
Abstract
Ralstonia solanacearum is the causal organism of bacterial wilt of more than 200 species representing 50 families of plants in tropical, subtropical, and warm temperate regions in the world. Traditionally classified into five races based on differences in host range, R. solanacearum has also been grouped into six biovars on the basis of biochemical properties. With recent developments in molecular biology, various DNA-based analyses have been introduced and used to confirm that this binary system does not completely represent the diversity within R. solanacearum strains. Therefore, a new hierarchical classification scheme has been suggested, which defines R. solanacearum as a species complex and reorganized the concept of the species as a monophyletic cluster according to a phylogenetic analysis based on genomic sequence data. Here we discuss the current bacterial wilt situation and genetic relationships based on the recent classification system of Japanese R. solanacearum strains as well as worldwide strains. We also review the genetic, biochemical, and pathological characteristics of R. solanacearum strains, in particular, those affecting potato and Zingiberaceae plants as distinctly important pathogens in relation to continuously problematic and recent emergent diseases in Japan.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Bacterial wilt diseases caused by Ralstonia solanacearum (Smith) (Yabuuchi et al. 1996) is a lethal vascular disease of more than 200 species representing 50 families of plants, including economically important crops such as potato, tomato, eggplant, pepper, ginger, and banana (Hayward 1991, 1994a), and the disease is very difficult to control. The importance of the disease has been widely recognized in tropical, subtropical and warm temperate regions of the world, including Japan and other Asian countries.
R. solanacearum embraces a diverse array of populations that differ in host range, geographical distribution, pathogenicity, epidemiological characters, and physiological properties. To describe this intra-specific variability of R. solanacearum, many classification schemes have been used for the identification and evolution of this bacterium (Buddenhagen and Kelman 1964; Denny and Hayward 2001; Okabe 1965). R. solanacearum has been traditionally classified into races based on differences in host range and into biovars on the basis of biochemical properties (Denny and Hayward 2001; Hayward 1964, 1994b).
Over the last 3 decades, molecular biological studies have been introduced to classify and identify R. solanacearum strains from diverse origins. Various genetic analyses also have been conducted to assess genetic variability between and within the population of R. solanacearum (Cook and Sequeira 1994; Cook et al. 1989; Fegan et al. 1998; Gillings and Fahy 1993, 1994; Horita and Tsuchiya 2001; Poussier et al. 1999, 2000a, b; Taghavi et al. 1996; Villa et al. 2005). These investigations have confirmed that the traditional dichotomy system (race and biovar) does not completely represent the diversity within R. solanacearum. On the basis of results obtained by molecular analyses, Fegan and Prior (2005) and Remenant et al. (2011) then defined this pathogen as a species complex and suggested a new classification system (Table 1).
In this review, we describe the development of both classical (biological) and modern (molecular) classification schemes on the genetic diversity of R. solanacearum Japanese strains as well as worldwide strains. We also describe the genetic, biochemical, and pathological characters of R. solanacearum strains, in particular, bacterial wilt of potato and Zingiberaceae plants as distinct pathogens in relation to continuously present or newly emergent diseases in Japan.
Classification of R. solanacearum strains based on biological properties and biochemical characters
For more than 4 decades, researchers of R. solanacearum mainly employed a binary system using two approaches, race and biovar, one emphasizing pathogenic specialization (host range or hosts primarily affected) and the other using selected biochemical characters (metabolism of different carbon sources) (Hayward 1991), respectively. Thus five races (Buddenhagen and Kelman 1964; Denny and Hayward 2001; He et al. 1983) and six biovars (Hayward 1964, 1994b; He et al. 1983) have been described and designated so far.
Race 1 affects tobacco, tomato, many solanaceous and other weeds and certain diploid bananas, race 2 affects triploid bananas (Moko disease) and Heliconia, race 3 affects potato and tomato, race 4 and race 5 affect ginger and mulberry, respectively. On the other hand, six biovars (1, 2, N2, 3, 4, and 5) were differentiated on the basis of the utilization and oxidation of six kinds of sugars (maltose, lactose, cellobiose, trehalose, m-inositol, and d-ribose) and three kinds of hexose alcohols (mannitol, sorbitol, and dulcitol).
Although this binary classification system has been useful in assessing geographical and pathological differences within R. solanacearum strains worldwide, it does not completely differentiate and characterize each strain as in the case of biovar 2, which has been further subdivided (Gillings and Fahy 1993; Hayward 1994b). It is therefore important to investigate genetic variability among the strains using various kinds of techniques to better understand the distribution and population dynamics of this bacterium not only under global environments but also in agricultural plants and soils.
The geographical distribution and diversity of Ralstonia solanacearum strains in Japan and Asia
In Japan, more than 46 species representing 24 families of plants have been reported as hosts so far, and new hosts continue to be found (Horita and Tsuchiya 2009, 2012). Distribution of races 1, 3, and 4 (Horita and Tsuchiya 2001; Katayama and Kimura 1986; Morita et al. 1996; Tsuchiya 2008; Tsuchiya et al. 1999; Yano et al. 2005) and biovars N2, 3, and 4 have been reported (Horita and Tsuchiya 2001; Horita et al. 2005, 2010). Race 1 strains have been isolated mainly from solanaceous crops including tomato, tobacco, eggplant, and sweet pepper. Race 3 has been isolated from potato and race 4 from Zingiberaceae plants. Ozaki and Kimura (1992) reported that Japanese R. solanacearum strains that affect Solanaceae crops can be divided into five groups (I–V) based on differences in their pathogenicity on several Solanum species, which have been used as resistant rootstocks for eggplant. Bacterial wilt of ginger caused by race 4 has recently emerged in Japan, as described later, and in many parts of Asia (including China, India, Malaysia, the Philippines, northern Thailand, and Indonesia) in the last decade.
The most important crop affected is tomato in the major production areas of Asian countries. Bacterial wilt is widely ranked among the top five diseases of other solanaceous crops, including eggplant, sweet pepper, and chili (Elphinstone 2005). Bacterial wilt significantly affects groundnut in Vietnam, the Philippines, Indonesia, and India. R. solanacearum has been reported on banana in several Asian countries, although further investigation is required to substantiate suspected cases reported from India, Malaysia, Sri Lanka, Taiwan, Thailand, and Vietnam (Elphinstone 2005). In the Philippines, R. solanacearum is still widespread throughout the country and is currently among the top five pathogens affecting banana and plantain (Elphinstone 2005). Moko disease and Bugtok disease is commonly found in commercial banana and cooking banana cultivars, respectively. Although the symptoms differ, both diseases are caused by race 2 strains, which are indistinguishable genetically. In Indonesia, the banana blood disease bacterium (BDB) (a closely related species to R. solanacearum) has been reported specifically (Eden-Green and Sastraatmadja 1990) and is spreading throughout the country and threatening banana production in Southeast Asia (Elphinstone 2005).
R. solanacearum strains belonging to biovars 3 and 4 are widespread and affect a very wide range of economically important crops from at least 20 Asian and Middle Eastern countries (Elphinstone 2005). On the contrary, biovar 2 and N2 strains primarily affect potato production in most Asian countries (Elphinstone 2005; Hayward et al. 1998; Horita et al. 2010; Xu et al. 2009).
Classification of R. solanacearum strains based on molecular analyses
The development of molecular biology has shifted the types of approaches used to characterize, identify plant pathogens and develop disease management strategies (Schaad et al. 2003). Consequently, more efficient approaches have been devised; the most popular and possibly the most accurate is DNA analysis.
Palleroni and Doudoroff (1971) clarified that DNA–DNA homology between R. solanacearum strains each belonging to distinct biovar is below the same species level (<70 %) and suggested the possibility that this bacterium originally consisted of multiple species. Accordingly, a classification scheme, based on analysis of restriction fragment length polymorphisms (RFLP) at the hypersensitive response and pathogenicity (hrp) locus and additional loci from the core genome (Cook and Sequeira 1994; Cook et al. 1989) revealed the existence of two evolutionary divisions: division I, strains mainly isolated from Asia and Australia; division II, strains mainly originating from South and Central America. The genetic classification based on geographical origin, thus correlates nicely with the biovar classification because the strains from division I match biovars 3, 4, and 5 and strains from division II match biovars 1, 2, and N2. The diversification of R. solanacearum into two major divisions later was confirmed using additional molecular criteria addressing various elements of the core genome [e.g., 16S ribosomal RNA gene [rDNA] sequences and polymerase chain reaction [PCR]-RFLP analysis of hrp genes (Poussier et al. 1999; Taghavi et al. 1996)].
Genomic fingerprint analyses using repetitive sequence-based PCR (rep-PCR) and amplified fragment length polymorphism (AFLP) are useful methods for assessing the diversity and genetic relationships among R. solanacearum strains (Fegan and Prior 2005; Horita and Tsuchiya 2001; Horita et al. 2005; Jaunet and Wang 1999; Lewis Ivey et al. 2007; Norman et al. 2009; Nouri et al. 2009; Thwaites et al. 1999; Xue et al. 2011; Yu et al. 2003). Recently, sequence information for the R. solanacearum genome has rapidly accumulated, providing an efficient tool to identify and discriminate individual strains (Castillo and Greenberg 2007; Fegan et al. 1998; Horita and Tsuchiya 2000; Lewis Ivey et al. 2007; Liu et al. 2009; Poussier et al. 2000a, b; Prior and Fegan 2005; Villa et al. 2005; Xu et al. 2009). Based on the results obtained from these analyses, Fegan and Prior (2005) defined this pathogen as a species complex and suggested a new classification system (phylotype, sequevar) according to the genomic DNA sequence information (Table 1).
Phylotype
In recent years, phylogenetic relationships of worldwide strains of R. solanacearum and related species (R. syzygii and BDB) have been assessed intensively based on their genomic DNA sequence information. Pathogenicity-related genes (hrpB, egl, peh, fliC), housekeeping genes (e.g., mutS, gyrB, gdhA), and rDNA (16S, ITS), which have been used for taxonomic analysis of bacterial species, were employed for the DNA sequence analyses (Castillo and Greenberg 2007; Fegan and Prior 2005; Fegan et al. 1998; Poussier et al. 2000a, b; Prior and Fegan 2005; Villa et al. 2005; Wicker et al. 2012). As shown in the phylogenetic analysis using nucleotide sequences of egl (Fig. 1), the results suggested that R. solanacearum strains can be divided into four major clades.

Phylogenetic relationships of Ralstonia solanacearum, R. syzygii, and blood disease bacterium of banana strains based on the partial endoglucanase gene sequences. The phylogenetic tree was constructed by the neighbor-joining method. Numbers in parentheses near the strain names indicate sequevars previously reported (Fegan and Prior 2005; Wicker et al. 2012). Values on the branches represent the results of bootstrap analysis with 1,000 iterations. The bar represents one nucleotide change per 100-nucleotide position
Fegan and Prior (2005) called these major clades phylotypes (I–IV) and suggested that each phylotype should be treated as a specific taxonomic group that corresponds almost completely to separate species or subspecies. This new classification scheme reorganized the concept of division and subdivision postulated on the basis of RFLP analysis (Cook and Sequeira 1994; Cook et al. 1989) and 16S rDNA sequence (Poussier et al. 2000a; Taghavi et al. 1996).
Each phylotype is closely related to its geographic origin (Fegan and Prior 2005). Strains in phylotype I originated in Asia, whereas strains in phylotype II are predominantly from the Americas. Phylotype III comprises strains from Africa and nearby islands, while phylotype IV contains strains isolated primarily from Indonesia and has also been found in some Asian countries (including Japan) and Australia. Phylotype IV also contains the two close relatives of R. solanacearum, P. syzygii and BDB.
Moreover, each phylotype correlated with specific races and biovars (Fegan and Prior 2005). Phylotype II contains race 2 (affecting triploid banana and Heliconia)/biovar 1 and race 3 (primarily affecting potato)/biovar 2 strains, while phylotype I includes race 4 (affecting ginger)/biovar 3, 4 and race 5 (affecting mulberry)/biovar 5 strains (Table 1).
Fegan and Prior (2005) also used a multiplex-PCR method to differentiate each phylotype based on differences in the nucleotide sequences of the rDNA-ITS region. This simple method is applicable to worldwide strains including Japanese strains. However, some primers used in the phylotype-aspecific multiplex-PCR amplify a specific band common to a related soil microorganism, R. pickettii, as well as to all strains of R. solanacearum, BDB, and P. syzygii, so that it needs to be handled carefully (Horita and Tsuchiya 2009).
Recently, Remenant et al. (2011) identified the whole genome sequence of three distinct phylotype IV strains (R. solanacearum, R. syzygii, and BDB) and compared their genomic structure with those previously reported for five R. solanacearum strains in phylotypes I, II, and III (Remenant et al. 2010). Therefore, this species complex could be divided into three genomic species. Phylotype I and III strains were differentiated from phylotype II strains (average nucleotide identity is below 92 %, less than for the same species level) (Goris et al. 2007). Phylotype IV strains consisted a single genomic species (average nucleotide identity is above 98 %, equivalent to more than 70 % DNA–DNA homology), though they showed clear phenotypic differences among the strains (Denny and Hayward 2001).
Thus, Remenant et al. (2011) suggested the revision of taxonomy of R. solanacearum species complex shown in Table 1. Except for phylotype II (which was maintained as R. solanacearum), phylotype I and III strains were assigned a new species name, R. sequeirae. Phylotype IV strains were proposed as one new species (R. haywardii) with three subspecies names (subsp. solanacearum, syzygii, and celebensis), which correspond to the former R. solanacearum, R. syzygii, and BDB, respectively (Table 1).
Sequevar
Each phylotype consists of a number of sequevars (Fegan and Prior 2005; Wicker et al. 2012). The sequevar is determined on the basis of the differences of the conserved region of endoglucanase gene (egl) sequence. Strains in the same sequevar show more than 99 % similarity in their sequences and cluster together with high confidence (>50 %) in bootstrap trials (Wicker et al. 2012). Until now, 52 sequevars have been suggested (Fegan and Prior 2005; Mahbou Somo Toukam et al. 2009; Wicker et al. 2012; Xu et al. 2009) (Table 1; Fig. 1). Some of the sequevars are correlated with specific races. For example, strains belonging to sequevar 3, 4, 6, 24 and sequevar 1 in phylotype II correspond to race 2 and race 3, respectively. Moreover, sequevar 16 and sequevar 12, 48 in phylotype I consist of race 4 and race 5 strains, respectively.
Multilocus sequence analysis/typing (MLSA/MLST)
In addition to the phylotype and sequevar, multilocus sequence analysis/typing (MLSA/MLST) has been used to discriminate and classify R. solanacearum strains (Castillo and Greenberg 2007; Lewis Ivey et al. 2007; Liu et al. 2009; Poussier et al. 2000b; Villa et al. 2005; Wicker et al. 2012), and Almeida et al. (2010) constructed an MLSA/MLST database (http://www.pamdb.org/) for this species as well as for other plant pathogenic bacteria (Pseudomonas syringae pathovars, Xanthomonas spp., and Acidovorax citrulli). As we described already, sequevar is determined on the basis of the single gene (egl) sequences, so that the presence of bias possibly appears in the analysis (Wicker et al. 2012). By targeting multilocus sequences (principally multiple housekeeping genes), strains can be distinguished from each other under universal criteria (Maiden 2006). MLSA/MLST also has been used to assess the relative contribution of mutation and recombination to the evolution of the species (Castillo and Greenberg 2007; Pérez-Losada et al. 2006; Wicker et al. 2012), and work on its systematic use for classification is ongoing.
Classification of Japanese strains of R. solanacearum based on molecular analysis
Phylotype (proposal name by Remenant et al. 2011) and sequevar of Japanese R. solanacearum strains and their correlation with host, geographic origin, race and biovar are shown in Table 2. Japanese strains can be divided into two phylotypes (I and IV) and eleven sequevars (8, 13–17, 34, 44, unknown 1–3) (Horita and Tsuchiya 2012; Waki et al. 2013). Phylotypes I and IV consist of nine (13–17, 34, 44, unknown 1–2) and two (8, unknown 3) sequevars, respectively. Strains in phylotype II or III have not been isolated so far.
Phylotype of the Japanese strain is closely related to biovar and its original host (Horita and Tsuchiya 2009). Biovar N2 strains from potato belong to phylotype IV, whereas biovar 3 and biovar 4 strains from potato and other kind of plant isolates are all included in phylotype I. Phylotype I strains have been isolated from a wide range of crops and areas where the disease occurred (from Hokkaido to Okinawa). On the other hand, phylotype IV strains have been isolated only from potato cultivation areas.
Some of the sequevars are also correlated with specific race, original host and/or geographic origin (Horita and Tsuchiya 2009, 2012; Horita et al. 2010; Waki et al. 2013). Race 3 strains, which primarily affect potato belong to two specific sequevars (8, unknown 3). Similarly, race 4 strains affecting Zingiberaceae plants (ginger, mioga, curcuma) belong to three specific sequevars (14, 16, unknown 2), and two of which (16, unknown 2) consisted of ginger and Zingiberaceae plant isolates only, respectively. On the other hand, most isolates of Solanaceae crops and other plants (except Zingiberaceae plants) belong to either sequevar 13–15, 17, 34, 44, or unknown 1 belonging to race 1, and two of these (13, 17) have been locally isolated from Okinawa and Kyushu, respectively.
Genetic, biochemical, and pathological characters of potato strains of Ralstonia solanacearum in Japan
In Japan, potato bacterial wilt has been recognized as a serious, emergent disease since World War II, and its area of occurrence is still expanding (Horita and Ooshiro 2002; Horita et al. 2010; Katayama and Kimura 1986; Narita 1958; Ooshiro 2008; Suga 2000; Suga et al. 2013). Geographic distribution and genetic, biochemical, and pathological characters of potato bacterial wilt pathogen in Japan have been widely assessed (Horita et al. 2010; Katayama and Kimura 1986; Suga 2000; Suga et al. 2013). Regarding the distribution of potato bacterial wilt, although the disease was first reported in the Hokkaido region, the northern district of Japan (Narita 1958), the potato disease has not been a serious problem there in the last 5 decades. On the contrary, Katayama and Kimura (1986) reported that potato bacterial wilt had frequently occurred in Nagasaki Prefecture, Kyushu District, since the 1960s, and they collected more than 200 strains from numerous potato-cultivation fields between 1981 and 1983. Horita et al. (2010) also surveyed and isolated about 100 strains from 25 areas in the same district between 1993 and 1999, which suggested that bacterial wilt disease had been widespread and continuous in potato-cultivating fields in such warm, temperate, southwestern regions during more than last 4 decades. Many strains of R. solanacearum from seriously damaged fields have been isolated since 1999, and recently, an outbreak occurred in Okinawa Island, the southernmost prefecture (Ooshiro 2008). On the other hand, a small number of R. solanacearum strains from potato fields in other comparatively warmer regions in Japan during specific years have also been reported.
Horita et al. (2010) determined the biovar and phylotype of the Japanese potato strains as belonging to three biovars (3, 4, N2) and two phylotypes (I and IV). Both biovar 4 (phylotype I) and biovar N2 (phylotype IV) strains were common and were widely isolated from Hokkaido to Okinawa, whereas biovar 3 (phylotype I) strains were found in limited areas (Nagasaki and Okinawa Prefectures) in Japan. Compared with the foreign potato bacterial wilt strains, biovar 2 (phylotype II) strains are predominant not only on American and European continents but also in some African and Asian countries, and most strains are thought to disseminate via infected seed potatoes worldwide (Elphinstone 2005; Hayward 1991; Hayward et al. 1998; Nouri et al. 2009; Swanson et al. 2005). Thus, the distribution of potato R. solanacearum strains in Japan seems to be quite different from those of other countries.
When the relationship between pathogenic characters and phylotypes of the potato strains in Japan was investigated (Suga et al. 2013), the tested strains were divided into 17 types based on differences in their pathogenicity to eggplant, S. integrifolium (rootstock for eggplant), tomato, and others. In particular, almost all phylotype I strains were pathogenic to eggplant, whereas all phylotype IV strains were nonpathogenic to eggplant but some of them were pathogenic to S. integrifolium. Consequently, phylotype IV strains did not belong to any pathogenic group reported by Ozaki and Kimura (1992).
Differences in pathogenic specificity to tomato cultivars have also been shown. Strains belonging to phylotype IV as well as phylotype I have affected tomato cultivation in Korea, Indonesia, and Australia (Jeong et al. 2007; Wicker et al. 2012), so we may need to pay attention to infection of tomato plants by phylotype IV strains in Japan as well.
Suga et al. (2013) also assessed pathogenic differences between potato phylotype I and IV strains against several Japanese potato varieties and breeding lines and indicated that phylotype IV strains show high virulence to the breeding lines carrying bacterial wilt resistance conferred from the wild potato Solanum phureja, which is resistant to phylotype I strains. Several wild species of potato such as S. phureja, S. stenostomum, and S. commersonii have been used as genetic resources to breed for resistance to bacterial wilt worldwide, and certain new potato varieties with a high level of resistance have been recommended (Boshou 2005). However, the high levels of resistance of these new potato varieties have been confirmed against only phylotype I or phylotype II strains. Phylotype IV strains have not been commonly used as targets for breeding of bacterial wilt resistance so far, since it has recently been categorized as one of the taxonomic groups within R. solanacearum and has been reported only from some Asian countries (Japan, Korea, the Philippines, and Indonesia) and Australia (Fegan and Prior 2005; Horita et al. 2010; Jeong et al. 2007). So further investigations using suitable R. solanacearum strains (include phylotype IV strains) are needed before practical introduction of new potato varieties to the field as well as to explore other new genetic resources for breeding with resistance to Japanese potato phylotype IV strains in the future.
From a plant quarantine point of view, phylotype II strains have not yet been reported in Japan (Horita et al. 2010) because only domestic seed potatoes have been distributed for cultivation and the import of the seed potatoes from foreign countries has been strictly prohibited by a plant protection law (Horita et al. 2010). Such a seed potato management system may have prevented the dissemination of phylotype II strains inside Japan, although both phylotypes I and IV strains are present. A supply of clean seed potato and elimination of the pathogen from infested fields are important factors for controlling the disease. Clearly then, an accurate diagnosis system against bacterial wilt pathogen needs to be developed for domestic seed potato management.
Emergence and dissemination of ginger bacterial wilt in Japan
Zingiberaceae plants (including ginger) are traditionally cultivated from rhizomes and used as condiments, for medicinal and ornamental purposes, and in food preparation in Africa, Asia, and the Americas. Bacterial wilt has affected Zingiberaceae plants, and R. solanacearum has been isolated in several countries or areas of Asia-Pacific region, including Hawaii, Mauritius, and several Asian countries (Elphinstone 2005; Hayward 1991; He 1986; Kumar et al. 2005; Lum 1973; Quinon et al. 1964; Titatarn 1986; Uematsu et al. 1983; Xu et al. 2009; Zehr 1969). In Australia, bacterial wilt of ginger has been present since 1955, but the disease has not been severe since 1970 (Hayward 1991; Hayward and Pegg 2013; Hayward et al. 1967).
In Japan, bacterial wilt of Zingiberaceae plants was first reported in cultivation fields of Curcuma alismatifolia (curcuma, introduced from Thailand as a planting material for cut flowers in 1989) at Kochi Prefecture in 1995 (Morita et al. 1996). Since 1997, outbreaks of the disease have occurred in ginger and mioga cultivation fields throughout neighboring areas of the same prefecture (Tsuchiya et al. 1999, 2005; Yano et al. 2005). Although the pathogen has been present in the prefecture for some time, since 2005, the the pathogen has been spreading to successive ginger fields prefectures such as Tochigi outside Kochi (Waki et al. 2013), such as Wakayama and Shimane Prefectures in Honshu district and in some prefectures in the Kyushu District (Tsuchiya et al. unpublished data). In most cases, the disease has emerged in an area after the introduction of infected planting materials (ginger rhizomes) originating from Kochi Prefecture where it was first introduced (Tsuchiya et al. 2005) or from other Asian countries (supplemental Fig. 1).
Causal pathogens were identified as race 4 based on their specific pathogenicity to Zingiberaceae crops (Morita et al. 1996; Tsuchiya et al. 1999, 2005; Yano et al. 2005, 2011) and biovar classification of the isolates from Kochi Prefecture revealed that all of the strains were identical to biovar 4 by comparing them with strains from Thailand and Indonesia (biovars 3 and 4) and from Australia and China (biovar 4) (Tsuchiya et al. 1999, 2005). Later, Waki et al. (2013) confirmed that the ginger strains from Tochigi Prefecture belonged to biovars 3 and 4. So far biovar 4 strains are found in these five countries, while biovar 3 strains have only been isolated from Japan, Thailand, and Indonesia. Most of the Zingiberaceae plant isolates from several other countries that were assessed with regard to their pathogenic characters were classified as biovar 3 or biovar 4 (Hayward 1994a).
All of the investigated Japanese and foreign Zingiberaceae strains belong to phylotype I (Waki et al. 2013). Genetic diversity analyzed by rep-PCR revealed that the Japanese race 4 strains are subclassified into two DNA fingerprint types; type I showed high similarity to that of Zingiberaceae strains from Thailand, and type II strains are homogeneous to ginger strains from China and Australia (Tsuchiya et al. 2005). Based on the fingerprint analysis, two PCR primer sets were designed to discriminate each strain representing type I or type II DNA fingerprints, respectively (Horita et al. 2004). The analysis revealed that strains from Thailand and Japan (Kochi, Tochigi, and some prefectures in Kyushu district) were included in type I, whereas strains from China, Australia, and some strains from Japan (Kochi and Kagoshima prefectures) were included in type II.
Phylogenetic analysis based on the egl sequences, Zingiberaceae plant isolates in phylotype I were further divided into several major groups each corresponding closely to six sequevars (14,16–18, 47, and unknown 2 in Tables 1 and 2) (Waki et al. 2013). These groups were closely correlated with the host species and/or geographic origin. Most of the Zingiberaceae isolates of R. solanacearum from Japan were genetically homogeneous to the Zingiberaceae strains from Thailand (represent type I DNA fingerprints and correspond to a specific sequevar, unknown 2) and the ginger strains from China and Australia (representing type II DNA fingerprints and sequevar 16). A few exceptional Japanese ginger strains are included in sequevar 14 with the Chinese and the Philippine ginger strains. Thus, the pathogen originated from different Asian countries and disseminated by infested seed rhizomes, thereby dispersing the pathogen within Japan and causing disease epidemics through separate routes.
Pathogenic characters of some Japanese strains from Zingiberaceae plants were assessed after root-injuring inoculation (Tsuchiya et al. 2005; Yano et al. 2011). Zingiberaceae strains (ginger, mioga, and curcuma isolates) representing type I DNA fingerprints were strongly pathogenic to ginger, mioga, and curcuma. By contrast, the rest of the ginger strains representing type II fingerprints were strongly pathogenic to ginger, but weakly pathogenic or nonpathogenic to mioga and curcuma. These findings indicate that differences exist between type I and type II strains in terms of host range.
In cross-inoculation tests with Japanese strains of R. solanacearum, isolates from plants other than Zingiberaceae plant were not pathogenic to ginger; however, the Zingiberaceae plant isolates in type I were strongly virulent to potato, but weakly virulent or avirulent on other Solanaceae crops (e.g., tomato, tobacco, eggplant, pepper) and peanuts (Yano et al. 2011). Some Thai ginger strains in type I had similar pathogenic characteristics (Titatarn 1986; Uematsu et al. 1983). On the other hand, the pathogenic characteristics of the Japanese strains in type II were the same as those of the ginger strains from China and Australia (both are in the same DNA type II), which were strongly pathogenic to most Solanaceae crops, but weakly virulent or avirulent on tobacco and peanuts (Hayward et al. 1967; He 1986; Pegg and Moffett 1971). These results thus suggested that the DNA-based groupings of the strains are closely correlated with the pathogenic characteristics such as host range and virulence on specific plants). However, further studies on pathogenicity tests would also be desirable on the strains in other genetic groups (sequevars).
As described, the bacterial wilt pathogens affecting Zingiberaceae plants can be divided into several genetically distant groups, that differ in terms of geographic origin, pathogenicity (host range, virulence on specific plants), and biovar, for example, suggesting that some strains might have disseminated locally and globally via transplanting material. Efficient countermeasures must therefore be independently considered and implemented according to the local situation (i.e., whether the pathogen is established, economic importance and degree of damage to the host crop, pathogenic character of the pathogen). Newly introduced (or imported) seed rhizomes should be subject to a quarantine check, and the host cultivation history of the rhizome-producing area (i.e., whether bacterial wilt disease has previously been recorded) would be investigated as in the case of potato.
Conclusion
A DNA-based classification system of R. solanacearum species complex has several advantages. (1) It can clarify the phylogenetic relationship among the strains and can differentiate the strains more precisely and exactly than traditional conventional methods (pathogenicity and biochemical tests). (2) Many strains that differed in host, geographic origins and phenotypic characters, some of which were difficult to collect and test in the same laboratory, can be compared and discriminated at one time under common criteria using the DNA sequences deposited in a public database. (3) It is basically compatible with traditional classification system (e.g., worldwide race 3/biovar 2 strains is equivalent to phylotype II/sequevar 1). Thus, this system (phylotype and sequevar) has been well received and become popular worldwide.
However, the system does have some critical problems. (1) Phylotype and sequevar are informal classifications under species and subspecies level as well as former infrasubspecific system (race and biovar) (as mentioned, each phylotype may be treated as one formal taxonomic group in the near future). (2) As more strains are analyzed, novel and unique DNA sequences might be found that do not belong to any designated sequevar (Hong et al. 2012; Horita and Tsuchiya 2012; Waki et al. 2013). However, the basic criteria to establish a new sequevar are not clearly evident based on previous reports. (3) The correlation of the sequevars with other phenotypic characters (e.g., host range, virulence, biovar) have not been assessed precisely yet except for some special cases. Further systematic analysis regarding these points is needed.
Regarding the proposed names of R. sequierae for phylotype I and phylotype III and R. haywardii for phylotype IV by Remenant et al. (2011) as mentioned earlier, these names should remain as proposals at present, even though a paper using those names has already been published but without proper validation required by the International Code of Nomenclature of Bacteria (Lapage et al. 1992) or proposed in the indispensable International Journal of Systematic and Evolutionary Microbiology (IJSEM). Nor have the phylotypes/new species yet been examined for their correlation to bacteriological/phenotypic properties in the usual phenotypical classification system.
For newly emergent diseases such as ginger bacterial wilt in Japan, the movement of infected, generally asymptomatic, planting material represents the most significant route by which the disease spreads globally and domestically. Seed and plants for planting are thus generally rigorously inspected to ensure that material is pathogen-free. Methods range from visual inspection of material to sampling backed by with laboratory diagnosis or the imposition of post-entry quarantine measures to ensure freedom from the pathogen and disease.
References
Almeida NF, Yan S, Cai R, Clarke CR, Morris CE, Schaad NW, Schuenzel EL, Lacy GH, Sun X, Jones JB, Castillo JA, Bull CT, Leman S, Guttman DS, Setubal JC, Vinatzer BA (2010) PAMDB, a multilocus sequence typing and analysis database and website for plant-associated microbes. Phytopathology 100:208–215
Boshou L (2005) A broad review and perspective on breeding for resistance to bacterial wilt. In: Allen C, Prior P, Hayward AC (eds) Bacterial wilt disease and the Ralstonia solanacearum species complex. APS Press, St. Paul, pp 225–238
Buddenhagen I, Kelman A (1964) Biological and physiological aspects of bacterial wilt caused by Pseudomonas solanacearum. Annu Rev Phytopathol 2:203–230
Castillo JA, Greenberg JT (2007) Evolutionary dynamics of Ralstonia solanacearum. Appl Environ Microbiol 73:1225–1238
Cook D, Sequeira L (1994) Strain differentiation of Pseudomonas solanacearum by molecular genetic methods. In: Hayward AC, Hartman GL (eds) Bacterial wilt: the disease and its causative agent, Pseudomonas solanacearum. CAB International, Wallingford, pp 77–93
Cook D, Barlow E, Sequeira L (1989) Genetic diversity of Pseudomonas solanacearum: detection of restriction fragment length polymorphisms with DNA probes that specify virulence and the hypersensitive response. Mol Plant Microbe Interact 2:113–121
Denny TP, Hayward AC (2001) Gram-negative bacteria: Ralstonia. In: Schaad NW, Jones JB, Chun W (eds) Laboratory guide for identification of plant pathogenic bacteria, 3rd edn. APS Press, St. Paul, pp 151–174
Eden-Green SJ, Sastraatmadja H (1990) Blood disease of banana present in Java. FAO Plant Protect Bull 38:49–50
Elphinstone JG (2005) The current bacterial wilt situation: a global overview. In: Allen C, Prior P, Hayward AC (eds) Bacterial wilt disease and the Ralstonia solanacearum species complex. APS Press, St. Paul, pp 9–28
Fegan M, Prior P (2005) How complex is the “Ralstonia solanacearum species complex”? In: Allen C, Prior P, Hayward AC (eds) Bacterial wilt disease and the Ralstonia solanacearum species complex. APS Press, St. Paul, pp 449–461
Fegan M, Taghavi M, Sly LI, Hayward AC (1998) Phylogeny, diversity and molecular diagnostics of Ralstonia solanacearum. In: Prior P, Allen C, Elphinstone J (eds) Bacterial wilt disease: molecular and ecological aspects. Springer, Berlin, pp 19–33
Gillings MR, Fahy P (1993) Genetic diversity of Pseudomonas solanacearum biovar 2 and N2 assessed using restriction endonuclease analysis of total genomic DNA. Plant Pathol 42:744–753
Gillings MR, Fahy P (1994) Genomic fingerprinting: towards a united view of the Pseudomonas solanacearum species complex. In: Hayward AC, Hartman GL (eds) Bacterial wilt: The disease and its causative agent, Pseudomonas solanacearum. CAB International, Wallingford, pp 95–112
Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P, Tiedje JM (2007) DNA–DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol 57:81–91
Hayward AC (1964) Characteristics of Pseudomonas solanacearum. J Appl Bacteriol 27:265–277
Hayward AC (1991) Biology and epidemiology of bacterial wilt caused by Pseudomonas solanacearum. Annu Rev Phytopathol 29:65–87
Hayward AC (1994a) The hosts of Pseudomonas solanacearum. In: Hayward AC, Hartman GL (eds) Bacterial wilt: the disease and its causative agent, Pseudomonas solanacearum. CAB International, Wallingford, pp 9–24
Hayward AC (1994b) Systematics and phylogeny of Pseudomonas solanacearum and related bacteria. In: Hayward AC, Hartman GL (eds) Bacterial wilt: the disease and its causative agent, Pseudomonas solanacearum. CAB International, Wallingford, pp 123–135
Hayward AC, Pegg KG (2013) Bacterial wilt of ginger in Queensland: reappraisal of a disease outbreak. Aust Plant Pathol 42:235–239
Hayward AC, Moffet ML, Pegg KG (1967) Bacterial wilt of ginger in Queensland. Qld J Agric Anim Sci 24:1–5
Hayward AC, Elphinstone JG, Caffier D, Janse J, Stefani E, French ER, Wright AJ (1998) Round table on bacterial wilt (brown rot) of potato. In: Prior P, Allen C, Elphinstone J (eds) Bacterial wilt disease: molecular and ecological aspects. Springer, Berlin, pp 420–430
He LY (1986) Bacterial wilt in the People’s Republic of China. In: Persley GJ (ed) Bacterial wilt disease in Asia and the South Pacific. Australian Centre for International Agricultural Research (ACIAR) Proc No. 13, Canberra, pp 40–48
He LY, Sequeira L, Kelman A (1983) Characteristics of strains of Pseudomonas solanacearum from China. Plant Dis 67:1357–1361
Hong JC, Norman DJ, Reed DL, Momol MT, Jones JB (2012) Diversity among Ralstonia solanacearum strains isolated from the southeastern US. Phytopathology 102:924–936
Horita M, Ooshiro A (2002) Genetic diversity of Ralstonia solanacearum strains isolated from potato and balsam pear in Okinawa Island. Bact Wilt Newsl 17:23
Horita M, Tsuchiya K (2000) Comparative analysis of Japanese and foreign strains of Ralstonia solanacearum based on 16S ribosomal RNA gene sequences. J Gen Plant Pathol 66:132–137
Horita M, Tsuchiya K (2001) Genetic diversity of Japanese strains of Ralstonia solanacearum. Phytopathology 91:399–407
Horita M, Tsuchiya K (2009) Current status and future prospects of the classification system for the bacterial wilt pathogen Ralstonia solanacearum species complex (in Japanese). Jpn J Phytopathol 75:297–306
Horita M, Tsuchiya K (2012) MAFF microorganism genetic resource manual no. 12 (ver. 2) Ralstonia solanacearum (in Japanese). National Institute of Agrobiological Sciences, Tsukuba, pp 1–32
Horita M, Yano K, Tsuchiya K (2004) PCR-based specific detection of Ralstonia solanacearum race 4 strains. J Gen Plant Pathol 70:278–283
Horita M, Tsuchiya K, Ooshiro A (2005) Characteristics of Ralstonia solanacearum biovar N2 strains in Asia. J Phytopathol 153:209–213
Horita M, Suga Y, Ooshiro A, Tsuchiya K (2010) Analysis of genetic and biological characters of Japanese potato strains of Ralstonia solanacearum. J Gen Plant Pathol 76:196–207
Jaunet TX, Wang JF (1999) Variation in genotype and aggressiveness of Ralstonia solanacearum race 1 isolated from tomato in Taiwan. Phytopathology 89:320–327
Jeong Y, Kim J, Kang Y, Lee S, Hwang I (2007) Genetic diversity and distribution of Korean isolates of Ralstonia solanacearum. Plant Dis 91:1277–1287
Katayama K, Kimura S (1986) Ecology and protection of bacterial wilt of potato: 1. Ecology and strains of Pseudomonas solanacearum (in Japanese with English summary). Bull Nagasaki Agric Forest Exp Stn 14:1–30
Kumar A, Anandaraj M, Sarma YR (2005) Rhizome solarization and microwave treatment: ecofriendly methods for disinfecting ginger seed rhizomes. In: Allen C, Prior P, Hayward AC (eds) Bacterial wilt disease and the Ralstonia solanacearum species complex. APS Press, St. Paul, pp 185–195
Lapage SP, Sneath PHA, Lessel EF, Skerman VBD, Seeliger HPR, Clark WA (eds) (1992) International code of nomenclature of bacteria. ASM Press, Washington (DC)
Lewis Ivey ML, McSpadden Gardener BB, Opina N, Miller SA (2007) Diversity of Ralstonia solanacearum infecting eggplant in the Philippines. Phytopathology 97:1467–1475
Liu Y, Kanda A, Yano K, Kiba A, Hikichi Y, Aino M, Kawaguchi A, Mizoguchi S, Nakaho K, Shiomi H, Takikawa Y, Ohnishi K (2009) Molecular typing of Japanese strains of Ralstonia solanacearum in relation to the ability to induce a hypersensitive reaction in tobacco. J Gen Plant Pathol 75:369–380
Lum KY (1973) Cross inoculation studies of Pseudomonas solanacearum from ginger. MARDI Res Bull 1:15–21
Mahbou Somo Toukam G, Cellier G, Wicker E, Guilhaud C, Kahane R, Allen C, Prior P (2009) Broad diversity of Ralstonia solanacearum strains in Cameroon. Plant Dis 93:1123–1130
Maiden MC (2006) Multilocus sequence typing of bacteria. Annu Rev Microbiol 60:561–588
Morita Y, Yano K, Tsuchiya K, Kawada Y (1996) Bacterial wilt of Curcuma alismatifolia caused by Pseudomonas solanacearum (in Japanese). Proc Assoc Pl Protec Shikoku 31:1–6
Narita T (1958) Studies on the bacterial diseases of potato in Hokkaido (in Japanese with English summary). Rep Hokkaido Pref Agric Exp Stn 8:1–80
Norman DJ, Zapata M, Gabriel DW, Duan YP, Yuen JMF, Mangravita-Novo A, Donahoo RS (2009) Genetic diversity and host range variation of Ralstonia solanacearum strains entering North America. Phytopathology 99:1070–1077
Nouri S, Bahar M, Fegan M (2009) Diversity of Ralstonia solanacearum causing potato bacterial wilt in Iran and the first record of phylotype II/biovar 2T strains outside South America. Plant Pathol 58:243–249
Okabe N (1965) Strains of Pseudomonas solanacearum E. F. Smith (in Japanese). Ann Phytopath Soc Japan 31:152–158
Ooshiro A (2008) Control of bacterial wilt by Geranium carolinianum L. (in Japanese). Plant Protect 62:90–95
Ozaki K, Kimura T (1992) Grouping of Pseudomonas solanacearum on the basis of pathogenicity to Solanum plants (in Japanese). Bull Chugoku Natl Agric Exp Stn 10:49–58
Palleroni NJ, Doudoroff M (1971) Phenotypic characterization and deoxyribonucleic acid homologies of Pseudomonas solanacearum. J Bacteriol 107:690–696
Pegg KG, Moffett ML (1971) Host range of the ginger strain of Pseudomonas solanacearum in Queensland. Austral J Exp Agric Anim Hus 11:696–698
Pérez-Losada M, Browne EB, Madsen A, Wirth T, Viscidi RP, Crandall KA (2006) Population genetics of microbial pathogens estimated from multilocus sequence typing (MLST) data. Infect Genet Evol 6:97–112
Poussier S, Vandewalle P, Luisetti J (1999) Genetic diversity of African and worldwide strains of Ralstonia solanacearum as determined by PCR-restriction fragment length polymorphism analysis of the hrp gene region. Appl Environ Microbiol 65:2184–2194
Poussier S, Prior P, Luisetti J, Hayward C, Fegan M (2000a) Partial sequencing of the hrpB and endoglucanase genes confirms and expands the known diversity within the Ralstonia solanacearum species complex. Syst Appl Microbiol 23:479–486
Poussier S, Trigalet-Demery D, Vandewalle P, Goffinet B, Luisetti J, Trigalet A (2000b) Genetic diversity of Ralstonia solanacearum as assessed by PCR-RFLP of the hrp gene region, AFLP and 16S rRNA sequence analysis, and identification of an African subdivision. Microbiology 146:1679–1692
Prior P, Fegan M (2005) Recent developments in the phylogeny and classification of Ralstonia solanacearum. Acta Hortic 695:127–136
Quinon VL, Aragaki M, Ishii M (1964) Pathogenicity and serological relationship of three strains of Pseudomonas solanacearum in Hawaii. Phytopathology 54:1096–1099
Remenant B, Coupat-Goutaland B, Guidot A, Cellier G, Wicker E, Allen C, Fegan M, Pruvost O, Elbaz M, Calteau A, Salvignol G, Mornico D, Mangenot S, Barbe V, Medigue C, Prior P (2010) Genomes of three tomato pathogens within the Ralstonia solanacearum species complex reveal significant evolutionary divergence. BMC Genom 11:379
Remenant B, de Cambiaire J-C, Cellier G, Jacobs JM, Mangenot S, Barbe V, Lajus A, Vallenet D, Medigue C, Fegan M, Allen C, Prior P (2011) Ralstonia syzygii, the blood disease bacterium and some Asian R. solanacearum strains form a single genomic species despite divergent lifestyles. PLoS One 6:e24356
Schaad NW, Frederick RD, Shaw J, Schneider WL, Hickson R, Petrillo MD, Luster DG (2003) Advances in molecular-based diagnostics in meeting crop biosecurity and phytosanitary issues. Annu Rev Phytopathol 41:305–324
Suga Y (2000) Ecology and control of bacterial wilt of potato (in Japanese). Phytopathol Soc Jpn Soilborne Dis Workshop Rep 20:174–179
Suga Y, Horita M, Umekita M, Furuya N, Tsuchiya K (2013) Pathogenic characters of Japanese potato strains of Ralstonia solanacearum. J Gen Plant Pathol 79:110–114
Swanson JK, Yao J, Tans-Kersten J, Allen C (2005) Behavior of Ralstonia solanacearum race 3 biovar 2 during latent and active infection of geranium. Phytopathology 95:136–143
Taghavi M, Hayward C, Sly LI, Fegan M (1996) Analysis of the phylogenetic relationships of strains of Burkholderia solanacearum, Pseudomonas syzygii, and the Blood disease bacterium of banana based on 16S rRNA sequences. Int J Syst Bacteriol 46:10–15
Thwaites R, Mansfield J, Eden-Green S, Seal S (1999) RAPD and rep PCR-based fingerprinting of vascular bacterial pathogens of Musa spp. Plant Pathol 48:121–128
Titatarn V (1986) Bacterial wilt in Thailand. In: Persley GJ (ed) Bacterial wilt disease in Asia and the South Pacific. Australian Centre for International Agricultural Research (ACIAR) Proc No. 13, Canberra, pp 65–67
Tsuchiya K (2008) Occurrence and spread of bacterial wilt diseases of Zingiberaceae plants caused by foreign strains (in Japanese). Plant Protect 62:72–75
Tsuchiya K, Yano K, Horita M, Morita Y, Kawada Y, d’Ursel CM (1999) Occurrence of bacterial wilt of ginger in Japan (abstract in Japanese). Ann Phytopathol Soc Jpn 65:363
Tsuchiya K, Yano K, Horita M, Morita Y, Kawada Y, d’Ursel CM (2005) Occurrence and epidemic adaptation of new strains of Ralstonia solanacearum associated with Zingiberaceae plants under agro-ecosystem in Japan. In: Allen C, Prior P, Hayward AC (eds) Bacterial wilt disease and the Ralstonia solanacearum species complex. APS Press, St. Paul, pp 463–469
Uematsu T, Chutenchitt S, Karnjanarat S, Vivithajinda S, Nabheerong N, Benjathikul S, Nilmanee S, Dhirabhava W, Buangsuwon D (1983) Bacterial diseases on economic crops in Thailand. Tropical Agricultural Research Center, Ministry of Agriculture, Forestry, and Fisheries. Japan and Department of Agriculture, Ministry of Agriculture and Cooperatives, Thailand, pp 1–266
Vaneechoutte M, Kämpfer P, De Baere T, Falsen E, Verschraegen G et al (2004) Wautersia gen. nov., a novel genus accommodating the phylogenetic lineage including Ralstonia eutropha and related species, and proposal of Ralstonia [Pseudomonas] syzygii (Roberts et al. 1990) comb. nov. Int J Syst Evol Microbiol 54:317–327
Villa JE, Tsuchiya K, Horita M, Natural M, Opina N, Hyakumachi M (2005) Phylogenetic relationships of Ralstonia solanacearum species complex strains from Asia and other continents based on 16S rDNA, endoglucanase, and hrpB sequences. J Gen Plant Pathol 71:39–46
Waki T, Horita M, Kurose D, Mulya K, Tsuchiya K (2013) Genetic diversity of Zingiberaceae plant isolates of Ralstonia solanacearum in the Asia-Pacific Region. JARQ 47:283–294
Wicker E, Lefeuvre P, de Cambiaire J-C, Lemaire C, Poussier S, Prior P (2012) Contrasting recombination patterns and demographic histories of the plant pathogen Ralstonia solanacearum inferred from MLSA. ISME J 6:961–974
Xu J, Pan ZC, Prior P, Xu JS, Zhang Z, Zhang H, Zhang LQ, He LY, Feng J (2009) Genetic diversity of Ralstonia solanacearum strains from China. Eur J Plant Pathol 125:641–653
Xue Q-Y, Yin Y-N, Yang W, Heuer H, Prior P, Guo J-H, Smalla K (2011) Genetic diversity of Ralstonia solanacearum strains from China assessed by PCR-based fingerprints to unravel host plant- and site-dependent distribution patterns. FEMS Microbiol Ecol 75:507–519
Yabuuchi E, Kosako Y, Yano I, Hotta H, Nishiuchi Y (1996) Transfer of two Burkholderia and an Alcaligenes species to Ralstonia gen. nov.: proposal of Ralstonia pickettii (Ralston, Palleroni and Doudoroff 1973) comb. nov., Ralstonia solanacearum (Smith 1896) comb. nov. and Ralstonia eutropha (Davis 1969) comb. nov. Microbiol Immunol 39:897–904
Yano K, Kawada Y, Tsuchiya K, Horita M (2005) First report of bacterial wilt of mioga (Zingiber mioga) caused by Ralstonia solanacearum in Japan (in Japanese with English summary). Jpn J Phytopathol 71:179–182
Yano K, Kawada Y, Horita M, Hikichi Y, Tsuchiya K (2011) Phylogenetic discrimination and host ranges of Ralstonia solanacearum isolates from Zingiberaceae plants (in Japanese with English summary). Jpn J Phytopathol 77:88–95
Yu Q, Alvarez AM, Moore PH, Zee F, Kim MS, de Silva A, Hepperly PR, Ming R (2003) Molecular diversity of Ralstonia solanacearum isolated from ginger in Hawaii. Phytopathology 93:1124–1130
Zehr EI (1969) Bacterial wilt of ginger in the Philippines. Philipp Agricst 53:224–227
Author information
Authors and Affiliations
Corresponding author
Additional information
M. Horita and K. Tsuchiya have contributed equally to preparation of the manuscript.
Electronic supplementary material
Below is the link to the electronic supplementary material.
10327_2014_537_MOESM1_ESM.ppt
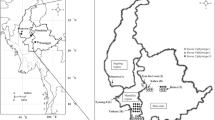
Distribution and presumable invasion routes of bacterial wilt pathogen of Zingiberaceae plants in Japan. Yellow and blue arrowheads indicate presumable invasion routes of the pathogen representing DNA type I (yellow circle) and type II (blue circle) fingerprints for strains suggested to originate from Thailand and China, respectively (Tsuchiya 2008; Tsuchiya et al. 2005). These strains were first disseminated to Kochi Prefecture through latently infected seed rhizomes, and the first outbreak of the disease occurred in a curcuma field in 1995 (type I) and subsequently in a ginger field in 1997 (type II). Thereafter, the disease has been found continuously in several prefectures since 2005. The yellow dotted arrowheads indicate suspected routes of spread. The number in parentheses near the prefecture name indicates the year when the disease was first found there. (PPT 176 kb)
Rights and permissions
About this article

Cite this article
Horita, M., Tsuchiya, K., Suga, Y. et al. Current classification of Ralstonia solanacearum and genetic diversity of the strains in Japan. J Gen Plant Pathol 80, 455–465 (2014). https://doi.org/10.1007/s10327-014-0537-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10327-014-0537-z