
Abstract
CBH1 (cellobiohydrolase) comprises the majority of secreted proteins by Trichoderma reesei. For expression of Talaromyces thermophilus lipase gene in T. reesei, a self-designed CBH1 promoter was applied to drive the lipase gene expression cassette which was bracketed by flanking sequences of cbh1 gene for homologous recombination. Protoplast and Agrobacterium-mediated plasmid transformations were performed and compared, resultantly, transformation mediated by Agrobacterium was overall proved to be more efficient. Stable integration of lipase gene into chromosomal DNA of T. reesei transformants was verified by PCR. After shaking flask fermentation, lipase activity of transformant reached 375 IU mL−1, whereas no cellobiohydrolase activity was detected. SDS-PAGE analysis further showed an obvious protein band about 39 kDa and no CBH1 band in fermentation broth, implying lipase gene was successfully extracellularly expressed in T. reesei via homologous recombination at cbh1 locus. This study herein would benefit genetic engineering of filamentous fungi and industrial application of thermo-alkaline lipase like in paper making and detergents addition.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
With more than half a century development, Trichoderma reesei, the sexual anamorph of the well-characterized Hypocrea jecorina, is hitherto the workhorse for biomass conversion industry [9, 43]. Due to the excellent capacity of protein expression, proper post-translational modifications and simplified downstream processing, it would continue to play an irreplaceable role in producing cellulolytic enzymes in the foreseeable future [40].
Compared with outstanding homologous protein secretion in T. reesei [2], heterologous gene expression in this fungus is less than satisfactory [32]. To increase the productivity of heterologous proteins, researches may be taken out from these two aspects, suppression of its own protein expression especially the main secreted component CBH1 (accounting for more than 50% of all proteins secreted) on one hand, and optimization of its secretion pathway for better extracellular transportation using genetic engineering on the other [13]. For downregulation of cellulolytic enzymes or research of key genes involved in protein secretion, RNA technologies such as antisense RNA, RNA interference and hammerhead ribozymes had been proved to be valuable tools in T. reesei [11]. Additionally, homologous recombination (HR) seems to be a more attractive option for T. reesei bioengineering and is frequently applied for enhancing enzyme production [34, 40]. However, HR is greatly hampered by its low transformation frequencies despite vast efforts have been made to create NHEJ (nonhomologous end joining pathways) pathway defective strains [28] or development of an efficient marker recycling system [10].
With the aim to increase transformation efficiency and pave the way for gene engineering in T. reesei, the prerequisite is establishing an efficient and convenient transformation platform [21]. There are chemical, physical and biological strategies for T. reesei transformation, namely the protoplast-mediated transformation (PMT), Agrobacterium tumefaciens-mediated transformation (ATMT), electroporation (EP) and biolistic transformation (BT) [24]. PMT is frequently considered for its handy procedure, needlessness of special equipment [12], and multi-copies of gene transformation [21], while, low transformation rate and some uncertain conditions like concentrations of PEG and protoplasts impede its application [44]. Standing out from its counterparts, the decisive advantage of ATMT is the high efficiency of transferring multitudinous genes randomly and mainly in single copy into genomes of numerous materials including mycelium, protoplasts, germling and spores [41]. Furthermore, ATMT could benefit homologous recombination [25] and genetic analysis of disrupted sequences [26]. Nevertheless, measures should be further taken to optimize and compare the various parameters during PMT and ATMT so as to make themselves better qualified.
Thermo-alkaline lipase from Talaromyces thermophilus showed great potential in detergents addition, paper making and biodiesel conversion, whereas it was constrained by the low protein expression capacity of the fungus itself [37, 38]. In this work, we set up a homologous integration platform for expressing this industrially important lipase gene in T. reesei. Vectors containing a self-designed cbh1 promoter were constructed for ensuring homologous recombination at cbh1 locus. PMT and ATMT methodologies were studied and compared with the aim to provide competent transformation protocols for T. reesei gene engineering. T. thermophilus lipase gene was then successfully expressed in recombinant T. reesei via this expression system, aiming to boost the application of thermo-alkaline lipase.
Materials and methods
Reagents and equipment
All the enzymes used in this research and PCR-related reagents and markers were purchased from TaKaRa Biotech Corporation in China. Antibiotics and various kits were from Sangon Biotech Corporation in China. All the chemicals applied here including PEG 6000 were bought from China National Pharmaceutical Group Corporation.
Experiments were carried out based on the platform set up in Biomass Chemical Engineering of Ministry of Education, Zhejiang University [6, 44]. PCR cycler was from Hangzhou LongGene Corporation and the gel imaging and analysis system was purchased from Shanghai Peiqing Science & Technology Co., Ltd.
Strains and media
Four strains of A. tumefaciens (AGL1, EHA105, LBA4404 and GV3101) were used for plasmid transformation by ATMT. T. reesei ZU-02 was used for preparing genome DNA and making recipients. All strains of A. tumefaciens and T. reesei ZU-02 were obtained from the strain collection of lab of Biomass Chemical Engineering of Ministry of Education, Zhejiang University [8, 14].
Medium for PMT and ATMT were based on Wang et al. [44] and Gu et al. [8], respectively. Seed and fermentation medium for lipase production were the same as described by Zhang et al. [46].
Plasmid construction
The terminator (0.6 kb) and promoter with the secretion sequence (1.4 kb) of cbh1 were cloned from genome of T. reesei using primers T1, T2 and P1, P2 (Table 1), respectively. The promoter sequence from −805 to −606 was then synthesized and quadruply repeated. An optimized T. thermophilus lipase gene (TTL, primer L1 and L2) was ligated between the promoter and terminator to construct the expression cassette. Flanking sequences of cbh1 were further cloned (using primer LA1, LA2 and RA1, RA2) and constructed into the both end of expression cassette for assurance of homologous recombination (Fig. 1).

Construction of recombinant plasmid pCB-hER. L, left flanking sequence of cbh1 gene; R, right flanking sequence of cbh1 gene; P, newly designed CBH1 promoter with quadruple repeats; S, signal sequence of CBH1 promoter; T, terminator of CBH1 promoter; H, hygromycin-resistant maker driven by PtrpC gene promoter; TTL, Talaromyces thermophilus lipase gene. Other abbreviations are inherent elements of pCAMBIA1300. Specific loci on scaffold 29 and dashed arrows are PCR primer binding sites for cloning
The hygromycin B-resistant maker driven by PtrpC gene promoter was amplified from pDESTR (GenBank AB218275.1) with primers H1 and H2 (Table 1).
Taking the binary vector pCAMBIA1300 as backbone, the expression cassette with homologous arms and hygromycin-resistant marker was afterwards applied to construct the final vector pCB-hER with restriction enzymes BamHI and SpeI, BstXI and XhoI, respectively (Fig. 1).
Protoplast-mediated transformation
For making protoplasts, fresh T. reesei spores were inoculated in 50 mL of YPD for 4–12-h germination at 200 rpm, 30 °C. These germinated spores were further collected and washed with 0.9% NaCl for one time and PM (10 mM sodium phosphate buffer, pH 5.8; 1.2 M magnesium sulfate) for two times. PM with 10 mg mL−1 snailase was then employed for partially lysing the spores for 0.5–1.5 h at 30 °C to release protoplasts, which were further collected by centrifugation and resuspended in 200 μL of TS (10 mM Tris–HCl, pH 7.5; 1 M sorbitol). This protoplast suspension was then centrifuged and washed once with TSC (10 mM Tris–HCl, pH 7.5; 1 M sorbitol; 20 mM CaCl2). The protoplast was finally resuspended in TSC with supplementing 10% of the final volume of TPC (10 mM Tris–HCl, pH 7.5; 1 M sorbitol; 20 mM CaCl2; 60% polyethylene glycol 6000) at the concentration from 107 to 109 protoplasts mL−1.
100 μL of protoplast suspension was added to 2 μg of plasmid pCB-hER before their incubation at 0 °C for 20 min. 500 μL TPC was then supplemented into this protoplast–plasmid mixture with maintaining at 25 °C for 20 min. This mixture was further blended with 600 μL of TSC and finally sprayed onto PDASH (PDA; 1 M sorbitol; 100 μg mL−1 hygromycin B) for regeneration and formation of transformants by keeping at 30 °C.
Agrobacterium-mediated transformation
Culturing of A. tumefaciens and transferring plasmid pCB-hER into various A. tumefaciens species were based on previous publications [14, 24]. When OD660 of recombinant A. tumefaciens reached 0.8, it was blended with 3-h pre-germinated T. reesei spores (107 spores mL−1) [46]. 100 μL of A. tumefaciens–T. reesei mixture was sprayed onto the nitrocellulose filter of induction medium with varied acetosyringone (0–250 μM) for a 36–48-h cultivation at pH 5.3 and 24 °C. The filter was then tiled reversely on PDA for producing transformant at 30 °C, where 150 μg mL−1 hygromycin B and 200 μg mL−1 cefotaxime were added for selection.
Screening of transformants
Promising transformants that formed on antibiotic PDA plates of PMT and ATMT were picked out by cutters (diameter: 5 mm), and subcultured on PDA with 200 μg mL−1 hygromycin B for two rounds. Afterwards, transformants were selected based on their colony diameters as further lipase-producing candidates.
Detection of lipase gene in recombinant T. reesei
To evaluate mitotic stability, T. reesei transformants were subcultured on PDA without hygromycin for five generations. The genomes of each monoconidia of transformants and original T. reesei were prepared via the method of CTAB [14]. Taking these genomes as templates, PCR using primer L1, L2 and C1, C2 was implemented to verify the existence of lipase and part of cbh1 gene in genomes. The PCR program was as follows: 94 °C 10 min; 94 °C × 50 s, 66 °C × 45 s, 72 °C × 1 min (30 circles); 72 °C 10 min. Amplified PCR products were determined with agarose gel electrophoresis.
Lipase production
Lipase production was carried out according to Zhang et al. [46]. 250-mL Erlenmeyer flask with 50 mL of fermentation medium was used, and the inoculum ratio was 10% (v/v). Fermentation was implemented at 30 °C, 180 rpm for 1 week. All the experiments were triply repeated.
Assay methods and data analysis
For transformants diameters, colonies which were grown on antibiotic PDA for 3 days were measured from two different directions, and the diameters equaled the average number.
Transformation efficiency meant the number of transformants attained per μg DNA in PMT and per 107 conidia in ATMT, respectively. In transformation optimization, relative transformation efficiency was calculated, and the transformation efficiency attained at the optimal conditions was taken as 100%. Experiments were carried out in triplicate and mean values were figured out.
Cellobiohydrolase activity was assayed according to Fang et al. [6]. One unit of cellobiohydrolase activity was defined as the amount of enzyme required for generating 1 mg of reducing sugars in 1 h. For determination of lipase activity, titration of olive oil emulsion was applied [46], and one unit of lipase activity was defined as the amount of lipase required for releasing 1 μmol of free fatty acids per minute. Assays for cellobiohydrolase and lipase activity were replicated three times.
For enzyme concentration, Bradford method was applied and it was detected as described in Zhang et al. [46].
Sodium dodecyl sulphate–polyacrylamide gel electrophoresis (SDS-PAGE) was used for protein separation. The fermentation broth of transformants was first centrifuged and 8% polyacrylamide was applied. The electrophoresis was then performed at 10 mA for 50 min and 20 mA for 120 min. After staining the gel with 0.27% Coomassie Brilliant Blue, it would be detained overnight using acid–methanol–water (10:20:70, v/v/v) and photographed for analysis.
Results and discussion
Construction of plasmid pCB-hER
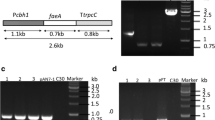
An efficient and convenient expression platform is the cornerstone for genetic engineering of T. reesei [45], which seems to be more urgent in the era of functional genomics since sequencing of T. reesei genome [20]. To increase the promotion effect of CBH1 promoter, upstream sequence (from −805 to −606) of this promoter which contained numerous binding sites for cis-transcription factors like Hap2/3/5, XYRI and ACEII [19] was quadruply repeated in the plasmid pCB-hER. Lipase gene was ligated between the CBH1 promoter (with signal sequence) and its terminator to construct the expression cassette (Fig. 1). This cassette was bracketed by the 5′ and 3′ flanking sequences of cbh1 gene, which were about 1.5 kb and could enable homologous integration of this expression cassette into genome of T. reesei [22]. PCR verifications of the homologous arms and the expression cassette with flanking sequences (6.0 kb, primer EA1 and EA2) were shown in Fig. 2.

PCR verification of homologous arms (a) and the expression cassette with flanking sequences (b). a Primer LA1, LA2 and RA1, RA2 were used to amplify the homologous arms from T. reesei genome. Lane M, DNA molecular mass maker; Lane 1, 5′ flanking sequence of cbh1; Lane 2, 3′ flanking sequence of cbh1. b Lane M, the DNA molecular mass maker; Lane 1, the lipase gene expression cassette with flanking sequences amplified by primer EA1 and EA2
Approaches from transcriptional level should be foremost considered for genetic manipulation of T. reesei [21]. With strong promotion of the self-designed promoter and screening easement by the hygromycin-resistant marker, this plasmid favoring homologous recombination would be widely adopted for gene expression in T. reesei.
Protoplast-mediated transformation
Due to recalcitrance of the sick cell wall, there is no general rule for implementing efficient transformation in filamentous fungi of interest [21]. Because of the handy protocol and needlessness of special equipment, PMT is one of the most common strategies for T. reesei transformation [28, 30], during which protoplasts making is of great importance. To ensure single genome transformation, T. reesei spores were separated from multicellular hyphae through a pre-germination treatment. We could see from Fig. 3 that 8 h was the optimal incubation time endowing the best growth phase for cells. With time increase, the efficiency decreased due to the bad effect of hyphae concentrating on following enzyme lysis.

Effects of spore pre-germination (black square), enzymolysis (black circle) and protoplast concentration (filled triangle) on efficiency of PMT. Fresh T. reesei spores were collected for pre-germination (4–12 h) and followed by 0.5–1.5-h lysis via 10 mg mL−1 snailase before making protoplasts. TSC with supplementing 10% of the final volume of TPC was used to suspend the protoplasts at the concentration from 107 to 109 protoplasts mL−1. These protoplasts were used for genetic transformation. The efficiency of optimal conditions was taken as 100%. Bars indicate standard deviations of three replicates
On account of the differing cell wall components and varied lytic effectiveness [27], it is critical to study the enzyme treatment of spores. Germinated spores were lysed by 10 mg mL−1 snailase in this research, and the best lytic time was 1 h (Fig. 3). This time doubled that used in lysing brewer’s yeast by 1% snailase, suggesting the unneglectable effect of cell type on lysis [5]. Longer treatment by snailase would destroy the cell wall structure and further decrease transformation rate. Concentration of protoplast was further investigated here, with the result that 5 × 108 protoplasts mL−1 (Fig. 3) was far more preferable than others. Under the optimal conditions, 105 transformants per μg DNA in PMT could be attained, and the average colony diameter could reach 45 mm after grown on antibiotic PDA for 3 days.
Since the fragility of protoplasts contributed to high mortality, protoplast making and enzyme lysis had been proved to be key factors in PMT in many papers [39, 41]. Compared with other expensive lysing enzymes [18], the effectiveness of 1-h snailase enzymolysis after spore pre-germination in YPD for 8 h was comparable and more promising in PMT.
Agrobacterium tumefaciens-mediated transformation
Constrained by the complex protoplast making processes in PMT, ATMT is increasingly preferred for its high efficiency and simplicity [36]. However, transferring T-DNA bracketing the target gene and selectable maker remains troublesome [42], especially the co-cultivation of bacteria and fungi [24]. It was found that the optimal OD600 of Agrobacterium, co-culture pH and temperature during ATMT were 0.8, 5.3 and 24 °C, respectively [46]. For decreasing spores cell recalcitrance could boost efficiency, pre-germination of spores would be remarkably beneficial in ATMT [1].
Due to the different Ti plasmid types, Agrobacterium species affect the transformation a lot [3]. Agrobacterium AGL-1 was found to be the most proper strain for ATMT of T. reesei, followed by EHA105 and LBA4404, and GV3101 could lead to nearly no positive transformants (Fig. 4a). This is in consistency with the result of Park [29] and Krishnamohan et al. [15], indicating the importance of vir gene expression on ATMT [15].

Effects of Agrobacterium species (a) and acetosyringone concentration (b) on efficiency of ATMT. After being transformed with recombinant plasmids, Agrobacterium was mixed with T. reesei spores (107 spores mL−1). This mixture was further cultured on nitrocellulose filter of induction medium (IM) supplemented with 0–250 µM Acetosyringone at pH 5.3 and 24 °C. The efficiency of AGL-1 was taken as 100%. Bars indicate standard deviations of three replicates
As the inducer of vir gene expression, acetosyringone represented phenolic compounds are indispensable in ATMT [4]. In AGl-1 transformation, results indicated that 200 μM was the optimal acetosyringone concentration (Fig. 4b), within which 3400 transformants per 107 conidia could be obtained and the colony diameter reached 55 mm after grown on antibiotic PDA for 3 days. The acetosyringone usage was much lower than that of Leclerque et al. [16], suggesting that varied Agrobacterium species may change the acetosyringone requirement. A similar result was reported by Chen et al. [4] in Nicotiana tabacum cv. SR1 transformation, and they pointed out that improper acetosyringone concentration would decrease the efficiency.
Comparison of protoplast and Agrobacterium-mediated transformation
Though PMT and ATMT have their own strengths and drawbacks [41], and individual studies occasionally puzzle us when making decisions [23], both are versatile strategies for genetic engineering of fungi and their applications are increasing [27].
It could be seen from Table 2 that the efficiency and operation cycle of ATMT are much more advantageous than PMT, this may due to the complex process but low generation rate of protoplasts during PMT [23]. Ruiz-Dı´ez [39] also suggested that not all protoplasts were competent for taking up exogenous DNA. Additionally, the average colony diameters of ATMT transformants were larger than those of PMT, which laid the foundation of higher lipase expression capacity [6] (Table 2). There was nearly no risk of losing heterologous lipase gene in ATMT transformants, nevertheless, about 14% of PMT transformants may lose that after subculture for five generations (Table 2).
Differences of PMT and ATMT may come from the varied mechanisms of DNA transferring [21], and conclusions may be drawn that ATMT was overall superior to PMT in transformation of heterologous genes into T. reesei genome.
Verification of lipase gene in recombinant T. reesei transformants
Recombinant T. reesei transformants were subcultured on PDA without hygromycin for five generations to assay the mitotic stability, for that some genetic traits such as antibiotic resistance may be lost during subculture [14]. Contrast to the original T. reesei ZU-02 (Fig. 5a), a band about 0.8 kb that corresponded with size of TTL (Fig. 5b) was detected in all genomes of transformants; however, the cbh1 gene was lost (Fig. 5a), meaning that heterologous lipase gene had been integrated into the cbh1 locus and could be stably inherited by recombinant T. reesei transformants.

PCR verification of TTL gene (a, b) and SDS-PAGE analysis (c) of recombinant and original T. reesei. a Lane M, DNA molecular marker; Lane 1–2, using genomic DNA of PMT transformants as templates with primer L1, L2 and C1, C2; Lane 3–4, using genomic DNA of ATMT transformants as templates with primer L1, L2 and C1, C2; Lane 5, using genomic DNA of T. reesei ZU-02 as template with primer L1, L2 and C1, C2. b Lane M, DNA molecular marker; Lane 1, using plasmid pCB-hER as templates with primer L1, L2. c Lane M, the protein molecular mass marker; Lane 1, supernatant of original T. reesei ZU-02 fermentation; Lane 2, lipase from the fermentation broth of transformants HT3
Lipase production by recombinant T. reesei
Lipase production by T. reesei transformants was performed at lab scale to validate the applicability of this expression system. It could be seen from Fig. 6 that pH of fermentation broth decreased to 4.3 in the first 24 h, and then went up to 5.5 afterwards. This obvious two-stage growth had been previously reported by Giese [7] and Li et al. [17], indicating the importance of preliminary mycelia growth for protein expression [31, 46].

Fermentation course of original and recombinant T. reesei HT3. Lipase production was carried out at lab scale using 250-mL Erlenmeyer flasks. Lipase activity (black square), pH (black diamond) and CBH1 activity (white circle) in the medium were measured every 24 h. Bars indicate standard deviations of three replicates
Lipase activity of ATMT transformant HT3 increased drastically during the first 3 d, and then gradually rose to 375 IU mL−1 after 120 h (Fig. 6). Lipase concentration simultaneously reached 50.6 mg L−1 in culture supernatant, where no cellobiohydrolase activity could be detected. By contrast, there was no lipase activity in broth of original T. reesei ZU-02, while its cellobiohydrolase activity increased quickly to 23.5 IU mL−1 after fermentation for 144 h (Fig. 6). Lipase activity attained here was 1.55-fold higher than that produced by T. reesei from random integration [46], 6.25 times the level secreted by T. thermophilus itself [37] or without RNAi-mediated gene silencing in Qin et al. [33], and was about 88 times as high as basic lipase production by T. reesei itself in submerge fermentation using olive oil as inducer [35].
In coincidence with lipase assay, there was a clear band about 39 kDa in broth of T. reesei HT3, the molecular mass expected for T. thermophilus lipase, and no band corresponding to CBH1 could be detected. On the contrary, original ZU-02 could secrete the 65-kDa CBH1 and there was no TTL band in its culture supernatant (Fig. 5c).
Results argue that lipase gene had been stably integrated into the cbh1 locus of T. reesei, leading to increased lipase expression level and declined CBH1 production. Hence, the expression system set up in this research was efficient, convenient and reproducible, and it could be exploited to develop expression platforms for other fungi of interest, which may greatly benefit further genetic manipulations of filamentous fungi.
Conclusions
In conclusion, T. thermophilus lipase gene was successfully expressed in recombinant T. reesei by homologous recombination. The plasmid containing flanking sequences of cbh1 was constructed, of which the expression cassette was driven by a self-designed CBH1 promoter. Multiple key factors of PMT and ATMT were studied and compared so as to develop an efficient and convenient transformation protocol. Lipase gene was further extracellularly expressed in recombinant T. reesei by means of this expression system, promoting the gene engineering of T. reesei and application of this industrially important lipase.
References
Abuodeh RO, Orbach MJ, Mandel MA, Das A, Galgiani JN (2000) Genetic transformation of Coccidioides immitis facilitated by Agrobacterium tumefaciens. J Infect Dis 181:2106–2110
Amore A, Faraco V (2012) Potential of fungi as category I Consolidated BioProcessing organisms for cellulosic ethanol production. Renew Sustain Energy Rev 16:3286–3301. doi:10.1016/j.rser.2012.02.050
Campoy S, Pérez F, Martín JF, Gutiérrez S, Liras P (2003) Stable transformants of the azaphilone pigment-producing Monascus purpureus obtained by protoplast transformation and Agrobacterium-mediated DNA transfer. Curr Genet 43:447–452. doi:10.1007/s00294-003-0417-0
Chen C-F, Chan K-G, Tan B-C, Khalid N (2015) Enhancement of Agrobacterium-mediated transformation efficiency of model plant using quorum sensing molecule, N-3-oxo-octanoyl-l-homoserine-lactone. Plant Cell Tissue Organ Cult (PCTOC) 121:481–487. doi:10.1007/s11240-015-0718-2
Ding WJ, Qian QF, Hou XL, Feng WY, Chen CY, Chai ZF, Zhang BR, Wang K (2002) A preliminary study of chromium distribution in chromium-rich brewer’s yeast cell by NAA. Biol Trace Elem Res 88:193–199. doi:10.1385/BTER:88:2:193
Fang H, Xia L (2013) High activity cellulase production by recombinant Trichoderma reesei ZU-02 with the enhanced cellobiohydrolase production. Bioresour Technol 144:693–697. doi:10.1016/j.biortech.2013.06.120
Giese H, Kruithof P, Meier K, Sieben M, Antonov E, Hommes RWJ, Büchs J (2014) Improvement and scale-down of a Trichoderma reesei shake flask protocol to microtiter plates enables high-throughput screening. J Biosci Bioeng 118:702–709. doi:10.1016/j.jbiosc.2014.05.016
Gu B, Xia L (2013) High expression of a neutral endo-β-glucanase gene from Humicola insolens in Trichoderma reesei. J Ind Microbiol Biotechnol 40:773–779. doi:10.1007/s10295-013-1267-5
Gusakov AV (2011) Alternatives to Trichoderma reesei in biofuel production. Trends Biotechnol 29:419–425. doi:10.1016/j.tibtech.2011.04.004
Hartl L, Seiboth B (2005) Sequential gene deletions in Hypocrea jecorina using a single blaster cassette. Curr Genet 48:204–211. doi:10.1007/s00294-005-0011-8
He R, Guo W, Wang L, Zhang D (2015) Construction of an efficient RNAi system in the cellulolytic fungus Trichoderma reesei. J Microbiol Methods 108:70–73. doi:10.1016/j.mimet.2014.11.010
He R, Ma L, Li C, Jia W, Li D, Zhang D, Chen S (2014) Trpac1, a pH response transcription regulator, is involved in cellulase gene expression in Trichoderma reesei. Enzyme Microbial Technol 67:17–26. doi:10.1016/j.enzmictec.2014.08.013
Iwashita K (2002) Recent studies of protein secretion by filamentous fungi. J Biosci Bioeng 94:530–535. doi:10.1016/S1389-1723(02)80191-8
Jin X, Xia L (2011) Heterologous expression of an endo-β-1,4-glucanase gene from the anaerobic fungus Orpinomyces PC-2 in Trichoderma reesei. World J Microbiol Biotechnol 27:2913–2920. doi:10.1007/s11274-011-0774-7
Krishnamohan A, Balaji V, Veluthambi K (2001) Efficient vir gene induction in Agrobacterium tumefaciens requires virA, virG, and vir box from the same Ti plasmid. J Bacteriol 183:4079–4089
Leclerque A, Wan H, Abschütz A, Chen S, Mitina GV, Zimmermann G, Schairer HU (2003) Agrobacterium-mediated insertional mutagenesis (AIM) of the entomopathogenic fungus Beauveria bassiana. Curr Genet 45:111–119. doi:10.1007/s00294-003-0468-2
Li C, Yang Z, He Can Zhang R, Zhang D, Chen S, Ma L (2013) Effect of pH on cellulase production and morphology of Trichoderma reesei and the application in cellulosic material hydrolysis. J Biotechnol 168:470–477. doi:10.1016/j.jbiotec.2013.10.003
Li J, Wang J, Wang S, Xing M, Yu S, Liu G (2012) Achieving efficient protein expression in Trichoderma reesei by using strong constitutive promoters. Microb Cell Fact 11:1–10. doi:10.1186/1475-2859-11-84
Mach R, Zeilinger S (2003) Regulation of gene expression in industrial fungi: trichoderma. Appl Microbiol Biotechnol 60:515–522. doi:10.1007/s00253-002-1162-x
Martinez D, Berka RM, Henrissat B, Saloheimo M, Arvas M, Baker SE, Chapman J, Chertkov O, Coutinho PM, Cullen D, Danchin EGJ, Grigoriev IV, Harris P, Jackson M, Kubicek CP, Han CS, Ho I, Larrondo LF, de Leon AL, Magnuson JK, Merino S, Misra M, Nelson B, Putnam N, Robbertse B, Salamov AA, Schmoll M, Terry A, Thayer N, Westerholm-Parvinen A, Schoch CL, Yao J, Barabote R, Nelson MA, Detter C, Bruce D, Kuske CR, Xie G, Richardson P, Rokhsar DS, Lucas SM, Rubin EM, Dunn-Coleman N, Ward M, Brettin TS (2008) Genome sequencing and analysis of the biomass-degrading fungus Trichoderma reesei (syn. Hypocrea jecorina). Nat Biotech 26:553–560. http://www.nature.com/nbt/journal/v26/n5/suppinfo/nbt1403_S1.html
Meyer V (2008) Genetic engineering of filamentous fungi—progress, obstacles and future trends. Biotechnol Adv 26:177–185. doi:10.1016/j.biotechadv.2007.12.001
Meyer V, Arentshorst M, El-Ghezal A, Drews A-C, Kooistra R, van den Hondel CAMJJ, Ram AFJ (2007) Highly efficient gene targeting in the Aspergillus niger kusA mutant. J Biotechnol 128:770–775. doi:10.1016/j.jbiotec.2006.12.021
Meyer V, Mueller D, Strowig T, Stahl U (2003) Comparison of different transformation methods for Aspergillus giganteus. Curr Genet 43:371–377. doi:10.1007/s00294-003-0406-3
Michielse C, Hooykaas PJ, van den Hondel CMJJ, Ram AJ (2005) Agrobacterium-mediated transformation as a tool for functional genomics in fungi. Curr Genet 48:1–17. doi:10.1007/s00294-005-0578-0
Michielse CB, Arentshorst M, Ram AFJ, van den Hondel CAMJJ (2005) Agrobacterium-mediated transformation leads to improved gene replacement efficiency in Aspergillus awamori. Fungal Genet Biol 42:9–19. doi:10.1016/j.fgb.2004.06.009
Mullins ED, Chen X, Romaine P, Raina R, Geiser DM, Kang S (2001) Agrobacterium-mediated transformation of Fusarium oxysporum: an efficient tool for insertional mutagenesis and gene transfer. Phytopathology 91:173–180. doi:10.1094/PHYTO.2001.91.2.173
Nevalainen KMH, Te’o VSJ, Bergquist PL (2005) Heterologous protein expression in filamentous fungi. Trends Biotechnol 23:468–474. doi:10.1016/j.tibtech.2005.06.002
Ouedraogo JP, Arentshorst M, Nikolaev I, Barends S, Ram AFJ (2015) I-SceI-mediated double-strand DNA breaks stimulate efficient gene targeting in the industrial fungus Trichoderma reesei. Appl Microbiol Biotechnol 99:10083–10095. doi:10.1007/s00253-015-6829-1
Park S-M, Kim D-H (2004) Transformation of a filamentous fungus Cryphonectria parasitica using Agrobacterium tumefaciens. Biotechnol Bioprocess Eng 9:217–222. doi:10.1007/BF02942296
Penttila M, Nevalainen H, Ratto M, Salminen E, Knowles J (1987) A versatile transformation system for the cellulolytic filamentous fungus Trichoderma reesei. Gene 61:155–164. doi:10.1016/0378-1119(87)90110-7
Prasetyo J, Sumita S, Okuda N, Park E (2010) Response of cellulase activity in pH-controlled cultures of the Filamentous fungus Acremonium cellulolyticus. Appl Biochem Biotechnol 162:52–61. doi:10.1007/s12010-009-8826-2
Punt PJ, van Biezen N, Conesa A, Albers A, Mangnus J, van den Hondel C (2002) Filamentous fungi as cell factories for heterologous protein production. Trends Biotechnol 20:200–206. doi:10.1016/S0167-7799(02)01933-9
Qin L-N, Cai F-R, Dong X-R, Huang Z-B, Tao Y, Huang J-Z, Dong Z-Y (2012) Improved production of heterologous lipase in Trichoderma reesei by RNAi mediated gene silencing of an endogenic highly expressed gene. Bioresour Technol 109:116–122. doi:10.1016/j.biortech.2012.01.013
Rahman Z, Shida Y, Furukawa T, Suzuki Y, Okada H, Ogasawara W, Morikawa Y (2009) Application of Trichoderma reesei cellulase and xylanase promoters through homologous recombination for enhanced production of extracellular β-glucosidase I. Biosci Biotechnol Biochem 73:1083–1089. doi:10.1271/bbb.80852
Rajesh EM, Arthe R, Rajendran R, Balakumar C, Pradeepa N, Anitha S (2010) Investigation of lipase production by Trichoderma reesei and optimization of production parameters. Electron J Environ Agric Food Chem 9:1177–1189
Richardson T, Thistleton J, Higgins TJ, Howitt C, Ayliffe M (2014) Efficient Agrobacterium transformation of elite wheat germplasm without selection. Plant Cell. Tissue Organ Cult (PCTOC) 119:647–659. doi:10.1007/s11240-014-0564-7
Romdhane IB-B, Fendri A, Gargouri Y, Gargouri A, Belghith H (2010) A novel thermoactive and alkaline lipase from Talaromyces thermophilus fungus for use in laundry detergents. Biochem Eng J 53:112–120. doi:10.1016/j.bej.2010.10.002
Romdhane IB-B, Romdhane ZB, Gargouri A, Belghith H (2011) Esterification activity and stability of Talaromyces thermophilus lipase immobilized onto chitosan. J Mol Catal B Enzym 68:230–239. doi:10.1016/j.molcatb.2010.11.010
Ruiz-Díez B (2002) Strategies for the transformation of filamentous fungi. J Appl Microbiol 92:189–195
Singh A, Taylor Ii LE, Vander Wall TA, Linger J, Himmel ME, Podkaminer K, Adney WS, Decker SR (2015) Heterologous protein expression in Hypocrea jecorina: a historical perspective and new developments. Biotechnol Adv 33:142–154. doi:10.1016/j.biotechadv.2014.11.009
Su X, Schmitz G, Zhang M, Mackie RI, Cann IKO (2012) Heterologous gene expression in filamentous fungi. Adv Appl Microbiol 81:1–61. doi:10.1016/b978-0-12-394382-8.00001-0
Sugui JA, Chang YC, Kwon-Chung KJ (2005) Agrobacterium tumefaciens-mediated transformation of Aspergillus fumigatus: an efficient tool for insertional mutagenesis and targeted gene disruption. Appl Environ Microbiol 71:1798–1802
Sun A, Peterson R, Te’o J, Nevalainen H (2016) Expression of the mammalian peptide hormone obestatin in Trichoderma reesei. New Biotechnol 33:99–106. doi:10.1016/j.nbt.2015.08.004
Wang B, Xia L (2011) High efficient expression of cellobiase gene from Aspergillus niger in the cells of Trichoderma reesei. Bioresour Technol 102:4568–4572. doi:10.1016/j.biortech.2010.12.099
Weld RJ, Plummer KM, Carpenter MA, Ridgway HJ (2006) Approaches to functional genomics in filamentous fungi. Cell Res 16:31–44
Zhang X, Li X, Xia L (2015) Heterologous expression of an alkali and thermotolerant lipase from Talaromyces thermophilus in Trichoderma reesei. Appl Biochem Biotechnol 176:1722–1735. doi:10.1007/s12010-015-1673-4
Acknowledgements
This work was supported by the National High-tech R&D Program (2007AA05Z401) and the Program for Zhejiang Leading Team of S&T Innovation (2011R50002) of China.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Zhang, X., Xia, L. Expression of Talaromyces thermophilus lipase gene in Trichoderma reesei by homologous recombination at the cbh1 locus. J Ind Microbiol Biotechnol 44, 377–385 (2017). https://doi.org/10.1007/s10295-016-1897-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-016-1897-5