
Abstract
A new acidophilic xylanase (XYN11A) from Penicillium oxalicum GZ-2 has been purified, identified and characterized. Synchronized fluorescence spectroscopy was used for the first time to evaluate the influence of metal ions on xylanase activity. The purified enzyme was identified by MALDI TOF/TOF mass spectrometry, and its gene (xyn11A) was identified as an open reading frame of 706 bp with a 68 bp intron. This gene encodes a mature protein of 196 residues with a predicted molecular weight of 21.3 kDa that has the 100 % identity with the putative xylanase from the P. oxalicum 114-2. The enzyme shows a structure comprising a catalytic module family 10 (GH10) and no carbohydrate-binding module family. The specific activities were 150.2, 60.2, and 72.6 U/mg for beechwood xylan, birchwood xylan, and oat spelt xylan, respectively. XYN11A exhibited optimal activity at pH 4.0 and remarkable pH stability under extremely acidic condition (pH 3). The specific activity, K m and V max values were 150.2 U/mg, 30.7 mg/mL, and 403.9 μmol/min/mg for beechwood xylan, respectively. XYN11A is a endo-β-1,4-xylanase since it release xylobiose and xylotriose as the main products by hydrolyzing xylans. The activity of XYN11A was enhanced 155 % by 1 mM Fe2+ ions, but was inhibited strongly by Fe3+. The reason of enhancing the xylanase activity of XYN11A with 1 mM Fe2+ treatment may be responsible for the change of microenvironment of tryptophan residues studied by synchronous fluorescence spectrophotometry. Inhibition of the xylanase activity by Fe3+ was first time demonstrated to associate tryptophan fluorescence quenching.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Hemicellulose biomass is an important source of renewable energy. Hemicellulose is the second most abundant natural polysaccharide and primarily consists of xylan, mannan and galactan. Xylans, which are heteropolymers made up of a linear chain of β-d-xylopyranose residues with β-(1,4) linkages substituted with sugars (arabinose, xylose, galactose, etc.), glucuronic acids, and other groups (e.g., acetyl, feruloyl, p-coumaryl), are among the most important hemicellulose components of the plant cell wall [43]. It is widely known that the complete hydrolysis of xylan requires the synergistic activities of several hydrolytic enzymes, primarily xylanase (EC 3.2.1.8) and β-xylosidase (EC 3.2.1.37), and additional enzymes such as α-l-arabinofuranosidase, α-d-glucuronidase, and acetyl xylan esterase [2]. Among these enzymes, the most important is endo-β-1,4-xylanase (EC 3.2.1.8), which cleaves internal glycosidic bonds in xylan to generate short xylo-oligosaccharides [4]. The majority of identified xylanases belong to glycoside hydrolase (GH) families 10 and 11, and others belong to GH families 5, 7, 8, 16, 26, 43, 52 and 62 [7]. Xylanases have received much attention because of their potential wide applications in agro-industrial processes, such as the bioconversion of hemicellulosic biomass into fermentative sugars, pulp bleaching, the improvement of baking properties in bread making, the enhancement of the digestibility of animal feed, the clarification of fruit juices and xylitol production [7].
Many microorganisms, such as Aspergillus spp., Trichoderma spp., Penicillium spp., and Bacillus spp., as well as other bacteria, fungi and yeasts, have been found to produce xylanases, and several studies have been performed to characterize these enzymes [9, 34, 39]. The industrial application enzymes have a strong desire for extremozymes with especial characteristics such as acidic- and thermo-stable. So far, most commercially available xylanases are obtained from Trichoderma and Aspergillus that lack stability at acidic pH, thus limiting their applications in animal feed, food, and bioenergy industries [5, 7]. To date, several acidophilic xylanases from fungi have been reported [18, 27, 45]. Acidophilic and acidic-stable xylanases would be more beneficial to those processes where low pH condition is required to avoid microbial contamination [16]. In bioconversion processes, acidic pretreatments are always used prior to or simultaneous to enzyme treatment, so using the acidophilic and acidic-stable xylanases in the low pH condition exhibited more advantages. For example, adjustment of the pH to acidic conditions can be avoided so as to reduce cost [7]. Furthermore, the wide industrial applications of xylanases in the world were still limited because of their low catalytic activities and high production costs. Use of the dumped agricultural residue instead of commercial xylan to produce xylanase will greatly decrease the production cost. There are many reports on xylanase production, purification, and properties. However, few studies focus on uses agricultural residue to high effectively produce acidophilic and acidic-stable xylanases. The previous study found that the fungus Penicillium oxalicum GZ-2 produces high levels of acidophilic xylanolytic activity (115.3 U/mL) when using agricultural waste (wheat straw) as a substrate [24]. In this study, a high level production of acidophilic and acidic-stable xylanases was obtained from P. oxalicum GZ-2, which used the agricultural waste (corncob) as substrate. We also purified and characterized an acidophilic and acidic-stable xylanase (XYN11A) from P. oxalicum GZ-2 and the gene encoding the xylanase was cloned and sequenced. In addition, synchronized fluorescence spectroscopy was used for the first time to evaluate the influence of metal ions on xylanase activity. Our study demonstrated that the enzyme XYN11A was potentially valuable in biotechnological applications, especially in the animal feed, food, and bioconversion industries required in low pH condition.
Materials and methods
Strains and culture conditions
The P. oxalicum GZ-2 used in this study was isolated and identified as previously reported [24] and has been deposited at the China General Microbiological Culture Collection Center (CGMCC 7527). For xylanase enzyme production by submerged fermentation, the basal culture medium consisted of 2.0 g of KH2PO4, 1.4 g of (NH4)2SO4, 1.0 g of tryptone, 0.3 g of urea, 0.4 g of CaCl2 ·2H2O, 0.3 g of MgSO4 ·7H2O, 7.5 mg of FeSO4 ·7H2O, 2.5 mg of MnSO4 ·H2O, 2.0 mg of ZnSO4, and 3.0 mg of CoCl2 in 1,000 mL of water, pH 5.0 [6]. Escherichia coli TOP 10 (Invitrogen, Carlsbad, USA), grown in Luria–Bertani medium at 37 °C, was used as the host cell for plasmid cloning.
Xylanase assay and protein concentrations determination
The xylanase activity was determined using 1 % beechwood xylan as substrate (w/v, Sigma, Louis, Missouri, USA) according to Bailey et al. [1]. One unit of activity was defined as the activity of enzyme releasing 1 μmol of reducing sugars per minute under these conditions. Reducing sugars were quantified by the DNS method with d-xylose as the standard [32]. The concentrations of soluble proteins in the filtered fermentation culture samples were determined using a BCA protein assay kit (Dingguo Changsheng Biotechnology Co., Ltd, Beijing, China) with bovine serum albumin as standard.
Production and purification of xylanase from P. oxalicum GZ-2 culture
For xylanase production in submerged fermentation, 100 mL of a basal culture medium (2.0 g of KH2PO4, 1.4 g of (NH4)2SO4, 1.0 g of tryptone, 0.3 g of urea, 0.4 g of CaCl2 ·2H2O, 0.3 g of MgSO4 ·7H2O, 7.5 mg of FeSO4 ·7H2O, 2.0 mg of MnSO4 ·H2O, 2.0 mg of ZnSO4, and 3.0 mg of CoCl2 in 1,000 mL of water, pH 5.0) was supplemented with 2 % (w/v) of the agricultural residue (corncob) in 500 mL Erlenmeyer flasks. Agricultural residue corncob was obtained locally (Nanjing, China), dried at room temperature, milled using a pulverizer and passed through a 40-mesh sieve.
Conidia suspensions (1 %, 1 × 107 conidia/mL) of strain GZ-2 were inoculated in the medium and incubated at 30 °C for 7 days (170 rpm) in triplicate. After 7 days of cultivation, the xylanase activity of submerged cultures was reached at the highest level. All purification procedures were performed at 4 °C. Fermentation broths were centrifuged at 10,000 rpm for 20 min and then filtered through a 0.45-μm membrane to obtain the culture supernatant. Solid ammonium sulfate was slowly added to the culture supernatant (200 mL) in an ice bath under slow constant stirring to achieve 80 % saturation. The crude protein obtained after centrifugation (20 min at 10,000 rpm) was dissolved in 10 mL of 20 mM acetate buffer, pH 4.0. The protein solution was dialyzed against the same buffer (1,000 mL) overnight using dialysis tubing (MWCO: 10,000) with three intermittent changes of the buffer at 4 °C. The dialyzed enzyme solution (5 mL) was loaded onto a Sephacryl S-100 column (1.6 × 100 cm, GE Healthcare, Uppsala, Sweden) previously equilibrated with the same buffer at a rate of 0.8 mL/min. Fractions (4 mL) were collected and the xylanase activity was determined. The active fractions were pooled, lyophilized, and dialyzed against the Tris–HCl buffer (20 mM, pH 8.0). The pooled proteins were applied to a Q-Sepharose fast flow (FF) (1.6 × 20 cm, GE Healthcare, Uppsala, Sweden) column that was previously equilibrated with ten column volumes of 20 mM Tris–HCl buffer (pH 8.0), and eluted with a linear gradient of 0–1 M NaCl in the same buffer at a flow rate of 1.5 mL/min for elution.
Biochemical characterization of XYN11A
Substrate specificity and kinetic parameters
Xylanase specific activity was measured using celluloses (carboxymethyl cellulose, Sigma, Louis, Missouri, USA and Whatman No. 1 filter paper) and hemicelluloses (beechwood xylan, birchwood xylan, and oat spelt xylan, Sigma, Louis, Missouri, USA) substrates (1 %, w/v). To determine the reaction rate of XYN11A, various xylan substrate concentrations were used to react at 50 °C in acetate buffer (pH 4.0, 50 mM). The Michaelis–Menten constant (K m ) and the maximum velocity (V max) were calculated using Lineweaver–Burk plots.
Optimal pH and temperature and stability of xylanase
The optimal pH for XYN11A activity was tested at 50 °C for 10 min with different substrates (beechwood, birchwood, and oat spelt) in different buffer solutions (50 mM): glycine–HCl buffer (pH 2.0), citrate buffer (pH 3.0–6.0), and sodium phosphate buffer (pH 7.0–8.0). The optimal temperature for XYN11A activity was determined by incubating the enzyme with xylan substrates (beechwood, birchwood, and oat spelt) at optimal pH buffer (50 mM) at different temperatures (30–80 °C) for 10 min. To determine the pH stability, the XYN11A enzyme was incubated in different pH buffers from 2.0 to 8.0 (as mentioned above) at 50 °C for 30 min. The thermal stability was determined at temperatures of 40, 45, 50, and 55 °C by incubation of suitably diluted enzyme samples in the absence of substrate for 0, 15, 30, 45, and 60 min. The remaining enzyme activity after each treatment was measured by incubating with 1 % (w/v) beechwood xylan solution at 50 °C for 10 min in acetate buffer (pH 4.0, 50 mM) using the DNS method.
Effect of metal ions and chemical reagents on XYN11A activity
Xylanase activity was measured using the enzyme assay described above with beechwood xylan as the substrate in the presence of different metal salts (BaCl2, CuSO4, FeCl3, MnSO4, CaCl2, CdSO4, FeSO4, CrCl3, CoCl2, LiCl, NiSO4, MgSO4 and HgCl2) at different concentrations (0.1, 1 and 10) or other reagents (β-mercaptoethanol (10 mM), DTT (DL-dithiothreitol, 10 mM)) and 1 % surfactant (Tween-80, Tween-20, and Triton X-100).
Analysis of enzyme–metal ions interactions using synchronous fluorescence spectrophotometry
Synchronous fluorescence spectroscopy can provide information about the molecular environment in the vicinity of the chromophore [12]. Synchronous fluorescence spectra were recorded at room temperature with a Varian Cary Eclipse fluorescence spectrophotometer (Varian Inc., Palo Alto, CA, USA) with 1.0-cm quartz cells. A 2.0-mL xylanase solution (2 μM) in acetate buffer (pH 4.0, 50 mM) solution containing 0.1–10 mM metal ions was titrated by successive additions of 1 M stock solution of metal ions. The synchronous fluorescence spectra were measured from 240 to 400 nm at Δλ = 60 (scanning interval, Δλ = λ em–λ ex). The spectrum only showed the spectroscopic behavior of tryptophan residues when Δλ was maintained a constant value (Δλ = 60) in the synchronous fluorescence [26]. Scanning interval and velocity were Δλ = 60 and 600 nm/min, respectively. The excitation and emission slit widths were 10 nm.
Thin-layer chromatography analysis of the hydrolysis products
For the analysis of the hydrolysis products, 9 mL of 1 % different xylans (beechwood xylan, birchwood xylan, and oat spelt xylan, Sigma, Louis, Missouri, USA) and 200 µL of XYN11A (0.43 μg/μL) or 400 μL of 1 mg/mL xylo-oligosaccharides (xylobiose, xylotriose, xylotetraose, xylopentaose, and xylohexaose, Megazyme, Wicklow, Ireland) and 10 μL of XYN11A were used for enzymatic hydrolysis in acetate buffer (pH 4.0, 50 mM). The reaction mixture was incubated at 50 °C in a water bath for 24 h. Samples were collected after 0, 10, 30 min, 2, 4, 8, 12, and 24 h of incubation. The reaction was stopped by boiling for 10 min and the samples were frozen. Then, 10 μL of each aliquot was spotted onto a thin-layer chromatography (TLC) plate, and the plates were developed with a solvent system consisting of chloroform–acetic acid–water (3:6:1, v/v). The plates were subsequently air dried and visualized by spraying with a 9:1 (v/v) mixture of methanol and sulfuric acid containing 0.2 % orcinol and heating at 85 °C for 5 min. Xylose (X1) and xylo-oligosaccharides (Megazyme, Wicklow, Ireland) xylobiose (X2), xylotriose (X3), xylotetraose (X4), xylopentaose (X5), xylohexaose (X6) were used as standards.
SDS-PAGE, zymogram analysis and protein identification
The fractions containing xylanase activity were analyzed by SDS-PAGE and zymography as described by Liao, et al. [24]. SDS-PAGE was performed using an 11 % (w/v) polyacrylamide gel with a 5 % stacking gel with the Mini-Protean II system (Bio-Rad, Hercules, CA) according to Laemmli [21]. The protein bands were visualized by staining with Coomassie Brilliant Blue R-250 (Bio-Rad, Hercules, USA). For the zymogram analysis, briefly, after the separation of the enzyme samples by SDS-PAGE using gels containing 0.1 % beechwood xylan, the zymogram gel was soaked for 1 h in 2.5 % (v/v) Triton X-100 to remove the SDS and re-nature the proteins in the gel, which was then washed thoroughly in MilliQ water (Millipore, Bedford, MA, USA) and incubated at 50 °C for 20 min in 50 mM acetate buffer (pH 4.8). The gel was submerged in 0.1 % (w/v) Congo red solution for 30 min and destained with l M NaCl until pale-red hydrolysis zones appeared against a red background. The reaction was then stopped by dipping the gel into 5 % acetic acid solution. The purified XYN11A was identified by MALDI TOF/TOF mass spectrometry (Applied Biosystems, Foster City, CA, USA) according to the method of Ravalason et al. [36]. To identify the protein sequence, a homology search was performed using Mascot (http://www.matrixscience.com). The partial amino acid sequence was used to identify analogous proteins through a BLAST search of the NCBI protein sequence database. Amino acid homology alignment of the predicted XYN11A with other highly homologous known xylanases was carried out.
Molecular manipulations
Isolation of genomic DNA and total RNA and first-strand cDNA synthesis
Mycelia of P. oxalicum GZ-2 were filtered from the culture medium with 2 % corncob powder after incubation at 30 °C for 72 h. The fresh mycelia were ground using a mortar in liquid nitrogen. The genomic DNA of P. oxalicum GZ-2 was extracted as described by Möller et al. [30]. For total RNA isolation, 100 mg of powdered mycelia was suspended in Trizol reagent (Invitrogen, Carlsbad, USA), and total RNA was isolated in accordance with the manufacturer’s protocol. The synthesis of cDNA and the reverse transcriptase (RT) reactions were performed using the PrimeScript™ RT reagent Kit with the gDNA Eraser Kit (Takara, RR047A, Dalian, China).
Cloning of the xylanase gene xyn11A
The degenerate primers xyn11Adf and xyn11-dr were designed using the iCODEHOP (http://blocks.fhcrc.org/codehop.html) program based on the multiple sequence alignment of analogous amino acid sequence using ClustalW2 program. The amplification reaction mixture (25 μL) was composed of 2.5 μL of 10 × PCR buffer, 2.5 μL of 25 mM Mg2+, 2 μL of 10 mM dNTPs, 1 μL of each primer (10 mM), 100 ng of DNA template, and 0.5 U of Taq DNA polymerase. The PCR cycling parameters were an initial denaturation step at 95 °C for 5 min, 30 cycles of amplification (denaturation at 94 °C for 30 s, annealing at 55 °C for 30 s, extension at 72 °C for 60 s), and a final elongation step at 72 °C for 10 min.
To obtain the 5′-end and the 3′-end of xyn11A, self-formed adaptor PCR (SEFA-PCR) was performed to amplify the core regions of xyn11A according to the protocol developed by Wang et al. [42] with the primers listed in Table 1. By aligning the sequences of the 5′-end and 3′-end PCR products, the full-length cDNA sequence of xyn11A was deduced and obtained through RT-PCR using the following specific primers: xyn11-f (ATGAAGGTCTTCGCAACCTT) and xyn11-r (TTAACCAGAGACCTGGACGC). The resulting PCR fragment was purified and ligated into the pMD-19T (Takara, Dalian, China) vector for sequencing and subjected to BLAST analysis using the NCBI database (http://blast.ncbi.nlm.nih.gov/Blast.cgi).
Sequence analysis
Geneious software (http://www.geneious.com) was employed to assemble and analyze the DNA sequence. Putative signal peptides were predicted using SignalP (http://www.cbs.dtu.dk/services/SignalP/). The exon–intron structure of the full-length gene was predicted using the online software FGENESH (http://linux1.softberry.com/berry.phtml). The nucleotide sequence for the xylanase gene (xyn11A) was deposited in GenBank (http://www.ncbi.nlm.nih.gov/genbank/) under the accession number KF233756.
Results
Production and purification of the xylanase
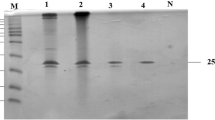
After 7 day of growth in the media supplemented with corncob powder, the production of xylanase from P. oxalicum in the culture media was reached the maximal level (85.0 U/mL), as shown in Fig. S1. Fractionation with ammonium sulfate precipitation (80 % saturation) increased the specific activity approximately 1.3-fold, with 63.2 % recovery of the xylanase activity. The protein was loaded onto a Sephacryl S-100 column and two peaks (XYN11A-peak-1 and XYN11A-peak-2) containing proteins with xylanase activity were detected (Fig. S2A). This purification step resulted in a 3.3-fold special activity (purification fold) increase with 8.7 % recovery with regard to XYN11A-peak-2; however, the first peak (XYN11A-peak-1) had greater xylanase activity than the second xylanase peak (XYN11A-peak-2). Two fractions were further purified at the same time, but we failed to obtain any purified xylanases from the XYN11A-peak-1. The protein of XYN11A-peak-2 was further purified with a Q-Sepharose FF column eluted with a NaCl gradient (0–1 M). The protein with xylanase activity was not bound to anion-exchange column (Fig. S2B). After the final purification step, the enzyme was purified 6.4-fold, with a recovery of 6.9 % and a specific activity of 150.2 U/mg of protein (Table 2). The purified enzyme preparation contained a single band of approximately 20 kDa on SDS-PAGE and zymogram gels, which was approached the calculated molecular weight of 21.3 kDa (Fig. 1). The purified protein spots were excised from the gels for analysis by MALDI TOF/TOF tandem mass spectrometry (Fig. S3). A Mascot search revealed that XYN11A has a high similarity to a glycoside hydrolase family 11 protein from Thielavia terrestris NRRL 8126 (GenBank accession no. XP_003651760) with a probability-based score of 176, and one peptide matched with this protein: YNQPSITGTSTFTQFFSVR.

SDS-PAGE (a) and zymograms (b) analysis of xylanase fractions purified from the P. oxalicum GZ-2 culture supernatant. Lane 1, concentrated culture supernatant by the precipitation of ammonium sulfate; lane 2, the pooled proteins after Sephacryl S-100; lane 3, the purified xylanase. M standard protein molecular weight markers
Effects of pH and temperature on XYN11A activity and stability
The activity of XYN11A was investigated at a series of pH values using three different xylan sources (beechwood, birchwood, and oat spelt) as substrates (Fig. 2a). Purified xylanase XYN11A exhibited highest xylanase activity at pH 4.0, irrespective of the source of the xylan. Beechwood xylan was the most suitable substrate at pH 3.0 conditions compared with other two xylans. However, at pH 5, xylanase XYN11A exhibited greater xylanase activity for oat spelt than the other two xylan sources. The pH stability profiles showed that XYN11A was highly stable (95 % activity remaining) under an extremely acidic pH range, ranging from 3.0 to 5.0 (Fig. 2b). We also investigated the effect of temperature on xylanase activity with different xylan sources as the substrate (Fig. 2c). The optimal temperature for XYN11A activity differed for various xylan sources. For beechwood xylan, the optimal temperature was 60 °C, but for birchwood xylan and oat spelt xylan, the optimal temperature was 50 °C. XYN11A was stable at 50 °C, with more than 95 % of the activity remaining after incubation at 50 °C for 60 min (Fig. 2d). After incubation at 55 °C for 15 min, the enzyme activity decreased rapidly and only retained 14 % of the initial activity.

The optimum pH (a) and temperature (c) for the activity of the purified xylanase XYN11A against beechwood xylan (Bexy), birchwood xylan (Bixy) and oat spelt xylan (Oxy). The pH stability (b) and thermal stability (d) of the purified xylanase XYN11A for beechwood xylan. Error bars present the standard deviation of three repeats
Effects of metal ions and reagents on the xylanase activity of XYN11A
The effects of various metal ions at different concentrations on the xylanase activity of XYN11A were evaluated, and the results are shown in Table 3. The enzyme activity was slightly inhibited in the presence of all metal ions at a low concentration (0.1 mM) (Ba2+, Cu2+, Fe3+, Mn2+, Ca2+, Cd2+, Cr3+, Co2+, Li+, Ni2+, Mg2+, and Hg2+) except Fe2+. For Fe3+ and Hg2+, the extent of the inhibition of enzyme activity was enhanced by the increase in the concentration of the metal ions from 0.1 mM to 10 mM. However, for other metal ions, the enzyme activity increased and then decreased with increasing concentration from 0.1 mM to 10 mM. Most of the metal ions strongly stimulated the activity of XYN11A at 1 mM. DTT (10 mM) and 1 % surfactants (Tween-80 and Tween-20) did not significantly influence the xylanase activity. However, triton X-100 and β-mercaptoethanol slightly inhibited the xylanase activity (Table 4).
Synchronous fluorescence analysis XYN11A interaction with metal ions
As depicted in Fig. 3a, the results show that the maximum emission wavelength of tryptophan residues is at 278 nm. The emission peaks of the synchronous fluorescence spectra of XYN11A with various amounts of metal ions exhibited different effects. At high concentration (10 mM) of metal ions, fluorescence intensity of the emission spectra of XYN11A was strongly enhanced as compared with the absence of metal ions for Ca2+, Fe2+, and Hg2+. From Fig. 3b, the maximum emission of tryptophan residues shows a red shift (from 278 to 281 nm), demonstrating that there is effect on the microenvironment of the tryptophan residues in XYN11A resulting in buried tryptophan exposes to the aqueous solvent. The results also showed that the fluorescence intensity of the emission spectra of XYN11A was decreased with increasing concentrations of Fe3+ (from 0.1 to 10 mM), which indicated that a concentration-dependant quenching of the intrinsic protein fluorescence had occurred (Fig. 3c). As shown in Fig. 3d, the fluorescence intensity of the emission spectra of XYN11A was strongly enhanced when XYN11A was added with high concentration of Hg2+ ions (10 mM), and a low concentration of Hg2+ ions (0.1 and 1 mM) had slight effect on tryptophan fluorescence intensity.

Synchronous fluorescence spectra of purified xylanase XYN11A in the absence and the presence of different concentrations of Ca2+ (a), Fe2+ (b), Fe3+(c), and Hg2+ (d) in acetate buffer pH 4.0 at room temperature (29 °C)
Substrate specificity and kinetic parameters
The xylanase activity of XYN11A was evaluated with various substrates at 50 °C (pH 4.0) for 10 min to determine the enzyme specificity (Table 5). XYN11A showed specificity toward polymeric xylan sources, but not to other substrates such as carboxymethyl cellulose and filter paper (data not shown). The activity of XYN11A with beechwood xylan as a substrate was twofold higher than the activity with other two xylan sources, birchwood xylan and oat spelt xylan. The kinetic analysis of XYN11A with different xylan sources as the substrate is presented in Table 5. The K m values of XYN11A for beechwood xylan, birchwood xylan, and oat spelt xylan were 30.7, 51.4, and 41.3 mg/mL, respectively, and V max values were 403.9, 610.2, and 475.1 μmol/min/mg, respectively.
Analysis of the hydrolysis products
The action of the enzyme XYN11A on various xylan sources and xylo-oligosaccharides was analyzed by TLC. XYN11A was able to efficiently degrade different types of xylan polymers, such as beechwood xylan, birchwood xylan, and oat spelt xylan. The products of the xylanase XYN11A hydrolysis of various xylan sources and xylo-oligosaccharides were mainly xylotriose and xylobiose (Fig. 4). There was no difference in the hydrolysis products between birchwood xylan and oat spelt xylan. However, an unknown hydrolysis product (Fig. 4a, underline) was found when beechwood xylan was used as the substrate. The concentrations of hydrolysis products were increased with the increasing incubation time from 10 min to 24 h, but the types of hydrolysis products did not change. The enzyme rapidly degraded xylotetraose (X4), xylopentose (X5), and xylohexaose (X6), but hydrolyzed xylobiose and xylotriose poorly (Fig. 4d).

TLC analysis of the products after hydrolysis of various xylans (1 %) and xylo-oligosaccharides (1 mg/mL) by XYN11A xylanase (0.43 μg/μL) for each reaction time of 0, 10, 30 min and 2, 4, 8, 12, and 24 h in 50 mM acetic buffer (pH 4.0) at 50 °C. S xylo-oligomer standards as substrate, a beechwood xylan as substrate, b birchwood xylan as substrate, c oat spelt xylan as substrate, d xylo-oligosaccharides as substrate
Cloning and sequence analysis of the xyn11A gene
A 437-bp gene fragment was amplified from the genomic DNA of strain GZ-2 using the degenerate primers xyn11A-df and xyn11A-dr. To amplify the full-length xyn11A gene, SEFA-PCR was used (Table 1), and 5′ and 3′ flanking regions of approximately 1,128 and 712 bp, respectively, were obtained. All fragments were assembled with core regions to create a 1,840-bp sequence containing a complete chromosomal gene of 705 bp predicted by FGENESH (http://linux1.softberry.com/berry). The full-length cDNA sequence of the xylanase gene was cloned from the synthesized cDNA using the xyn11A-f and xyn11A-r primers, which contained a 636 bp open reading frame (ORF). One intron of 68 bp interrupted the xylanase coding sequence according to the alignment of the DNA and cDNA sequences. A putative 16-residue signal peptide at the N-terminus was found by SignalP analysis with the SignalP server system (http://www.cbs.dtu.dk/services/SignalP/). The deduced mature protein of xyn11A consisted of 196 residues with a calculated molecular mass of 21.3 kDa and a theoretical pI of 6.18 (http://web.expasy.org/compute_pi/). Based on the analysis of the deduced amino acid sequence using the NCBI BLAST tool, the xylanase contains a catalytic domain typical of GH11 enzymes and no carbohydrate-binding domain (http://pfam.sanger.ac.uk/search/sequence). In xyn11A, the two putative catalytic residues Glu 106 and Glu 199 were found in highly conserved regions of the active site (Fig. 5) (http://pfam.sanger.ac.uk/search/sequence).

Alignment of sequences deduced from XYN11A with other xylanases from Aspergillus niger (AAZ66797), Bispora sp. (ADZ99365), Penicillium sp. (BAA88421), and Thielavia terrestris (XP_003651760) using ClustalW2 program. The predicted signal peptide sequence is shown before the perpendicular. Identical and similar amino acids are indicated by asterisks and single and double dots. The conserved region used for the degenerate PCR primer is marked by a straight line; the putative catalytic (Glu) residues are boxed in black. Asp residues conserved in the acidophilic xylanases marked by plus. Four tryptophan residues were marked by red box
Discussion
The previous studies showed that the genus Penicillium was an alternative to Trichoderma reesei for biofuel production and a promising producer of biomass-degrading enzymes suitable for applications in various industries [14, 25, 31]. Our previous study found that P. oxalicum GZ-2 produces multiple acidophilic xylanolytic enzymes during submerged fermentation with agricultural residues [24]. In this study, a xylanase from P. oxalicum GZ-2 was purified, identified and characterized, and its gene was cloned and sequenced. The complete genome of P. oxalicum was recently unveiled [25], which was the first genome of a high lignocellulolytic enzymes-producing species in the genus Penicillium. A 100 % similar putative xylanase was predicted in the genome of Penicillium decumbens 114-2, however, its function and feature have not been verified and characterized to date. Our study is the first report on the purification and characterization of this enzyme in P. oxalicum. Analysis of the internal amino acid sequence of P. oxalicum GZ-2 xyn11A showed that it contains the conserved sequence of the glycoside hydrolase 11 catalytic domains without a carbohydrate-binding domain or linker. The deduced amino acid sequence of XYN11A exhibited significant identities with other known GH11 xylanases, as shown in Fig. 5.
Several xylanases have been purified from fungi, such as xynI from Acrophialophora nainiana [44], xynC from Penicillium capsulatum [37] and xylanase from Trichoderma viride [39]. However, only a few acidophilic and acidic-stable xylanases (pH 1–4) produced by low cost feedstock have been studied in P. oxalicum to date [10, 18]. In this study, an acidophilic and acidic-stable xylanase (XYN11A) from P. oxalicum GZ-2 was purified to homogeneity when agricultural waste was used as strongly effective substrate. After gel filtration with a Sephacryl S-100 column, two fractions (XYN11A-peak-1 and XYN11A-peak-2) with xylanase activity were detected. The first peak, with a high molecular mass, showed greater xylanase activity compared with the second peak, which contained the low-molecular-weight protein XYL11A. Many xylanase activity bands with molecular masses between 30 and 72 kDa were also observed in the zymogram gel (Fig. 1b) after ammonium sulfate precipitation and gel filtration. These numerous bands explain the high xylanase activity of the XYN11A-peak-1. There were various xylanases detected in the supernatant of GZ-2, and thus, more xylanases will be purified in the future.
XYN11A’s xylanase activity with birchwood xylan or oat spelt xylan as a substrate was significantly lower than with beechwood xylan at pH 3.0. At pH 5.0, XYN11A exhibited 94, 70 and 72 % of its maximal xylanase activity with oat spelt xylan, beechwood xylan and birchwood xylan, respectively. XYN11A exhibited maximal activity at pH 4.0 towards three types of xylans (beechwood xylan, birchwood xylan, and oat spelt xylan), exhibiting almost 80 % of xylanase activity at pH 3.0 for beechwood xylan, and pH stability retained more than 95 % of the activity over an acidic pH range (pH 3–5). The optimal pH characteristic of XYN11A was similar to acidic fungal xylanases (XYN10G5) from Phialophora sp. G5, but the extreme low pH (pH 2.0) stability of XYN11A (remained more than 50 % activity) was better than XYN10G5 (no activity) [45]. The pH stability of XYN11A showed more potential application in extreme acidic condition than the most of fungal xylanase, which was stable at pH 4.0–6.0 [3, 16]. These results indicated that XYN11A was a good candidate enzyme for applications in the animal feed, food, and bioconversion industries where are required in low pH condition. The three-dimensional structures of acidophilic xylanases from Aspergillus niger and Trichoderma reesei have been reported, which revealed that an Asp residue is important for the acidophilic nature of the protein [38]. An Asp residue (Asp64) was present in the conserved domain of our xylanase, as in other acidophilic xylanases (Fig. 5, marked with a plus), and this residue is replaced by Asn in other fungal xylanases. The Asp residue is thought to be a hydrogen-bond linked to the glutamic acid residues that can serve as general acid/base catalysts for the hydrolase activity [28].
XYN11A exhibited different optimal temperatures for various xylan sources. This enzyme has maximal xylanase activity at 60 °C and retained 40 % activity at 70 °C for beechwood xylan, and 50 °C was optimal temperature for other two xylans. These results are similar to most known fungal xylanases (optimal temperature between 40 and 60 °C) [7]. These results indicate that the reaction temperature used in different applications should be adjusted according to the type of substrate. The thermal stability profiles showed that the XYN11A was stable at temperature below 50 °C. However, the xylanase activity of XYN11A was significantly decreased from 100 to 14 % after 15 min incubation at 55 °C.
The highest specific activity was observed with beechwood xylan (150.2 U/mg), followed by oat spelt xylan (72.6 U/mg) and birchwood xylan (60.2 U/mg), similar to the results for the acidic xylanase XYLD produced by Bispora sp. [29]. In contrast, Zhang et al. [45] reported that the specific activity of an acidic GH10 xylanase for xylan polymers was in the following order: oat spelt xylan >birchwood xylan >beechwood xylan. GH11 xylanases usually have higher specific activity for fewer xylan sources; however, GH10 xylanases are active on a broader range of substrates.
The K m and V max values were also estimated for different xylan sources. The apparent K m value (30.7 mg/ml) was the lowest for the beechwood xylan among the three types of xylans, indicating that the enzyme has a higher affinity for beechwood xylan, similar to xyn1 form A. niger [8] and XYLD from Bispora sp. [29]. XYN11A had the highest V max value (610.2 μmol/min/mg) for birchwood xylan, which indicated that this enzyme has a higher catalytic efficiency for hydrolyzing birchwood xylan. Similarly, XYLD from Bispora sp. [29] and XYNIII from Acrophialophora nainiana [40] had the highest V max values for oat spelt xylan. The K m and V max values of XYN11A for beechwood xylan are in agreement with the values for other fungal xylanases, which range from 0.09 to 40.9 mg/mL for K m and from 0.106 to 6,300 μmol/min/mg for V max [2]. We found that the dissolvability of beechwood xylan was better than that of birchwood xylan or oat spelt xylan (data not shown), which might cause difference in K m values among birchwood xylan and beechwood xylan. However, the structures of beechwood xylan and birchwood xylan are similar. XYN11A lacks carbohydrate-binding domains, which increases the affinity of the enzymes for its substrates, and thus, the lack of carbohydrate-binding domains might result in high K m values for the minimally soluble birchwood and oat spelt xylans. Beechwood and birchwood xylans have few ramifications, containing approximately 94 % xylose, in contrast to the xylan from oat spelt, which is a more ramified arabinoxylan that contains approximately 70 % xylose. The main chain of this xylan is highly branched with arabinose residues. These structural differences between the beechwood and birchwood xylans and the oat spelt xylan might lead to the much lower K m value of XYN11A for beechwood xylan than for oat spelt xylan [13]. Vieira et al. [40] demonstrated that xynIII preferred soluble xylan sources as substrates, with higher affinity for soluble xylans than for insoluble xylan sources.
Most of the metal ions tested substantially enhanced the activity of XYN11A at a moderate concentration (1 mM). The enzyme activity was strongly inhibited by Fe3+ and Hg2+ at the concentrations of 0.1, 1, and 10. The inhibition of the enzyme activity by Hg2+ may be due to oxidation of specific residues containing sulfhydryl groups, like cysteine residue [41]. There were 21 cysteine residues in the amino acid sequence of XYN11A and Cys119 was near to the putative catalytic residues Glu106. In this study, the extent of the inhibition of enzyme activity was increased with the increases of Fe3+ concentration from 0.1 to 10 mM. In contrast, Fe3+ has been reported to be an enhancer [17, 35]. Tryptophan, the intrinsic fluorophore in proteins, is highly sensitive to the polarity of its surrounding environment [11]. The purpose is to investigate the interaction of the metal ions to XYN11A using synchronous fluorescence spectroscopy by simultaneous scanning of the excitation and emission wavelengths while maintaining a constant wavelength interval between them. The fluorescence of the indole chromophore is highly sensitive to its microenvironment making it an ideal choice for responding protein conformation changes and interactions with other molecules [11, 12]. When wavelength interval was maintained at a constant value (Δλ = 60), the synchronous fluorescence provided the characteristic information of tryptophan residues [26]. Many reports about using synchronous fluorescence spectrometry to study the interactions between bovine serum albumin and chemical materials are available [12, 22]. However, only a few references use fluorescence spectrometry to study xylanases interaction with their substrates [19, 33]. Nath and Rao studied the structural and functional role of tryptophan in Bacillus xylanases by fluorescence spectrometry [33]. Tryptophan fluorescence is highly sensitive to position and environment, which is directly influenced by the protein conformation [12]. The amino acid sequence of XYN11A contained four tryptophan residues (Fig. 5, marked in red box). The maximum emission of synchronous fluorescence shows a red shifted when tryptophan residues are exposed to a polar environment (exposed to solvent) and have high mobility. Tryptophan residues buried in the protein display blue-shifted emission. Treatment of XYN11A with Fe2+, the maximum emission of synchronous fluorescence has red shift (from 278 to 281 nm), illustrating that a typical tryptophan residue of XYN11A has moved from a hydrophobic environment to a more hydrophilic or polar environment. Therefore, the conformation of tryptophan residues of XYN11A may affect the enzyme activity. It can be seen from Fig. 3c the tryptophan fluorescence quenching of XYN11A with increased concentration of Fe3+ resulted in the enhancement of inhibition xylanase activity, so it may be essential for the structure of the active site responsible for the activity of enzyme. The emission peaks of the synchronous fluorescence spectra of XYN11A with various amounts of Fe3+ did not shift, which indicated that Fe3+ has little effect on the microenvironment of the tryptophan residues in xylanase. Based on the multiple alignments of xyn11A, tryptophan 83 maybe was an essential amino acid to be responsible for interaction between XYN11A and Fe3+ to change the xylanase activity, because it was closest to the putative catalytic residues Glu 106 (Fig. 5). Further insight into the tertiary structure of XYN11A may be obtained from more fluorescence quenching mechanism experiments.
The xylanase XYN11A hydrolyzed xylans randomly and rapidly degraded xylotetraose (X4), xylopentose (X5), and xylohexaose (X6), but hardly hydrolyzed xylotriose (X3) and xylobiose (X2). This enzyme yielded xylotriose and xylobiose as the main products. Its effect on xylans indicates that it is an endo-β-1,4-xylanase. Similarly, other xylanases from fungi such as Chaetomium sp. and Paecilomyces thermophila hydrolyze xylans to produce predominantly xylobiose and xylotriose [15, 23]. Hemicellulose degradation is a complex process involving the coordination of several enzymes, including xylanases, β-xylosidases, arabinosidases, and glucuronidases [2]. Thus, XYN11A, together with other cellulases, has the potential to increase the efficiency of the enzymatic pretreatment of lignocellulosic biomass during bioconversion. Kumar and Wyman reported the increase in glucose release with xylanase supplementation of cellulase digestion of corn stover [20]. In this study, a low-molecular weight acidophilic and acidic-stable xylanase (XYN11A) was purified to electrophoretic homogeneity from P. oxalicum GZ-2 grown on corncobs, and its gene and amino acid sequence were reported. XYN11A exhibited optimal activity at pH 4.0 and remarkable pH stability under extremely acidic condition (pH 3). XYN11A was identified as an endo-β-1,4-xylanase, releasing xylobiose and xylotriose as the main products when hydrolyzing various xylans. Our results suggested that the change of conformation and microenvironment of tryptophan by adding Fe3+ was a reason to decrease the xylanase activity of XYN11A. Taking advantage of its acidophilic and elevated acidic stability, this enzyme has potential applications in the various industries desiring a low pH condition.
References
Bailey MJ, Biely P, Poutanen K (1992) Interlaboratory testing of methods for assay of xylanase activity. J Biotechnol 23(3):257–270
Beg Q, Kapoor M, Mahajan L, Hoondal G (2001) Microbial xylanases and their industrial applications: a review. Appl Microbiol Biotechnol 56(3):326–338
Belancic A, Scarpa J, Peirano A, Diaz R, Steiner J, Eyzaguirre J (1995) Penicillium purpurogenum produces several xylanases: purification and properties of two of the enzymes. J Biotechnol 41(1):71–79
Biely P (1985) Microbial xylanolytic systems. Trends Biotechnol 3(11):286–290
Cai H, Shi P, Bai Y, Huang H, Yuan T, Yang P, Luo H, Meng K, Yao B (2011) A novel thermoacidophilic family 10 xylanase from Penicillium pinophilum C1. Process Biochem 46(12):2341–2346
Chand P, Aruna A, Maqsood A, Rao L (2005) Novel mutation method for increased cellulase production. J Appl Microbiol 98(2):318–323
Collins T, Gerday C, Feller G (2005) Xylanases, xylanase families and extremophilic xylanases. FEMS Microbiol Rev 29(1):3–23
Dobrev G, Zhekova B, Delcheva G, Koleva L, Tziporkov N, Pishtiyski I (2009) Purification and characterization of endoxylanase Xln-1 from Aspergillus niger B03. World J Microbiol Biotechnol 25(12):2095–2102
Dong X, Meinhardt SW, Schwarz PB (2012) Isolation and characterization of two endoxylanases from Fusarium graminearum. J Agric Food Chem 60(10):2538–2545. doi:10.1021/jf203407p
Driss D, Bhiri F, Elleuch L, Bouly N, Stals I, Miled N, Blibech M, Ghorbel R, Chaabouni SE (2011) Purification and properties of an extracellular acidophilic endo-1,4-β-xylanase, naturally deleted in the “thumb”, from Penicillium occitanis Pol6. Process Biochem 46(6):1299–1306. doi:10.1016/j.procbio.2011.02.022
Eftink (2006) Fluorescence techniques for studying protein structure. In: Suelter CH (ed) Methods of biochemical analysis: protein structure determination, vol 35. John Wiley & Sons, Inc, Hoboken. doi:10.1002/9780470110560.ch3
Engelborghs Y (2003) Correlating protein structure and protein fluorescence. J Fluoresc 13(1):9–16
Guo B, Chen X-L, Sun C-Y, Zhou B-C, Zhang Y-Z (2009) Gene cloning, expression and characterization of a new cold-active and salt-tolerant endo-β-1, 4-xylanase from marine Glaciecola mesophila KMM 241. Appl Microbiol Biotechnol 84(6):1107–1115
Gusakov A (2011) Alternatives to Trichoderma reesei in biofuel production. Trends Biotechnol 29(9):419–425
Jiang Z, Cong Q, Yan Q, Kumar N, Du X (2010) Characterisation of a thermostable xylanase from Chaetomium sp. and its application in Chinese steamed bread. Food Chem 120(2):457–462
Juturu V, Wu JC (2011) Microbial xylanases: engineering, production and industrial applications. Biotechnol Adv 30(6):1219–1227
Khandeparkar RDS, Bhosle NB (2006) Isolation, purification and characterization of the xylanase produced by Arthrobacter sp. MTCC 5214 when grown in solid-state fermentation. Enzyme Microb Technol. doi:10.1016/j.enzmictec.2005.12.008
Kimura T, Ito J, Kawano A, Makino T, Kondo H, Karita S, Sakka K, Ohmiya K (2000) Purification, characterization, and molecular cloning of acidophilic xylanase from Penicillium sp. 40. Biosci Biotech Bioch 64(6):1230–1237
Kumar AR, Hegde SS, Ganesh KN, Khan MI (2003) Structural changes enhance the activity of Chainia xylanase in low urea concentrations. Biochim Biophys Acta Proteins Proteomics 1645(2):164–171. doi:10.1016/s1570-9639(02)00530-7
Kumar R, Wyman CE (2009) Effect of xylanase supplementation of cellulase on digestion of corn stover solids prepared by leading pretreatment technologies. Bioresour Technol 100(18):4203–4213
Laemmli UK (1970) Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227(5259):680–685
Li D, Zhu M, Xu C, Ji B (2011) Characterization of the baicalein-bovine serum albumin complex without or with Cu2+ or Fe3+ by spectroscopic approaches. Eur J Med Chem 46(2):588–599
Li L, Tian H, Cheng Y, Jiang Z, Yang S (2006) Purification and characterization of a thermostable cellulase-free xylanase from the newly isolated Paecilomyces thermophila. Enzyme Microb Technol 38(6):780–787
Liao H, Xu C, Tan S, Wei Z, Ling N, Yu G, Raza W, Zhang R, Shen Q, Xu Y (2012) Production and characterization of acidophilic xylanolytic enzymes from Penicillium oxalicum GZ-2. Bioresour Technol 123(2):117–124
Liu G, Zhang L, Wei X, Zou G, Qin Y, Ma L, Li J, Zheng H, Wang S, Wang C (2013) Genomic and secretomic analyses reveal unique features of the lignocellulolytic enzyme system of Penicillium decumbens. PLoS ONE 8(2):e55185
Lu Y, Feng Q, Cui F, Xing W, Zhang G, Yao X (2010) Interaction of 3′-azido-3′-deamino daunorubicin with human serum albumin: investigation by fluorescence spectroscopy and molecular modeling methods. Bioorg Med Chem Lett 20(23):6899–6904
Luo H, Li J, Yang J, Wang H, Yang Y, Huang H, Shi P, Yuan T, Fan Y, Yao B (2009) A thermophilic and acid stable family-10 xylanase from the acidophilic fungus Bispora sp. MEY-1. Extremophiles 13(5):849–857. doi:10.1007/s00792-009-0272-0
Luo H, Wang Y, Li J, Wang H, Yang J, Yang Y, Huang H, Fan Y, Yao B (2009) Cloning, expression and characterization of a novel acidic xylanase, XYL11B, from the acidophilic fungus Bispora sp. MEY-1. Enzyme Microb Technol 45(2):126–133
Luo H, Yang J, Li J, Shi P, Huang H, Bai Y, Fan Y, Yao B (2010) Molecular cloning and characterization of the novel acidic xylanase XYLD from Bispora sp. MEY-1 that is homologous to family 30 glycosyl hydrolases. Appl Microbiol Biotechnol 86(6):1829–1839
Möller E, Bahnweg G, Sandermann H, Geiger H (1992) A simple and efficient protocol for isolation of high molecular weight DNA from filamentous fungi, fruit bodies, and infected plant tissues. Nucleic Acids Res 20(22):6115
Marjamaa K, Toth K, Bromann PA, Szakacs G, Kruus K (2013) Novel Penicillium cellulases for total hydrolysis of lignocellulosics. Enzyme Microb Technol 52(6–7):358–369
Miller GL (1959) Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal Chem 31(3):426–428
Nath D, Rao M (1998) Structural and functional role of tryptophan in xylanase from an extremophilic Bacillus: assessment of the active site. Biochem Biophys Res Commun 249(1):207–212
Pokhrel S, Yoo YJ (2009) Designing active site pK(a) values to shift optimum pH of Bacillus circulans xylanase. New Biotechnol 25:126
Rajagopalan G, Yew KW, He J, Yang KL (2013) Production, purification, and characterization of a xylooligosaccharides-forming xylanase from high-butanol-producing strain Clostridium sp, BOH3. Bioenerg Res 6(2):1–10
Ravalason H, Gwénaël J, Daniel M, Maryvonne P, Coutinho PM (2008) Secretome analysis of Phanerochaete chrysosporium strain CIRM-BRFM41 grown on softwood. Appl Microbiol Biotechnol 80(4):719–733. doi:10.1007/s00253-008-1596-x
Ryan SE, Nolan K, Thompson RN, Gubitz GM, Savage AV, Tuohy MG (2003) Purification and characterization of a new low molecular weight endoxylanase from Penicillium capsulatum. Enzyme Microb Technol 33(6):775–785. doi:10.1016/s0141-0229(03)00176-5
Torronen A, Rouvinen J (1995) Structural comparison of two major endo-1,4-xylanases from Trichoderma reesei. Biochem 34(3):847–856
Ujiie M, Roy C, Yaguchi M (1991) Low-molecular-weight xylanase from Trichoderma viride. Appl Environ Microbiol 57(6):1860–1862
Vieira COA, Filho EXF (2003) Purification and characterization of a novel cellulase-free xylanase from Acrophialophora nainiana. FEMS Microbiol Lett 223(2):309–314
Von Gal Milanezi N, Mendoza DPG, de Siqueira FG, Silva LP, Ricart CAO, Filho EXF (2012) Isolation and characterization of a xylan-degrading enzyme from Aspergillus niger van tieghem LPM 93 with potential for industrial applications. Bioenerg Res 5(2):363–371
Wang S, He J, Cui Z, Li S (2007) Self-formed adaptor PCR: a simple and efficient method for chromosome walking. Appl Environ Microbiol 73(15):5048–5051
Wong K, Tan L, Saddler JN (1988) Multiplicity of β-1,4-xylanase in microorganisms: functions and applications. Microbiol Mol Biol Rev 52(3):305–317
Ximenes FA, de Sousa MV, Puls J, da Silva Jr FG, Filho EXF (1999) Purification and characterization of a low-molecular-weight xylanase produced by Acrophialophora nainiana. Curr Microbiol 38(1):18–21
Zhang F, Shi P, Bai Y, Luo H, Yuan T, Huang H, Yang P, Miao L, Yao B (2011) An acid and highly thermostable xylanase from Phialophora sp. G5. Appl Microbiol Biotechnol 89(6):1851–1858
Acknowledgments
This research was financially supported by the Agricultural Ministry of China (2011-G27), Special Fund for Agro-scientific Research in the Public Interest (201203001), National key technology R&D program (L020130249), and National Undergraduate Innovative Experiment Program (201310307020).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Liao, H., Sun, S., Wang, P. et al. A new acidophilic endo-β-1,4-xylanase from Penicillium oxalicum: cloning, purification, and insights into the influence of metal ions on xylanase activity. J Ind Microbiol Biotechnol 41, 1071–1083 (2014). https://doi.org/10.1007/s10295-014-1453-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10295-014-1453-0