
Abstract
In Hypocrea jecorina (anamorph: Trichoderma reesei) multiple gene deletions are limited by the number of readily available selection markers. We have therefore constructed a blaster cassette which enables successive gene knock-outs in H. jecorina. This 3.5 kb pyr4 blaster cassette contains the H. jecorina pyr4 marker gene encoding orotidine-5′-monophosphate (OMP) decarboxylase flanked by two direct repeats of the Streptoalloteichus hindustanus bleomycin gene (Sh ble), which facilitate the excision of the blaster cassette by homologous recombination after each round of deletion. Functionality of this pyr4 blaster cassette was demonstrated by deletion of the glk1 encoding glucokinase and hxk1 encoding hexokinase. 1.4–1.8 kb of the non-coding flanking regions of both target genes were cloned into the respective blaster cassettes and transformation of a pyr4 negative H. jecorina strain with the two cassettes resulted in 10–13% of the transformants in the deletion of one of the two kinase genes. For excision of the pyr4 blaster cassettes, Δglk1 strains were selected for growth in the presence of 5-fluoroorotic acid. Recombination between the two Sh ble elements resulted in uridine auxotrophic strains which retained their respective glucokinase negative phenotype. Subsequent transformation of one of these auxotrophic Δglk1 strains with the hexokinase blaster cassette resulted in pyr4 prototrophic strains deleted in both glk1 and hxk1. Δglk1 strains showed reduced growth on d-glucose and d-fructose whereas Δhxkl strains showed reduced compact growth on d-glucose but were unable to grow on d-fructose as carbon source. The double Δglk1Δhxk1 deletion strain was completely unable to grow on either d-glucose or d-fructose.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The ascomycete Hypocrea jecorina (anamorph Trichoderma reesei) is industrially applied for the production of enzymes including a number of (hemi)cellulases and its strong cellobiohydrolase promoters are used for the expression of recombinant proteins (Penttilä 1998). Although a sexual cycle of H. jecorina has been described (Kuhls et al. 1996), most of the research and all of the industrial application are performed almost exclusively with a single asexual isolate H. jecorina QM6a from the Solomon Islands and its derivatives (Kubicek and Harman 1998). Functional genomic studies in H. jecorina depend on an efficient targeted gene manipulation system and the construction of defined mutants for the investigation of gene function. DNA mediated transformation in H. jecorina is integrative and relies on a limited number of dominant markers and auxotrophic markers. These include the Escherichia coli hph (hygromycin B phosphotransferase), the E. coli and Strepotalloteichus hindustanus ble (bleo/phleomycin resistance), the Aspergillus nidulans amdS (acetamidase) or the H. jecorina pyr4 (for a review see Mach 2004).
Traditional DNA mediated transformations are limited in terms of the number of marker genes which can be inserted. This fact restricts studies of e.g. the function of orthologous and paralogous genes or of whole gene families. Therefore the development of a versatile transformation system independent on the number of available markers would be beneficial. In yeasts, so called blaster cassettes were developed which allow the repeated use of the URA3 (the yeast pyr4 homologue) marker to construct multiple disrupted strains (Alani et al. 1987; Fonzi and Irwin 1993). Such blaster cassettes consist of the URA3 encoding the orotidine-5′-decarboxylase flanked by two direct repeats. Mutants which are defective in URA3 are auxotrophic for uridine (uracil), but are—in contrast to prototrophic strains—resistant to 5-fluoroorotic acid (5-FOA; Boeke et al. 1984), which is converted by orotidine-5′-monophosphate (OMP)-decarboxylase to the toxic intermediate 5-fluoro-UMP. Integration of the blaster is therefore selected via Ura3 function and excision of the URA3 marker is then forced in the presence of 5-FOA by recombination between the two direct repeats. As a consequence, this blaster cassette can be reused for successive rounds of gene deletions, allowing multiple deletions with a single cassette. This cassette has permitted successive disruption of C. albicans alleles (reviewed in Pla et al. 1996) and even families of genes (Mio et al. 1996; Muhlschlegel and Fonzi 1997; Sanglard et al. 1997) with a single auxotrophic marker. In filamentous fungi, a similar blaster cassette was successfully applied for the deletion of rodA in the opportunistic pathogen A. fumigatus (d’Enfert 1996) and aroC in A. nidulans (Krappmann and Braus 2003).
We developed a blaster cassette for multiple gene deletions in H. jecorina based on the H. jecorina pyr4 flanked by direct repeats of the S. hindustanus ble. The functionality of the blaster cassette for successive gene deletion is demonstrated by the construction of stable H. jecorina strains deleted in the gluco- or hexokinase encoding genes and the reuse of the pyr4 blaster to construct double knock-out strains.
Materials and methods
Strains and culture conditions
Hypocrea jecorina strain QM9414 (ATCC 26921) and its uridine auxotrophic pyr4 mutant TU-6 (ATCC MYA-256) (Gruber et al. 1990b) were maintained on malt extract agar (Merck, VWR International, Austria) or potato dextrose agar (Difco, BD Biosciences, Schwechat, Austria) supplemented with 10 mM uridine when necessary. Fungal cultures were grown at 30°C in a medium described by (Mandels and Andreotti 1978). Fungal growth on different carbon sources was determined by placing a small piece of agar (d=0.5 cm) in the centre of each agar plate. Escherichia coli strain JM109 (Promega, Madison, WI,USA.) was used for plasmid propagation.
Identification and sequence analysis of the H. jecorina glk1 (encoding glucokinase) and hxk1 (encoding hexokinase)
A tblastn search of the T. reesei/H. jecorina QM6a genome sequence (http://gsphere.lanl.gov/trire1/trire1.home.html) with the Aspergillus niger glucokinase (GenBank accession no. CAA67949) and hexokinase (GenBank accession no. CAA08922) proteins as query identified single orthologues for each gene. The deduced aa sequence of the H. jecorina glucokinase encoding gene (glk1) and the hexokinase encoding gene (hxk1) showed 58% sequence identity to the A. niger GlkA, and 73% to the A. niger HxkA, respectively. The two kinase genes were amplified by PCR with oligonucleotide pair Gluco5′F and Gluco3′R, as Hexo5′F as Hexo3′R respectively from H. jecorina QM9414 genomic DNA and sequenced (Table 1).
Plasmid constructions
The pyr4 blaster cassette was constructed by inserting the H. jecorina pyr4 (Gruber et al. 1990a) gene between two S. hindustanus Sh ble fragments orientated as direct repeats. Therefore the Sh ble was amplified twice from the plasmid pPICZB (Invitrogen, Vienna, Austria) using two primer pairs and introducing the following restriction sites (given in brackets): zeo1fw (XbaI) and zeo1rv (XhoI), respectively zeo2fw (XhoI) and zeo2rv (BamHI). The resulting 380 bp amplicons were digested with XbaI/XhoI and BamHI/XhoI respectively and ligated into an XbaI/BamHI digested pUC19 (Yanisch-Perron et al. 1985). The resulting vector containing the two Sh ble gene fragments as direct repeat was digested with XhoI to insert a 2.7 kb SalI H. jecorina pyr4 fragment resulting in the 6.2 kb blaster plasmid pLH1mb.
About 1.4 kb of the 5′ and 3′ region of glk1 were amplified using the primer pairs which introduced the following restriction sites: gluco5′F (EcoRI) and gluco5′R (BamHI), gluco3′F (XbaI) and gluco3′R (XbaI and EcoRI). The EcoRI/BamHI restricted 5′ region fragment was ligated into the EcoRI/BamHI sites of pLH1mb. Next, the XbaI restricted 3′ region of glk1 was inserted into the XbaI site resulting in the final vector pΔglk1.
About 1.4 kb of the 5′ and 1.8 kb of the 3′ region of hxk1 were amplified using the primers hexo5′F (EcoRI) and hexo5′R (BamHI), respectively hexo3′F (HindIII) and hexo3′R (HindIII plus a natural EcoRI site). The EcoRI/BamHI digested 5′ region was ligated into the respective sites in pLH1mb following the insertion of the 3′ region of glk1 into the HindIII site resulting in pΔhxk1.
Transformation of H. jecorina
Protoplast preparation and DNA mediated transformation was described by (Gruber et al. 1990b). For deletion of the glk1 and hxk1 the blaster cassettes (about 6.2 and 6.6 kb) were (1) excised from pΔglk1 and pΔhxk1 with EcoRI. Fragments were purified from agarose gels (QIAquick Gel Extraction Kit, VWR International, Vienna, Austria). After transformation protoplasts were stabilized and regenerated on minimal medium plates containing d-sorbitol (1 M). d-glucose as carbon source was replaced by glycerol or l-arabinose to prevent a negative selection for strains deleted in one of the kinase genes. After 4–5 days colonies were transferred to minimal medium without d-sorbitol for sporulation. Conidia were usually obtained after 3–4 days and purified on minimal medium plates containing the colony restrictor Triton X-100 (0.1% v/v) and peptone (0.1% w/v) which accelerates germination. After 1.5 days single colonies were picked and transferred to minimal medium for sporulation.
Excision of the pyr4 blaster cassette
Two to three day old spores were suspended in 0.9% (w/v) NaCl and 0.05% (w/v) Tween 80, filtered through glass wool to remove residual hyphae. 0.9×107 –1.5×107 conidia were plated on minimal medium plates containing 5-FOA (1.5 g/l; Fermentas, St. Leon-Rot, Germany), peptone (0.1 g/l) and 10 mM uridine. 5-FOA resistant colonies were obtained after 3–4 days and transferred to minimal medium containing uridine for sporulation. Purified conidia were then tested for uridine auxotrophy on minimal medium plates.
Fungal DNA isolation and hybridization
DNA was prepared from H. jecorina strains grown for about 24–30 h in 100 ml flasks on a rotary shaker (250 rpm) at 30°C. Mycelia were harvested by filtration, washed with cold sterile tap water, blotted dry between paper towels, and ground to a fine powder under liquid nitrogen. Powdered mycelia was suspended in buffer A (0.1 M Tris–HCl, pH 8.0, 1.2 M NaCl, 5 mM EDTA), incubated for 20 min at 65°C, cooled down on ice, mixed with 0.5 v phenol and 0.5 v chloroform and centrifuged (12,000 rpm, 15 min). Following a chloroform (1 v) extraction, DNA was precipitated with 1 v of isopropanol and washed with 70% (v/v) ethanol. Standard methods (Sambrook and Russel 2001) were used for DNA electrophoresis, blotting, and hybridization of DNA. Probes labelled with [α32 P]dCTP by random priming were: a 1.4 kb XbaI glk1, a 1.8 kb HindIII hxk1, a 2.7 kb Sal pyr4 fragment and a 380 bp Sh ble amplicon.
Results
Construction of a pyr4 blaster cassette for sequential targeted gene deletions
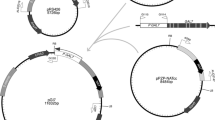
A blaster cassette containing the H. jecorina pyr4 gene flanked by two gene fragments of the S. hindustanus Sh ble was constructed (Fig. 1). The two Sh ble elements were orientated as direct repeats to facilitate the excision of the pyr4 marker by loop-out between the homologous regions after a successful gene deletion. On each side of the blaster cassette unique restriction sites (EcoRI, BamHI, XbaI, SalI and HindIII) were located to enable the insertion of the up- and downstream regions of the target genes. Starting from this pyr4 blaster cassette we constructed two different blaster cassettes for deletion of the H. jecorina glucokinase (glk1) and hexokinase (hxk1) encoding genes by amplification of their noncoding regions by PCR. Appropriate restriction sites were introduced at the end of each fragment by PCR to facilitate the ligation of these fragments into the pyr4 blaster cassette. The final glucokinase blaster pΔglk1 contained 1.4 kb of each flanking regions of the glk1 coding region, while the hexokinase blaster pΔhxk1 contained 1.4 kb of the up- and 1.8 kb of the downstream region of the hxk1 coding region. In addition, the final blaster cassettes for the two genes were constructed in such a way that they could easily be excised by a single EcoRI digest. Alternatively, the respective cassette can be amplified by PCR using the primer pair located in the up- and downstream regions.

Schematic representation of the pyr4 blaster cassette pLH1mb and gene replacement at the H. jecorina glk1 and hxk1 loci. pLH1mb contains the H. jecorina pyr4 gene flanked by two direct repeats of a S. hindustanus ble fragment. Orientation of the different genes is indicated by arrows. Important restriction enzyme sites which are useful for cloning of the up and downstream regions of the target genes into pLH1mb, for the release of the blaster cassettes from the vector or for the Southern analyses are also indicated. Positions of the probes for glk1 and hxk1 are indicated
Deletion of glk1 encoding glucokinase and hxk1 encoding hexokinase in H. jecorina
The functionality of the blaster approach for H. jecorina was tested by deletion of two genes encoding hexose phosphorylating enzymes in the uridine auxotrophic pyr4 negative strain TU-6. Although A. nidulans strains lacking hexokinase or glucokinase grew well on d-glucose containing media (Flipphi et al. 2003), we replaced d-glucose in the protoplast regeneration plates by either glycerol or l-arabinose to avoid any possible negative selection for homologous integrated blaster cassettes. Glycerol was chosen because it is channelled into glycolysis after the hexose phosphorylation steps while the pentose l-arabinose is catabolized by a path not involving glycolytic enzymes (Chiang and Knight 1961). H. jecorina TU-6 was transformed with the two blaster fragments and the resulting transformants were selected for uridine prototrophy on minimal medium. Purified transformants were tested for growth on a number of carbon sources including d-glucose, d-fructose, glycerol, and l-arabinose to select for putative gene knock-outs. Southern analysis confirmed the glk1 or hxk1 deletions (Fig. 2a, b). Thirteen percent of the total number of glucokinase and 10% of the hexokinase transformants showed a deletion at the respective gene locus. Hybridization with the coding region of the respective genes confirmed their complete removal. Growth tests on a number of carbon sources showed that Δglk1 strains showed reduced growth on d-glucose and d-fructose, whereas Δhxk1 strains showed reduced compact growth on d-glucose and were unable to grow on d-fructose (Fig. 3). However, growth of both deletion strains was also affected on glycerol or l-arabinose indicating a pleiotropic effect resulting from these deletions.

Southern analyses of H. jecorina gluco- and hexokinase negative strains. The endogenous non-functional copy of the pyr4 is marked by an arrow.a Genomic DNA of the parental strain TU-6, a Δhxk1 and a Δglk1Δhxk1 strain were digested with ClaI and probed with hxk1, pyr4 and Sh ble fragments. Insertion of the blaster cassette at the hxk1 locus leads to an increase of the hybridizing band from 2.7 kb in strain TU-6 to 8.7 kb in the Δhxk1 and Δglk1Δhxk1 using the hxk1 fragment as probe. This 8.7 kb band is also detected with the pyr4 or Sh bl fragments as probe. In the Δglk1Δhxk1 strain an additional weaker hybridizing fragment is found with the Sh ble fragment as probe which corresponds to a single Sh ble fragment resulting from the excision of the glucokinase blaster cassette. b Genomic DNA of strain TU-6, a Δglk1, a glucokinase blaster excised strain Δglk1-ex and a Δglk1Δhxk1 strain were digested with XmnI and probed with the respective fragments. Homologous insertion of the glucokinase blaster cassette leads to an increase of the hybridizing band from 7.1 kb in the TU-6 strain to 8.8 kb in the Δglk1 strain when probed with the glk1 fragment. This 8.8 kb band is also detected with the pyr4 or Sh ble probe. In strain Δglk1-ex the glk1 and Sh ble hybridizing band is reduced to 5.5 kb due the excision of the glucokinase blaster. In strain Δglk1Δhxk1 the glk1 band is also reduced to 5.5 kb, but an additional stronger Sh ble hybridizing band resulting from two Sh ble fragments of the hexokinase blaster and a pyr4 hybridizing band are found

Growth comparison of QM9414, a Δglk1, a Δhxk1 and a Δglk1Δhxk1 strain after 3.5 days on different carbon sources. The growth behaviour of QM9414 or TU-6 strains with an ectopically integrated hexokinase or glucokinase cassette was essentially the same. Abbreviations:Gly glycerol,Glc d-glucose, Fru d-fructose and Ara l-arabinose
Excision of the pyr4 blaster in the Δglk1 strain
A successful re-use of the blaster cassette depends on the excision of the pyr4 marker by recombination between the flanking Sh ble direct repeats. We chose three Δglk1 strains and plated their conidiospores on 5-FOA plates to force and select for the excision of the pyr4 blaster. 5-FOA resistant colonies appeared after 3–5 days of incubation with a frequency of about 1–2×10−4 . As these colonies did not sporulate, 5-FOA resistant colonies were transferred to minimal medium plates containing uridine which allowed also the growth of strains in which the blaster cassette did not loop out. Purified colonies were then tested for uridine auxotrophy on minimal medium. About 90% of the 5-FOA resistant strains picked were found to be uridine auxotroph while the remaining 10% were uridine prototroph. A Southern analysis showed that only a single copy of the Sh ble fragment was left in the auxotrophic strains and that the pyr4 from the blaster cassette was completely removed, but the Δglk1 genotype retained (Fig. 2b). Sequencing of a PCR fragment comprising the disrupted glk1 locus confirmed that only a single Sh ble fragment was left in these strains. The Sh ble fragment was bordered by two restriction enzyme sites for XbaI and BamHI which could only originate from a recombination between the two original Sh ble fragments from the blaster cassette.
Deletion of hxk1 in the Δglk1 strain
Following the successful excision of the blaster cassette, we demonstrated its reuse in a second round of gene deletion. We chose to construct a Δglk1Δhxk1 strain and transformed therefore five of the uridine auxotrophic Δglk1 strains – obtained after the blaster cassette excision – with the hexokinase blaster. All five Δglk1 strains could be transformed to uridine prototrophy, indicating that their auxotrophy was indeed a result of the excision of the pyr4 blaster and not due to other mutations. l-arabinose was used in the protoplast regeneration medium since we expected the double mutant to be unable to grow on d-glucose. Transformants were purified and subjected to growth tests and Southern analyses (Figs. 2, 3). About 12% of the transformants turned out to be double deleted Δglk1Δhxk1 strains. They were completely unable to grow on d-glucose and d-fructose, and showed a stronger reduced growth on glycerol and l-arabinose.
Discussion
Complementation of uridine auxotrophic pyr4 mutants to prototrophy is probably the most successful strategy for gene manipulation in filamentous fungi but is limited by the one-time use of the pyr4 as marker gene. Here, we successfully overcame this limitation by applying a blaster approach for successive gene knock-outs in H. jecorina using a single marker. The pyr4 blaster cassette was successfully excised by selection for resistance to 5-FOA in Δglk1 strains and could be re-used to construct Δglk1Δhxk1 strains. The frequency of recombination between the two 380 bp Sh ble fragments was with 1–2×10−4 in about the same range as reported for A. fumigatus (4×10−4 ; d’Enfert 1996) and for A. nidulans (2×10−4; Krappmann and Braus 2003), which allowed a straight forward selection of pyr4 negative strains resulting from the looping out of the blaster cassette. 5-FOA resistance can in principle result from mutations in at least two genes: orotate phosphoribosyltransferase (pyr2) and OMP decarboxylase (pyr4). Ninety percent of the obtained 5-FOA resistant colonies were uridine auxotrophic, while the remaining 10% were prototrophic. This was most probably the result of the transfer of the colonies to non-selective medium which was done to facilitate their sporulation. Transformation of five randomly chosen auxotrophic strains with the hexokinase blaster showed that all five strains could be complemented with the pyr4 gene proving that the uridine auxotrophy was the result of the pyr4 excision.
In H. jecorina, research is focused on its anamorph form T. reesei. The lack of research with the sexual form prevented characterization of auxotrophic mutants, and therefore transformation strategies that involve the conversion of auxotrophic mutants to prototrophy are only poorly developed in H. jecorina, while they are well established in other fungal species including S. cerevisiae or A. nidulans. The successful application of the blaster cassette system to the anamorph of H. jecorina, provides therefore an interesting opportunity to accelerate functional genomics in this fungus especially in the view of the recent release of a draft version of the H. jecorina genome (http://gsphere.lanl.gov/trire1/trire1.home.html). Although it is at the moment illusive to target all putative genes in H. jecorina, we think that our approach is especially valuable for the investigation of the function of fungal, specifically, H. jecorina specific genes. Our interest is directed towards paralogous genes which have developed during evolution from their ancestral genes by gene duplication and often tend to evolve toward functional diversification. It may also aid in the investigation of the function of whole gene families: H. jecorina is an excellent producer of extracellular enzymes secreting a high number of e.g. cellulases or xylanases most of which have not yet been functionally characterized. A search of the H. jecorina genome sequence database reveals the presence of a high number of additional biomass degrading enzymes including genes encoding for cellulases, xylanases, pectinases or chitinases.
The blaster system offers also an application for the construction of industrial Hypocrea/Trichoderma strains. As a producer of low cost enzymes and recombinant proteins for a number of applications, genetic transformation systems are desired which do not lead to the accumulation of antibiotic resistance marker. Although the strains used in this study still carry a single antibiotic resistance marker after excision of the pyr4 blaster, it should be possible to replace the Sh ble direct repeat by an autochthonous H. jecorina sequence.
Here, we applied the blaster system to H. jecorina, but it can easily be adapted to other Trichoderma or fungal species in general, especially for those in which classical genetic approaches are not practicable. The range of organisms seems to be limited only by the availability of OMP decarboxylase negative strains. Such strains can be obtained by classical mutagenesis approaches and selection on 5-FOA (Gruber et al. 1990b). Fungal OMP decarboxylase genes are highly conserved and work therefore also in heterologous systems (cf: Gruber et al. 1990b; d’Enfert 1996; Punt et al. 2001). It is therefore possible to construct multiple disrupted strains with this blaster cassette in any fungal species which is efficiently transformed by the H. jecorina pyr4 gene.
In A. nidulans, glucose-, hexokinase mutants and double mutants were obtained by classical mutagenesis (Roberts 1963; Flipphi et al. 2003). Although the growth phenotype of A. nidulans hxkA1 (hexokinase deficient, formerly designated frA1 for fructose non-utilizing) mutant (Roberts 1963; Ruijter et al. 1996) is comparable to the H. jecorina Δhxk1 by being unable to grow on d-fructose and that both H. jecorina and A. nidulans double mutants are unable to grow on d-glucose and d-fructose, we noted differences in the utilization of the other carbon sources tested. While the A. nidulans glucokinase and hexokinase single mutants exhibit no other nutritional deficiencies, we found that in H. jecorina both kinase genes are necessary for fast growth on a number of carbon sources in H. jecorina including l-arabinose or glycerol.
d-fructose inhibited the growth of the hxkA1 mutant on other sugars (Roberts 1963) and Ruijter et al. (1996) showed that d-fructose and d-mannitol inhibited growth of this mutant on l-arabinose. This observation could be explained by repression of enzymes involved in l-arabinose catabolism by d-fructose and d-mannitol: Although both are not metabolized in the absence of hexokinase, their accumulation might be able to at least partially repress the synthesis of enzymes necessary for metabolism of other carbon sources.
The availability of the three isogenic mutants constructed in this study will also allow the study of the role of the two hexose phosphorylating enzymes in the signalling of carbon catabolite repression and in a second path of d-galactose utilization besides the classical Leloir pathway in H. jecorina (Seiboth et al. 2004). In A. nidulans only the double mutant is impaired in d-glucose and d-fructose repression for ethanol and acetate catabolism and xylan degradation (Flipphi et al. 2003). In H. jecorina, so far only a single carbon catabolite derepressed mutant which has a truncated cre1 gene was described (Ilmen et al. 1996). With respect to d-galactose utlization in A. nidulans Fekete et al. (2004) showed that a double mutant in the galactokinase (which catalyzes the first step in the Leloir pathway of d-galactose) and hexokinase is unable to grow on d-galactose as single carbon source. Although differences in the catabolic pathways for d-galactose in these two fungi might exist, it is likely that the second pathway of d-galactose utlization in H. jecorina proceeds also via d-fructose involving hexokinase.
References
Alani E, Cao L, Kleckner N (1987) A method for gene disruption that allows repeated use of URA3 selection in the construction of multiply disrupted yeast strains. Genetics 116:541–545
Boeke JD, LaCroute F, Fink GR (1984) A positive selection for mutants lacking orotidine-5′-phosphate decarboxylase activity in yeast: 5-fluoro-orotic acid resistance. Mol Gen Genet 197:345–346
Chiang C, Knight SG (1961) L-Arabinose metabolism by cell-free extracts of Penicillium chrysogenum. Biochim Biophys Acta 46:271–278
d’Enfert C (1996) Selection of multiple disruption events in Aspergillus fumigatus using the orotidine-5′-decarboxylase gene, pyrG, as a unique transformation marker. Curr Genet 30:76–82
Fekete E, Karaffa L, Sandor E, Banyai I, Seiboth B, Gyemant G, Sepsi A, Szentirmai A, Kubicek CP (2004) The alternative D-galactose degrading pathway of Aspergillus nidulans proceeds via L-sorbose. Arch Microbiol 181:35–44
Flipphi M, van de Vondervoort PJ, Ruijter GJ, Visser J, Arst HN Jr, Felenbok B (2003) Onset of carbon catabolite repression in Aspergillus nidulans. Parallel involvement of hexokinase and glucokinase in sugar signaling. J Biol Chem 278:11849–11857
Fonzi WA, Irwin MY (1993) Isogenic strain construction and gene mapping in Candida albicans. Genetics 134:717–728
Gruber F, Visser J, Kubicek CP, de Graaf LH (1990a) Cloning of the Trichoderma reesei pyrG-gene and its use as a homologous marker for a high-frequency transformation system. Curr Genet 18:447–451
Gruber F, Visser J, Kubicek CP, de Graaff LH (1990b) The development of a heterologous transformation system for the cellulolytic fungus Trichoderma reesei based on a pyrG-negative mutant strain. Curr Genet 18:71–76
Ilmen M, Thrane C, Penttila M (1996) The glucose repressor gene cre1 of Trichoderma: isolation and expression of a full-length and a truncated mutant form. Mol Gen Genet 251:451–460
Krappmann S, Braus GH (2003) Deletion of Aspergillus nidulans aroC using a novel blaster module that combines ET cloning and marker rescue. Mol Genet Genomics 268:675–683
Kubicek CP, Harman GE (eds) (1998) Trichoderma and Gliocladium. Taylor and Francis Ltd, London
Kuhls K, Lieckfeldt E, Samuels GJ, Kovacs W, Meyer W, Petrini O, Gams W, Borner T, Kubicek CP (1996) Molecular evidence that the asexual industrial fungus Trichoderma reesei is a clonal derivative of the ascomycete Hypocrea jecorina. Proc Natl Acad Sci USA 93:7755–7760
Mach RL (2004) Transformation and gene manipulation in filamentous fungi: an overview. In: Arora DK (ed) Handbook of Fungal Biotechnology, 2nd edn. Marcel Dekker, New York, pp 109–119
Mandels MM, Andreotti RE (1978) The cellulose to cellulase fermentation. Proc Biochem 13:6–13
Mio T, Yabe T, Sudoh M, Satoh Y, Nakajima T, Arisawa M, Yamada-Okabe H (1996) Role of three chitin synthase genes in the growth of Candida albicans. J Bacteriol 178:2416–2419
Muhlschlegel FA, Fonzi WA (1997) PHR2 of Candida albicans encodes a functional homolog of the pH-regulated gene PHR1 with an inverted pattern of pH-dependent expression. Mol Cell Biol 17:5960–5967
Penttilä ME (1998) Heterologous protein production in Trichoderma. In: Harman GE, Kubicek CP (eds) Trichoderma and Gliocladium. Taylor and Francis Ltd., London, pp 356–383
Pla J, Gil C, Monteoliva L, Navarro-Garcia F, Sanchez M, Nombela C (1996) Understanding Candida albicans at the molecular level. Yeast 12:1677–1702
Punt PJ, Seiboth B, Weenink XO, van Zeijl C, Lenders M, Konetschny C, Ram AF, Montijn R, Kubicek CP, van den Hondel CA (2001) Identification and characterization of a family of secretion-related small GTPase-encoding genes from the filamentous fungus Aspergillus niger: a putative SEC4 homologue is not essential for growth. Mol Microbiol 41:513–525
Roberts CF (1963) The genetic analysis of carbohydrate utilization in Aspergillus nidulans. Gen Microbiol 31:45–58
Ruijter GJ, Panneman H, van den Broeck HC, Bennett JM, Visser J (1996) Characterisation of the Aspergillus nidulans frA1 mutant: hexose phosphorylation and apparent lack of involvement of hexokinase in glucose repression. FEMS Microbiol Lett 139:223–228
Sambrook J, Russel DW (2001) Molecular cloning. A laboratory manual, 3rd edn. Cold Spring Harbor Laboratory Press, Cold Spring Harbor
Sanglard D, Hube B, Monod M, Odds FC, Gow NA (1997) A triple deletion of the secreted aspartyl proteinase genes SAP4,SAP5, and SAP6 of Candida albicans causes attenuated virulence. Infect Immun 65:3539–3546
Seiboth B, Hartl L, Pail M, Fekete E, Karaffa L, Kubicek CP (2004) The galactokinase of Hypocrea jecorina is essential for cellulase induction by lactose but dispensable for growth on d-galactose. Mol Microbiol 51:1015–1025
Yanisch-Perron C, Vieira J, Messing J (1985) Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 33:103–119
Acknowledgements
L.H. and B.S. were supported by grant P16143 from the Austrian Science Foundation FWF and by the Hochschuljubiläumsstiftung der Stadt Wien. Sequence data were obtained from the Department of Energy Joint Genome Institute (http://www.jgi.doe.gov). The T. reesei genome sequencing project was funded by the United States Department of Energy. We thank Christian P. Kubicek for critically reading the manuscript.
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by U. Kück
Nucleotide sequence data reported are available in the DDBJ/EMBL/GenBank databases under the accession numbers DQ068384 (H. jecorina glk1) and DQ068385 (H. jecorina hxk1).
Rights and permissions
About this article
Cite this article
Hartl, L., Seiboth, B. Sequential gene deletions in Hypocrea jecorina using a single blaster cassette. Curr Genet 48, 204–211 (2005). https://doi.org/10.1007/s00294-005-0011-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00294-005-0011-8