et al. 2004; Antón et al. 2008; Ghai et al. 2011). However, not all the DGGE sequences obtained in this work matched with environmental clones previously recovered from the same ponds. This phenomenon has also been observed in studies with natural samples where the use of different sets of primers (for cloning and DGGE analyses) demonstrated that not all the sequences obtained by DGGE matched exactly with sequences in libraries and even some groups of microorganisms appeared only under one of the two techniques applied (Santos et al. 2010). In addition, in spite of the temporal stability of the samples, there are sequences that appear only at a given season and therefore may not have been recovered in the studies by Baati et al. (2008), and Trigui et al. (2011) that were based on a single sampling time.
Finally, although DGGE and FISH has a relatively low resolution mainly due to unspecificity in the used primers and probes (Daims et al. 1999), it provides a good snapshot of the most abundant and predominant microbial groups (Pedrós-Alió 2006) that in the case of hypersaline systems agree with data retrieved by high-throughput molecular techniques such as metagenomics (Rodriguez-Brito et al. 2010) and recently by single cell sequencing (Ghai et al. 2011), that indicates that halophilic square Archaea together with Bacteroidetes retrieved in the present study likely play a significant role in the ecosystem functioning.
Dynamics of the prokaryotic community
As shown in Fig. 3, both for Archaea and Bacteria, there is a succession of phylotypes along the salinity gradient that in the case of Bacteria is accompanied by a decrease in diversity. This decrease of bacterial diversity along salinity gradient in salterns has been previously reported (Benlloch et al. 2002). However, there is almost no data on the temporal stability of the prokaryotic community inhabiting the different ponds. In order to get a better understanding of this phenomenon, we analyzed two types of ponds (M1 and TS18), with two different degrees of salinity and different microbial communities (Baati et al. 2008; Trigui et al. 2011) along a complete year.
The crystallizer TS18 shows the well known dominance of Archaea in hypersaline environments, although a considerable number of Bacteria are also present. For both domains, the composition is very stable along the year as shown by DGGE patterns, with all the bacteria bands affiliated to Bacteroidetes. The only remarkable change is the absence, in the sample taken in November 2007, of band 1B that corresponds to an uncultured Bacteroidetes, which is present in all the M1 and in the rest of TS samples. One possible explanation is that this phylotype can not stand the high magnesium concentration present in this sample since it is present in all the ponds in all seasons, except the three samples with highest magnesium (R2MR08, TS18N07 and S6MR08). Notice that sample TSMY08 has higher salinity than R2MR08 but lower magnesium concentration.
The community inhabiting pond M1 experienced more changes along the year than that of TS18, both in numbers and “species” composition. From November 2007 to May 2008, the DGGE archaeal band pattern remained rather stable although they most likely represent inactive species since Archaea cannot be detected by FISH. In summer, the archaeal community undergoes a change that implies not only an increase in activity (as indicated by FISH numbers) but a shift in the “species” composition, with some new phylotypes appearing (e.g. 1A and 2A) while other disappear (24A), as shown in Fig. 1.
Bacteria in pond M1 undergo considerable changes along the year that affect specially the bands specific of this pond, as shown in Figs. 1 and 3, and also affects the overall bacterial diversity of the sample, as indicated by the Shannon index values (Table 1). M1 specific sequences (Fig. 3a) fall on the right side of the graph, apart from the rest of sequences that can be found in most of the other ponds. Their distribution, almost along a vertical line, indicate that their physicochemical conditions represented by horizontal vectors affect all these sequences in the same way. Thus, differences in their location in the diagram are due to their relationship with temperature (the vector with highest vertical component). In other words, these sequences are found in samples with similar chemical composition but different temperatures that are, obviously, due to the seasonal changes. As shown in Fig. 3b, some M1 specific sequences appear only in one of the seasons, while other are found only in certain periods of the year such as autumn–winter (as 9B and 7B) or spring-summer (as 13B).
Concluding remarks
The present study represents a step further in the knowledge of prokaryotic community dynamics in hypersaline environments, which is key to understand the structure of microbial communities and how they may respond to changes in abiotic factors of the environment. Our data indicate that salinity is not the only force structuring the halophilic community and that seasonal changes can affect greatly the composition of the microbial assemblage, specially the bacterial component, and thus have to be taken into account to describe properly the microbiota of this “low-diversity” systems.
References
Amann RI, Binder BJ, Olson RJ, Chisholm SW, Devereux R, Stahl DA (1990) Combination of 16S rRNA-targeted oligonucleotide probes with flow cytometry for analyzing mixed microbial populations. Appl Environ Microbiol 56:1919–1925
Amann RI, Ludwig W, Schleifer KH (1995) Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol Rev 59:143–169
Antón J, Llobet-Brossa E, Rodríguez-Valera F, Amann R (1999) Fluorescence in situ hybridization analysis of the prokaryotic community inhabiting crystallizer ponds. Environ Microbiol 1:517–523
Antón J, Rosselló-Mora R, Rodríguez-Valera F, Amann R (2000) Extremely halophilic bacteria in crystallizer ponds from solar salterns. Appl Environ Microbiol 66:3052–3057
Antón J, Oren A, Benlloch S, Rodríguez-Valera F, Amann R, Rosselló-Mora R (2002) Salinibacter ruber., gen. nov., sp. nov., a novel, extremely halophilic member of the Bacteria from saltern crystallizer ponds. Int J Syst Evol Microbiol 52:485–491
Antón J, Peña A, Santos F, Martínez-García M, Schmitt-Kopplin P, Rosselló-Mora R (2008) Distribution, abundance and diversity of the extremely halophilic bacterium Salinibacter ruber. Saline Syst 4:2
Baati H, Guermazi S, Amdouni R, Gharsallah N, Sghir A, Ammar E (2008) Prokaryotic diversity of a Tunisian multipond solar saltern. Extremophiles 12:505–518
Benlloch S, Acinas SG, Martínez-Murcia AJ, Rodríguez-Valera F (1996) Description of prokaryotic biodiversity along the salinity gradient of a multipond solar saltern by direct PCR amplification of 16S rDNA. Hydrobiologia 329:19–31
Benlloch S, López-López A, Casamayor EO, Øvreås L, Goddard V, Daae FL, Smerdon G, Massana R, Joint I, Thingstad F, Pedrós-Alió C, Rodríguez-Valera F (2002) Prokaryotic genetic diversity throughout the salinity gradient of a coastal solar saltern. Environ Microbiol 4:349–360
Burns DG, Camakaris HM, Janssen PH, Dyall-Smith ML (2004) Combined use of cultivation-dependent and cultivation-independent methods indicate that members of most haloarchaeal groups in an Australian crystallizer pond are cultivable. Appl Environ Microbiol 70:5258–5265
Casamayor EO, Massana R, Benlloch S, Øvreås L, Diez B, Goddard VJ, Gasol JM, Joint I, Rodriguez-Valera F, Pedrós-Alió C (2002) Changes in archaeal, bacterial and eukaryal assemblages along a salinity gradient by comparison of genetic fingerprinting methods in a multipond solar saltern. Environ Microbiol 4:338–348
Clementino MM, Vieira RP, Cardoso AM, Nascimento APA, Silveira CB, Riva TC, Gonzalez ASM, Paranhos R, Albano RM, Ventosa A, Martins OB (2008) Prokaryotic diversity in one of the largest hypersaline coastal lagoons in the world. Extremophiles 12:595–604
Daims H, Brühl A, Amann R, Schleifer KH, Wagner M (1999) The domain-specific probe EUB338 is insufficient for the detection of all bacteria: development and evaluation of a more comprehensive probe set. Syst Appl Microbiol 22:434–444
Dong H, Zhang G, Jiang H, Yu B, Chapman LR, Lucas CR, Fields MW (2006) Microbial diversity in sediments of saline Qinghai Lake, China: linking geochemical controls to microbial diversity. Microbiol Ecol 51:65–82
Elloumi J, Carrias JF, Ayadi H, Sime-Ngando T, Bouaïn A (2009) Communities structure of the planktonic halophiles in the solar saltern of Sfax, Tunisia. Estuar Coast Shelf Sci 81:19–26
Gasol JM, Zweifel UL, Peters F, Fuhrman JH, Hagström A (1999) Significance of size and nucleic acid content heterogeneity as measured by flow cytometry in natural planktonic bacteria. Appl Environ Microbiol 65:4475–4483
Ghai R, Pašić L, Fernández AB, Martin-Cuadrado AB, Mizuno CM, McMahon KD, Thane Papke R, Stepanauskas R, Rodruiguez-Brito B, Rodruiguez-Brito B, Rohwer F, Sánchez-Porro C, Ventosa A, Rodríguez-Valera F (2011) New abundant microbial groups in aquatic hypersaline environments. Sci Rep 1:135
Henriques IS, Alves A, Tacão M, Almeida A, Cunha A, Correia A (2006) Seasonal and spatial variability of free-living bacterial community composition along an estuarine gradient (Ria de Aveiro, Portugal). Estuar Coast Shelf Sci 68:139–148
Janse I, Bok J, Zwart G (2004) A simple remedy against artificial double bands in denaturing gradient gel electrophoresis. J Microbiol Methods 57:279–281
Jiang H, Dong H, Zhang G, Yu B, Chapman LR, Fields MW (2006) Microbial diversity in water and sediment of Lake Chaka, an athalassohaline lake in northwestern China. Appl Environ Microbiol 72:3832–3845
Jongman RH, ter Braak CJF, Van Tongeren OFR (1995) Data analysis in community and landscape ecology. Cambridge University Press, Cambridge 299
Koizumi Y, Kojima H, Oguri K, Kitazato H, Fukui M (2004) Vertical and temporal shifts in microbial communities in the water column and sediment of saline meromictic lake Kaiike (Japan), as determined by 16S rDNA-based analysis, and related to physicochemical gradients. Environ Microbiol 6:622–637
Lebaron P, Servais P, Baudoux AC, Bourrain M, Courties C, Parthuisot N (2002) Variation of bacterial specific activity with cell size and nucleic acid content assessed by flow cytometry. Aquat Microbiol Ecol 28:131–140
Litchfield CD, Gillevet PM (2002) Microbial diversity and complexity in hypersaline environments: a preliminary assessment. J Ind Microbiol Biotechnol 28:48–55
Martínez-García M, Díaz-Valdés M, Antón J (2010) Diversity of pufM genes, involved in aerobic anoxygenic photosynthesis, in the bacterial communities associated with colonial ascidians. FEMS Microbiol Ecol 71:387–398
Maturrano L, Santos F, Rosselló-Mora R, Antón J (2006) Microbial diversity in Maras salterns, a hypersaline environment in the Peruvian Andes. Appl Environ Microbiol 72:3887–3895
Moeseneder MM, Arrieta JM, Muyzer G, Winter C, Herndl GJ (1999) Optimization of terminal-restriction fragment length polymorphism analysis forcomplex marine bacterioplankton communities and comparison with denaturing gradient gel electrophoresis. Appl Environ Microbiol 65:3518–3525
Mutlu M, Martínez-García M, Santos F, Peña A, Guven K, Antón J (2008) Prokaryotic diversity in Tuz Lake, a hypersaline environment in Inland Turkey. FEMS Microbiol Ecol 65:474–483
Muyzer G, De Waal EC, Uitterrlinden AG (1993) Profiling in complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes coding for 16S rRNA. Appl Environ Microbiol 59:695–700
Muyzer G, Hottenträger S, Teske A, Wawer C (1996) Denaturing gradient gel electrophoresis of PCR-amplified 16S rRNA-a new molecular approach to analyse the genetic diversity of mixed microbial communities. In: Akkermans ADL, Van Elsas JD, De Brujn F (eds) Molecular microbial ecology manual. Kluwer Academic, Dordrecht, pp 1–23
Oren A (2002) Diversity of halophilic microorganisms: environments, phylogeny, physiology, and applications. J Ind Microbiol Biotechnol 28:56–63
Oren A (2008) Microbial life at high salt concentrations: phylogenetic and metabolic diversity. Saline Syst 4:2
Øvreås L, Daae FL, Torsvik V, Rodrıguez-Valera F (2003) Characterization of microbial diversity in hypersaline environments by melting profiles and reassociation kinetics in combination with terminal restriction fragment length polymorphism (T-RFLP). Microb Ecol 46:291–301
Pagaling E, Wang H, Venables M, Wallace A, Grant WD, Cowan DA, Jones BE, Ma Y, Ventosa A, Heaphy S (2009) Microbial biogeography of six salt lakes in Inner Mongolia, China, and a salt lake in Argentina. Appl Environ Microbiol 75:5750–5760
Pedrós-Alió C (2006) Marine microbial diversity: can it be determined? Trends Microbiol 14:257–263
Pernthaler A, Pernthaler J, Amann R (2004) Sensitive multi-color fluorescence in situ hybridization for the identification of environmental microorganisms. Mol Microb Ecol Manual 11:711–726
Rodriguez-Brito B, Li L, Wegley L et al (2010) Viral and microbial community dynamics in four aquatic environments. ISME Journal 4:739–751
Rodríguez-Valera F, Acinas SG, Antón J (1999) Contribution of molecular techniques to the study of microbial diversity in hypersaline environments. In: Oren A (ed) Microbiology and biochemistry of hypersaline environments. CRC Press, Boca Ratón, pp 27–38
Rosselló-Mora R, Lee N, Antón J, Wagner M (2003) Substrate uptake in extremely halophilic microbial communities revealed by microautoradiography and fluorescence in situ hybridization. Extremophiles 7:409–413
Santos F, Peña A, Nogales B, Soria-Soria E, García Del Cura MA, González-Martín JA, Antón J (2010) Bacterial diversity in dry modern freshwater stromatolites from ruidera pools natural park, Spain. Syst Appl Microbiol 33:209–221
Schäfer HL, Bernard L, Courties C, Lebaron F, Servais P, Pukall R, Stackebrandt E, Troussellier M, Guindulain T, Vives-Rego J, Muyzer G (2001) Microbial community dynamics in Mediterranean nutrient-enriched seawater mesocosms: changes in the genetic diversity of bacterial populations. FEMS Microbiol Ecol 34:243–253
Snaidr J, Amann R, Huber I, Ludwig W, Schleifer KH (1997) Phylogenetic analysis and in situ identification of bacteria in activated sludge. Appl Environ Microbiol 63:2884–2896
Sorensen KB, Canfield DE, Teske AP, Oren A (2005) Community composition of a hypersaline endoevaporitic microbial mat. Appl Environ Microbiol 71:7352–7365
Trigui H, Masmoudi S, Brochier-Armanet C, Barani A, Grégori G, Denis M, Dukan S, Maalej S (2011) Characterization of heterotrophic prokaryote subgroups in the Sfax coastal solar salterns by combining flow cytometry cell sorting and phylogenetic analysis. Extremophiles. doi:10.1007/s00792-011-0364-5
Tsiamis G, Katsaveli K, Ntougias S, Kyrpides N, Anderson G, Pyceno Y, Bourtzis K (2008) Prokaryotic community profiles at different operational stages of a Greek solar saltern. Res Microbiol 159:609–627
Walsh DA, Papke RT, Doolittle WF (2005) Archaeal diversity along a soil salinity gradient prone to disturbance. Environ Microbiol 7:1655–1666
Acknowledgments
This work was funded by projects A/6679/06 from the Spanish Agency for International Cooperation (AECI) and CGL2009-12651-C02-01 from the Spanish Ministry of Science and Innovation (MICINN). We also wish to acknowledge the Tunisian COTUSAL salt company in Sfax for allowing members of our research team to access to the saltern.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Boujelben, I., Gomariz, M., Martínez-García, M. et al. Spatial and seasonal prokaryotic community dynamics in ponds of increasing salinity of Sfax solar saltern in Tunisia. Antonie van Leeuwenhoek 101, 845–857 (2012). https://doi.org/10.1007/s10482-012-9701-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10482-012-9701-7